
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
α-Benzoyloxylation of β-keto sulfides at ambient temperature†
Enrico Pirasa,
Francesco Seccia,
Pierluigi Cabonib,
Maria Francesca Casula a and
Angelo Frongia*a
a and
Angelo Frongia*a
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Complesso Universitario di Monserrato, S.S. 554, Bivio per Sestu, I-09042, Monserrato, Cagliari, Italy. E-mail: afrongia@unica.it
bDipartimento di Scienze della Vita e dell’Ambiente, Università degli Studi di Cagliari, Via Ospedale 72, 09124, Cagliari, Italy
First published on 20th October 2017
Abstract
A facile and efficient protocol for the α-benzoyloxylation of β-keto sulfides is presented. This methodology provides a step-economical, mild and practical access to highly functionalized α-O-benzoyloxy substituted β-keto sulfides, including those with quaternary carbons, which are not easily obtained through currently available methods.
Introduction
Sulfur-containing compounds are key structural cores of bioactive products, medicinally important compounds, and organic materials and thus, of great chemical relevance.1 In particular, α-acyloxy sulfides and their analogues are frequently used in various synthetic applications: as valuable precursors in the preparation of complex synthetic intermediates2 and bioactive natural products.3 Well-established strategies to prepare these compounds include: (i) Pummerer-type rearrangement of alkyl sulfoxides;4 (ii) nucleophilic reaction of α-substituted alkyl sulfides;5 (iii) anti-Markovnikov addition of vinyl esters with arylthiols;6 (iv) addition of aryl disulfides with (acyloxymethyl)magnesium chlorides;7 (v) anodic oxidation of sulfides.8 Recent advances, by Shi et al. report that alkyl sulfides can be converted to α-acyloxy sulfides in the presence of hypervalent iodine reagents and tetra-n-butylammonium bromide.9 Alternatively, Yuan developed an elegant gold-catalyzed iodine(III)-mediated direct oxidative acyloxylation of C(sp3)–H bonds of methyl sulfides.10 It is interesting to point out that it is known that benzoyl peroxide could undergo decomposition in the presence of organic sulfides11 to form several products including: sulfoxides, sulfenic acid, olefins, disulfides and α-benzoyloxy substitution adducts. Surprisingly, despite some scattered procedures,12 typically requiring high reaction temperatures, the relevance of this latter transformation in organic synthesis has been highly overlooked so far. It is likely that this transformation is scarcely described from the point of view of preparation, due to the fact that α-benzoyloxy substituted products are generally obtained in poor chemical yield and/or with low selectivity in combination with significant amounts of the corresponding sulfoxides.In this work we address a general protocol for the synthesis of α-O-benzoyl substituted β-keto sulfides by the reaction strategy shown in Scheme 1. Based on literature, we speculated that the nucleophilic attack of the sulfur atom of a β-keto sulfide 1 on the O–O bond of benzoyl peroxide 2 would form an intermediate sulfonium salt A bearing fairly acidic hydrogens, due to the inductive effect of the positive sulfur and the presence of the carbonyl function. We hypothesize that intermediate A would be deprotonated at ambient temperature by the benzoate anion (Scheme 1, path a), previously formed during the course of the reaction, leading to a sulfur-stabilized carbonium ion B poised to undