
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailored fabrication of TiO2@carbon nanofibers composites via foaming agent migration
Xianhang Sui *,
Xianhua Li,
Liang Chen,
Guangyou Li,
Fengsen Lin and
Changbo Liu
*,
Xianhua Li,
Liang Chen,
Guangyou Li,
Fengsen Lin and
Changbo Liu
Weihai Guangwei Composites Co., Ltd, Weihai 264200, China. E-mail: suixianhang@163.com; Fax: +86 0631 5298266; Tel: +86 0631 5298266
First published on 23rd October 2017
Abstract
Based on the merits and demerits of conventional methods for fabricating TiO2@carbon nanofibers (TiO2@CNFs) composites, the composites were prepared via a foaming-assisted electrospinning strategy and subsequent thermal treatment. Betaine was used as the foaming agent and assembled with TiO2 before electrospinning. It was found that the foaming agent of betaine introduced within the solutions played a crucial role on the migration of TiO2 nanoparticles to the surface of CNFs, enabling their regular movement when the carbonization temperature increased from 400 to 1000 °C. The TiO2 loading amount on the surface of CNFs increased gradually with an enhanced temperature, resulting in an increase in specific surface area from 30.09 to 419.8 m2 g−1. In addition, the obtained TiO2@CNFs composites exhibited excellent hydrophilicity and high degree of graphitization. Accordingly, when the carbonization temperature increased, the composites presented an enhanced photocatalytic activity toward the degradation of rhodamine B. Hence, a novel method for preparing TiO2@CNFs composites with tailored TiO2 loading amount could be envisioned.
1. Introduction
Civilization dictates the progress of society, which results in more production from textile industry with increasing quantity of discharge of wastewater and organic dyes.1 As an effective approach, photocatalytic degradation has been employed for effluent treatment, where most of the photocatalysts were clean, economical and environment-friendly. Oxide semiconductor materials have been gradually developed into a promising approach to solve environmental protection issues, such as air purification, hazardous waste remediation and water purification.2–4 Among the various semiconductor photocatalysts, TiO2 has been extensively studied for the removal of toxic dyes due to its high chemical stability, controllable structure, nontoxicity and low cost.5 However, the large band gap prevents it from practical applications, i.e., the issue of recombination of photogenerated charge carriers has not been fully overcome.6To overcome such limitations, various approaches have been studied to improve the photocatalytic activity by extending the photoresponsive range and increasing the electron–hole extending pair separation efficiency.7–10 Lee et al. demonstrated that electrostatic layer-by-layer deposition effectively immobilized and stabilized TiO2 nanoparticles on polymer nanofibers for degradation of bisphenol A.11 One-dimensional carbon nanofibers (CNFs) have been used for the development of load-bearing nanoparticles due to their good conductive property, high adsorptive capacities and mechanical strength as well as strong fatigue resistance.12–15 Electrospinning was reported as a straightforward technique that could produce continuous fibers with fine flexibility, low density, high porosity, and ease of scalable synthesis.16–18 Therefore, electrospinning technique combined with thermal treatment provided a promising and cost-effective approach to prepare continuous CNFs loaded with TiO2 nanoparticles (TiO2@CNFs composites).19–21 In recent years, there have been two main approaches to prepare TiO2@CNFs composites via electrospinning technique: blended spinning22 and post-assembly pathways.23 It is well known that most of the TiO2 nanoparticles were embedded inside the CNFs in blended spinning method and part of TiO2 nanoparticles could fall off from CNFs in post-assembly pathways. Therefore, the preparation and optimization of TiO2@CNFs composites still remains a challenge.
Based on these considerations, the objective of this study is to demonstrate a novel TiO2@CNFs composite formed by the combination of foaming-assisted electrospinning strategy and thermal treatment involving pre-oxidation and carbonization. Betaine was used as the foaming agent and assembled with TiO2 before electrospinning. Further, on calcination, the abundant vapors released from betaine could drive TiO2 nanoparticles from within the surface of the CNFs, leading to formation of tailored TiO2 loading amount composites. The morphology, structure and photocatalytic activity of TiO2@CNFs composites with different heat treatment temperatures were then investigated.
2. Experimental section
2.1 Materials
Polyacrylonitrile (PAN, Mw = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was provided by Shaoxing Gimel Advanced Materials Technology Co., Ltd, China. TiO2 nanoparticles (20–50 nm) were purchased from Shanghai Titanium Dioxide Chemical Products Co., Ltd. Betaine was provided by Tianjin Tianzhi Fine Chemical Co., Ltd. N,N-Dimethylformamide was used as a solvent, purchased from Tianjin Fuyu Chemical Co., Ltd. Rhodamine B (RhB) was provided by Tianjin Fengchuan Chemical Reagent Science And Technology Co., Ltd. All chemicals were used as received.
000) was provided by Shaoxing Gimel Advanced Materials Technology Co., Ltd, China. TiO2 nanoparticles (20–50 nm) were purchased from Shanghai Titanium Dioxide Chemical Products Co., Ltd. Betaine was provided by Tianjin Tianzhi Fine Chemical Co., Ltd. N,N-Dimethylformamide was used as a solvent, purchased from Tianjin Fuyu Chemical Co., Ltd. Rhodamine B (RhB) was provided by Tianjin Fengchuan Chemical Reagent Science And Technology Co., Ltd. All chemicals were used as received.
2.2 Preparation of TiO2@CNFs composites
A certain amount of TiO2 nanoparticles were dispersed in betaine solution and sonicated for 1 h to obtain a homogeneous solution (10 wt%). Then, the resulting mixtures were stirred for 24 h and dried at 80 °C overnight.A homogeneous solution of PAN and betaine–TiO2 mixtures (12 wt%) dissolved in N,N-dimethylformamide was constantly stirred at room temperature for 24 h. Then, the nanofibers were electrospun onto the aluminium foil. The voltage, feed rate and collection distance were 25 kV, 1 mL h−1 and 15 cm, respectively. After electrospinning, the blend fibers were dried at 60 °C under vacuum overnight.
The dried blend fibers were stabilized in an air atmosphere at 270 °C for 4 h with a ramp rate of 2 °C min−1 in order to induce dimensional stability of the carbon nanofibers (CNFs). Then, the above stabilized fibers were carbonized in a tube furnace at 400, 600, 800 and 1000 °C for 2 h under N2 atmosphere with a ramp rate of 5 °C min−1, respectively. The preparation process of TiO2@CNFs composites is shown in Fig. 1.
 | ||
| Fig. 1 Schematic illustrations of the synthetic pathways of TiO2@CNFs composites. | ||
2.3 Characterization
The morphology of the as-prepared composites was observed using scanning electron microscope (SEM, Hitachi S-4800) and transmission electron microscope (TEM, JEM-2100). The Brunauer–Emmett–Teller (BET) specific surface areas were examined using N2 adsorption–desorption isotherms with Micromeritics Analytical Services (TriStar 3000, GA). X-Ray Diffraction (XRD) patterns of composites were recorded at a scanning speed of 5° min−1 from 3 to 80° with Cu Kα radiation (λ = 1.54059 Å). Raman spectrum was used to investigate the crystallinity of carbon nanofibers with an excitation wavelength of 514 nm. The water contact angles were tested using an image processing program based on the sessile-drop method (JYSP-180 Contact Angle Analizer). At least five contact angle values of different spots were collected to obtain the final contact angle value.24,252.4 Photocatalytic tests
The photocatalytic activity of the as-prepared composites was measured by degrading RhB under the irradiation of a 250 W ultraviolet lamp. The obtained composites (2 cm × 2 cm) were mixed with a solution of 10 mg L−1 RhB using a mechanical shaker in the dark for 60 min to ensure a good dispersion and establish adsorption–desorption equilibrium between the organic molecules and the catalyst surfaces.26 Then, the mixtures were exposed to UV light. After UV irradiation, 3 mL dye solution was withdrawn at given intervals of illumination (10 min). Finally, the concentrations of RhB aqueous solution were recorded by UV spectrophotometer (Shimadzu UV-2450, Japan) at the maximum adsorption wavelength (λ = 554 nm) according to Lambert–Beer's law.27 In the entire photodegradation process, the temperature of the solution was kept at 20 ± 2 °C.3. Results and discussion
3.1 Morphology and structure of TiO2@CNFs composites
Fig. 2 shows the SEM images of TiO2@CNFs composites prepared at different carbonization temperatures. Clearly, the content of TiO2 nanoparticles on the surface of CNFs could be viewed, indicating that the TiO2 loading amount on the surface of CNFs was enhanced when the carbonization temperature increased. During the carbonization process, betaine would be decomposed and rapidly volatilized. The outward diffusion of some gas generated a force to migrate the TiO2 nanoparticles outside. In this case, it could be assumed that the interface layer between the TiO2 and carbon nanofibers would change due to the increased temperature. Therefore, the composites with tailored TiO2 loading amount could be obtained. It is interesting to note that when the carbonization temperature was 1000 °C, the TiO2 nanoparticles were agglomerated and became larger (red circles in Fig. 2d), indicating that a small amount of the TiO2 nanoparticles could fall off. The SEM images not only presented a uniform distribution of TiO2 nanoparticles on the surface of CNFs with further increasing the carbonization temperature, but also showed that most of the carbon nanofibers were bonded to each other, which could affect the results of photocatalytic tests.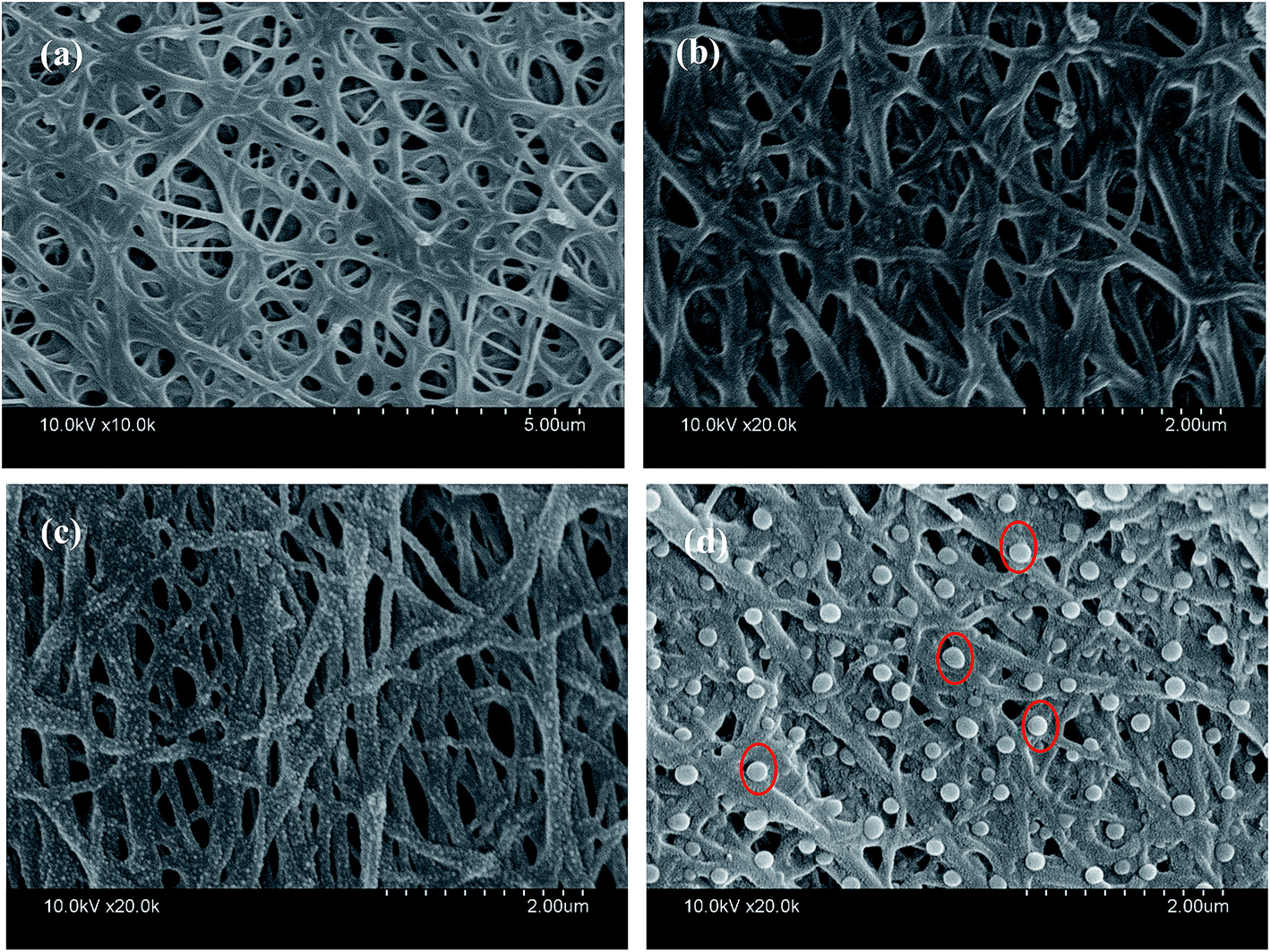 | ||
| Fig. 2 SEM images of TiO2@CNFs composites at (a) 400 °C, (b) 600 °C, (c) 800 °C, and (d) 1000 °C. | ||
The relevant N2 adsorption–desorption isotherm curves of the TiO2@CNFs composites are shown in Fig. 3, exhibiting a type IV isotherms with a variety of typical adsorptive behaviours, such a monolayer adsorption. This indicated that the mesoporous structure could exist within the composites,28 which were formed by the betaine volatilizing with the increasing carbonization temperature. Table 1 shows the calculated BET specific surface areas, presenting a dramatic increasing trend. Notably, the specific surface areas of composites increased from 30.09 to 419.8 m2 g−1 with the increase in carbonization temperature. Such large changes could be explained by the gradual migration of TiO2 nanoparticles to the surface of CNFs, which formed the rough structures, indicating that a small amount of photocatalytic activity would be expected.
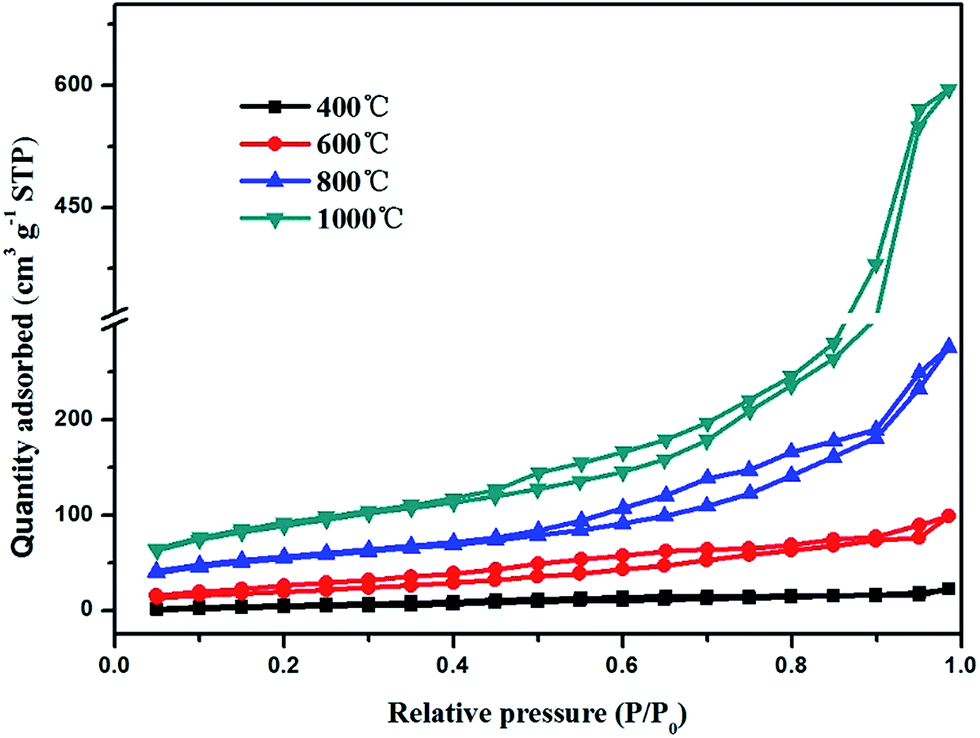 | ||
| Fig. 3 N2 adsorption–desorption isotherms of TiO2@CNFs composites. | ||
| 2θ (°) | d002 (nm) | ID/IG | Specific surface area (m2 g−1) | |
|---|---|---|---|---|
| 400 °C | 25.18 | 0.3496 | 1.339 | 30.09 |
| 600 °C | 25.30 | 0.3479 | 1.1913 | 113.2 |
| 800 °C | 25.33 | 0.3475 | 1.035 | 287.4 |
| 1000 °C | 25.38 | 0.3469 | 0.9833 | 419.8 |
The TEM images in Fig. 4 reveal clear lattice fringes of the nanocrystal with an interplanar space of 0.35 nm (ref. 29) when the carbonization temperature changed from 400 to 800 °C. The fringes correspond to the (101) crystal plane of anatase TiO2 nanoparticles. When the carbonization temperature was increased to 1000 °C, the interplanar space changed to 0.32 nm, indicating that part of the crystal type has transformed to rutile TiO2.30 In particular, for the composite prepared at high carbonization temperature, it could be seen that the ultrasonic process during the sample preparation for TEM examination hardly had an effect on the TiO2 nanoparticles; hence, they did not fall off the CNFs, implying that the interface between TiO2 nanoparticles and CNFs was strong. In addition, for the composite prepared at high carbonization temperature, the migration process of TiO2 nanoparticles could be viewed visually from the TEM pictures, which was consistent with the results of SEM tests.
 | ||
| Fig. 4 TEM showing the TiO2 nanocrystals in (101) orientation at (a) 400 °C, (b) 600 °C, (c) 800 °C, and (d) 1000 °C. | ||
The crystalline structure of composites could be analysed by the XRD method. Fig. 5a shows the XRD patterns of TiO2@CNFs composites at different carbonization temperatures, which were consistent with the TiO2 anatase phase (JCPDS no. 21-1272), indicating that the crystalline structure of TiO2 nanoparticles remained unchanged with the increasing carbonization temperature. Nevertheless, when the carbonization temperature was increased up to 1000 °C, the crystal type of TiO2 nanoparticles was transformed into rutile phase (JCPDS no. 21-1276), which was consistent with the results of TEM analyses. It is well known that anatase TiO2 is in favour of photocatalytic degradation, indicating that a slight decrease in photocatalytic activity of the composites would occur when the carbonization temperature was 1000 °C. The interlayer distance d002 of carbon fibers, which was calculated from XRD spectra using Bragg equations,31 is shown in Table 1. As it is well known, the carbon fiber has a broad peak at 2θ of 25.6°, which is attributed to the lattice plane of graphitized carbon (002).32,33 It was evident that the diffraction peaks of anatase TiO2 (101) and carbon fiber (002) were coincident. In addition, it was reported that the changes in diffraction peak at about 25.6° in TiO2@CNFs composites mainly resulted from the enhancement in degree of graphitization of CNFs.34 It could be observed from Table 1 that the d002 interlayer spacing, indicative of the degree of graphitization,35 decreased from 0.3496 to 0.3469 when the carbonization temperature increased from 400 to 1000 °C, indicating the improvement of graphitization degree. Hence, the improved photocatalytic activity of composites would be expected.
 | ||
| Fig. 5 XRD patterns (a) and Raman spectra (b) of TiO2@CNFs composites. | ||
Raman spectroscopy of TiO2@CNFs composites, which were prepared at different carbonization temperatures, is demonstrated in Fig. 5b. As shown in Fig. 5b and Table 1, the degree of graphitization of composites increased gradually due to the lower ID/IG value when the carbonized temperature ranged from 400 to 1000 °C, where the ID and IG represent the intensity of defect graphite structure and sp2 hybridized carbon,36 respectively. Notably, the ID/IG values of composites decreased, confirming the transformation of disordered carbon into graphitized carbon with an enhanced carbonization temperature,37 which matched well with the abovementioned XRD results.
3.2 Hydrophilicity of TiO2@CNFs composites
The water contact angle was an important parameter for testing the surface hydrophilicity of the as-prepared composites. The contact angle results are shown in Fig. 6, from which a clear trend could be drawn that the contact angle values of TiO2@CNFs composites decreased gradually when the carbonization temperature increased. Notably, when the carbonization temperature increased above 800 °C, the contact angle values were almost 0°. It should be ascribed to the hydrophilic TiO2 nanoparticles migrated to the surface of CNFs to reduce the interface energy with the increase in carbonization temperature, making the composite surface hydrophilic, which was consistent with the abovementioned SEM and TEM results. The results also presented that TiO2 nanoparticles were anchored firmly on the surface of the CNFs, confirming that the interface between TiO2 nanoparticles and CNFs was strong, indicating that the hydrophilic property of TiO2@CNFs composites could play a favourable role in elevating the photocatalytic activity.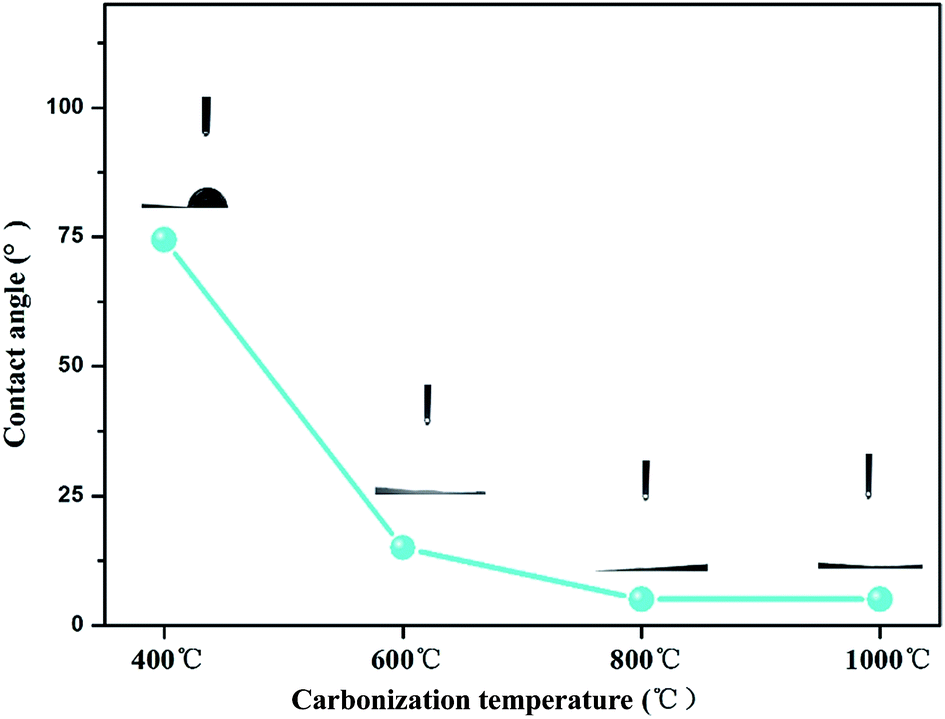 | ||
| Fig. 6 Contact angles of TiO2@CNFs composites at different carbonization temperatures. | ||
3.3 Photocatalytic activity of TiO2@CNFs composites
The photocatalytic activities of the TiO2@CNFs composites were evaluated by the degradation of RhB. Fig. 7a shows the plots of the time-dependent degradation efficiencies of RhB over the four photocatalysts, which clearly show the degradation trends. For the first 60 min, the degradation rate changed slightly, indicating that the absorption–desorption equilibrium between the RhB solution and the composite surfaces was achieved. The photocatalytic activities of TiO2@CNFs composites gradually increased with an increase in temperature, which was attributed to the gradual increase in degree of graphitization and TiO2 loading amount on the surface of CNFs. Fantastically, in spite of the transformation of TiO2 crystal structure, the photocatalytic activities of the composites improved, indicating that TiO2 loading amount on the surface of CNFs was vital for enhancing photocatalytic activities. However, when the carbonization temperature was 1000 °C, the degradation rate was only 87.9%, which could be owing to the fact that part of TiO2 nanoparticles had fallen off and most of the carbon nanofibers were bonded to each other. Hence, the parameters of thermal treatment and the addition of betaine–TiO2 mixture would be further investigated in our later study.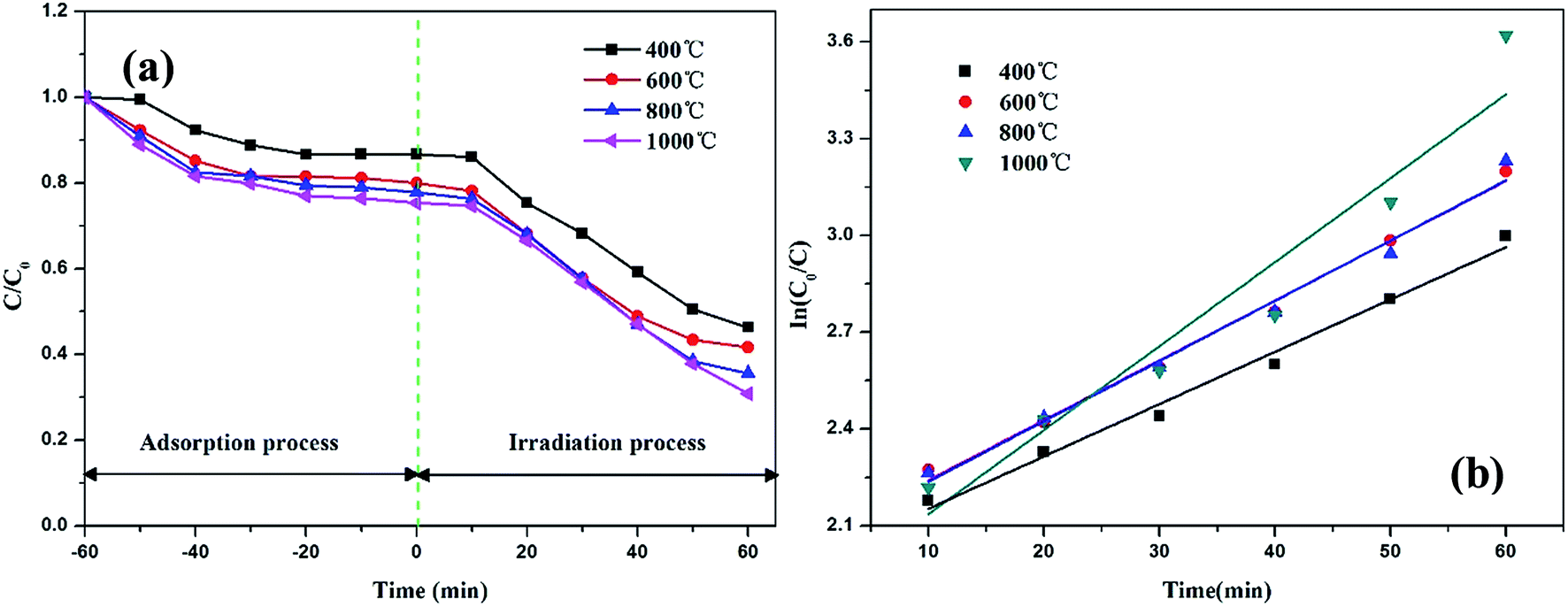 | ||
| Fig. 7 Photocatalytic degradation profiles (a) and kinetic linear fitting curves (b) of RhB over TiO2@CNFs composites. | ||
From the kinetic studies on the photocatalytic reactions over the four samples, their photocatalytic performances could be investigated by the Langmuir–Hinshelwood pseudo-first-order kinetics model38 using the equation ln(C0/C) = kt (where C0 and C are the concentrations of the initial and real-time RhB, respectively, and k is the apparent rate constant). From Fig. 7b, it can be observed that the corresponding reaction rate constants were calculated as 0.019, 0.025, 0.023 and 0.029 min−1 with correlation coefficients (R2) of 0.996, 0.994, 0.991 and 0.988, respectively. These results implied that the photocatalytic performances of TiO2@CNFs composites improved as the carbonization temperature increased, which matched well with the photocatalytic activities illustrated in Fig. 5a.
Fig. 8 shows the schematic illustration of the mechanism of photocatalytic activity of TiO2@CNFs composites for RhB solution. First, the RhB organics were adsorbed to the surface of CNFs, which was ascribed to the high specific area of composites. Second, when the TiO2 nanoparticles were illuminated by UV light with higher photon energy than the band gap of TiO2 nanoparticles, electrons in the valence band (VB) could be excited to the conduction band (CB), resulting in improvement in the photocatalytic activity of TiO2 nanoparticles.39 In addition, there have been some reports that CNFs could introduce UV-generated electrons (e−) into the conduction band (CB) of TiO2 nanoparticles,40,41 indicating that the electron–hole pair separation efficiency increased, which could improve the photocatalytic activity of composites. These electrons transferred between CNFs and TiO2 nanoparticles could react with O2, producing the highly reactive superoxide radical ions (˙O2−). Simultaneously, the positively charged holes (h+) could be generated due to the transfer of electrons from the valence band of TiO2 nanoparticles to the conduction band of CNFs. These positively charged holes (h+) might then react with the OH− produced by the dissociation of H2O, which could lead to the formation of the hydroxyl radical (˙OH). In conclusion, CNFs could play an important role in forming highly reactive superoxide radical ions (˙O2− and ˙OH).
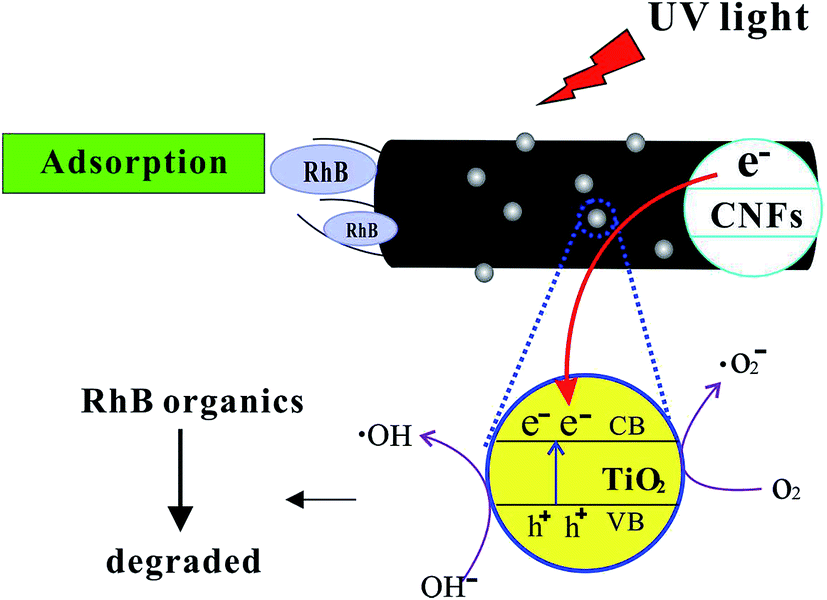 | ||
| Fig. 8 Proposed mechanism of photocatalytic degradation of RhB. | ||
4. Conclusions
We have reported a simple method to fabricate TiO2@CNFs composites as photocatalysts via the foaming-assisted electrospinning technique and a subsequent thermal treatment process. The introduced betaine used as the foaming agent in the initial solutions played an important role in the formation of the composites with tailored TiO2 loading amount, enabling effective migration of TiO2 to the surface of CNFs. When the carbonization temperature ranged from 400 to 1000 °C, the TiO2 loading amount on the surface of CNFs increased, resulting in an increase in specific surface area from 30.09 to 419.8 m2 g−1. In addition, the obtained TiO2@CNFs composites exhibited excellent hydrophilicity and high degree of graphitization. The investigation of the photocatalytic degradation of RhB disclosed that the composites presented an enhanced photocatalytic activity when the carbonization temperature increased. In addition, at a temperature of 1000 °C, the composites showed the best photocatalytic activity. It is promising that the present study could provide some useful insight for the exploration of high loading amount of photocatalysts with excellent performances.Conflicts of interest
The authors declare that they have no conflict of interest to this study.References
- Z. Xu, X. Li, K. Teng, B. Zhou, M. Ma, M. Shan, K. Jiao, X. Qian and J. Fan, J. Membr. Sci., 2017, 535, 94–102 CrossRef CAS.
- J. Li, W. Ma, C. Chen, J. Zhao, H. Zhu and X. Gao, J. Mol. Catal. A: Chem., 2007, 261, 131–138 CrossRef CAS.
- B. Cheng, Y. Le and J. Yu, J. Hazard. Mater., 2010, 177, 971–977 CrossRef CAS PubMed.
- D. Wang, X. Li, J. Chen and X. Tao, Chem. Eng. J., 2012, 198–199, 547–554 CrossRef CAS.
- X. Li, B. Zhou, W. Wang, Z. Xu, N. Li, L. Kuang, C. Li, W. Mai, H. Fu and H. Lv, J. Alloys Compd., 2017, 706, 103–109 CrossRef CAS.
- R. Sedghi, H. R. Moazzami, S. S. Hosseiny Davarani, M. R. Nabid and A. R. Keshtkar, J. Alloys Compd., 2017, 695, 1073–1079 CrossRef CAS.
- G. Zhou, X. Liu, C. Nan, Y. Liu, D. Wang and X. Chen, New J. Chem., 2013, 37, 2582–2588 RSC.
- H. Liang, D. Yu, J. Bai, C. Li and T. Ma, Compos. Interfaces, 2015, 22, 663–671 CrossRef CAS.
- S. Dutta, R. Sahoo, C. Ray, S. Sarkar, J. Jana, Y. Negishi and T. Pal, Dalton Trans., 2015, 44, 193–201 RSC.
- A. Morawski, M. Janus, B. Tryba, M. Toyoda, T. Tsumura and M. Inagaki, Pol. J. Chem. Technol., 2009, 11, 46–50 Search PubMed.
- J. A. Lee, Y. S. Nam, G. C. Rutledge and P. T. Hammond, Adv. Funct. Mater., 2010, 20, 2424–2429 CrossRef CAS.
- Y. Xiong, J. Qian, Y. Cao, X. Ai and H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 16684–16689 CAS.
- P. Zhang, C. Shao, Z. Zhang, M. Zhang, J. Mu, Z. Guo and Y. Liu, Nanoscale, 2011, 3, 2943–2949 RSC.
- S. A. Patil, S. Chigome, C. Hagerhall, N. Torto and L. Gorton, Bioresour. Technol., 2013, 132, 121–126 CrossRef CAS PubMed.
- M. Inagaki, Y. Yang and F. Kang, Adv. Mater., 2012, 24, 2547–2566 CrossRef CAS PubMed.
- X. Li, K. Teng, J. Shi, W. Wang, Z. Xu, H. Deng, H. Lv and F. Li, J. Taiwan Inst. Chem. Eng., 2016, 60, 636–642 CrossRef CAS.
- J. Liu, J. Li, A. Sedhain, J. Lin and H. Jiang, J. Phys. Chem. C, 2008, 112, 17127–17132 CAS.
- K. I. Choi, W. Lee, S. H. Lee and C. Lim, Mater. Lett., 2015, 158, 36–39 CrossRef CAS.
- X. Li, H. Lin, X. Chen, H. Niu, T. Zhang, J. Liu and F. Qu, New J. Chem., 2015, 39, 7863–7872 RSC.
- C. H. Kim, B.-H. Kim and K. S. Yang, Carbon, 2012, 50, 2472–2481 CrossRef CAS.
- T. J. Athauda, J. G. Neff, L. Sutherlin, U. Butt and R. R. Ozer, ACS Appl. Mater. Interfaces, 2012, 4, 6917–6926 CAS.
- F. Mehrpouya, H. Tavanai, M. Morshed and M. Ghiaci, J. Nanopart. Res., 2012, 14, 1074–1084 CrossRef.
- C. Su, X. Ran, J. Hu and C. Shao, Environ. Sci. Technol., 2013, 47, 11562–11568 CrossRef CAS PubMed.
- J. Zhang, Z. Xu, W. Mai, C. Min, B. Zhou, M. Shan, Y. Li, C. Yang, Z. Wang and X. Qian, J. Mater. Chem. A, 2013, 1, 3101 CAS.
- T. Wu, B. Zhou, T. Zhu, J. Shi, Z. Xu, C. Hu and J. Wang, RSC Adv., 2015, 5, 7880–7889 RSC.
- X. Wang, J. Choi, D. R. G. Mitchell, Y. B. Truong, I. L. Kyratzis and R. A. Caruso, ChemCatChem, 2013, 5, 2646–2654 CrossRef CAS.
- R. Zhang, X. Wang, J. Song, Y. Si, X. Zhuang, J. Yu and B. Ding, J. Mater. Chem. A, 2015, 3, 22136–22144 CAS.
- Y. Si, X. Wang, Y. Li, K. Chen, J. Wang, J. Yu, H. Wang and B. Ding, J. Mater. Chem. A, 2014, 2, 645–652 CAS.
- C. Zhao, L. Liu, Q. Zhang, J. Rogers, H. Zhao and Y. Li, Electrochim. Acta, 2015, 155, 288–296 CrossRef CAS.
- H. Hou, M. Shang, L. Wang, W. Li, B. Tang and W. Yang, Sci. Rep., 2015, 5, 15228 CrossRef CAS PubMed.
- J. Jin, Z. Q. Shi and C. Y. Wang, Electrochim. Acta, 2014, 141, 302–310 CrossRef CAS.
- X. Sui, Z. Xu, C. Hu, L. Chen, L. Liu, L. Kuang, M. Ma, L. Zhao, J. Li and H. Deng, Compos. Sci. Technol., 2016, 130, 46–52 CrossRef CAS.
- M. B. Vázquez-Santos, E. Geissler, K. László, J. N. Rouzaud, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2012, 116, 257–268 Search PubMed.
- K. Tang, Y. Li, H. Cao, C. Su, Z. Zhang and Y. Zhang, Electrochim. Acta, 2016, 190, 678–688 CrossRef CAS.
- H. Xiao, Y. Lu, M. Wang, X. Qin, W. Zhao and J. Luan, Carbon, 2013, 52, 427–439 CrossRef CAS.
- L. Liu, F. Wu, H. Yao, J. Shi, L. Chen, Z. Xu and H. Deng, Appl. Surf. Sci., 2015, 337, 241–248 CrossRef CAS.
- J. J. Wang, Y. Y. Shao, Z. B. Wang and Y. Z. Gao, J. Phys. Chem. C, 2008, 112, 5784–5789 CAS.
- Z. Guo, C. Shao, J. Mu, M. Zhang, Z. Zhang, P. Zhang, B. Chen and Y. Liu, Catal. Commun., 2011, 12, 880–885 CrossRef CAS.
- J. Yun, H. I. Kim and Y. S. Lee, J. Mater. Sci., 2013, 48, 8320–8328 CrossRef CAS.
- H. R. P. Bishweshwar Panta, H. Y. K. Yuri Choic and N. A. M. B. Mira Parkc, Ceram. Int., 2013, 39, 7029–7035 CrossRef.
- H. E. Unalan, D. Wei, K. Suzuki, S. Dalal, P. Hiralal, H. Matsumoto, S. Imaizumi, M. Minagawa, A. Tanioka, A. J. Flewitt, W. I. Milne and G. A. J. Amaratunga, Appl. Phys. Lett., 2008, 93, 133116–133119 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |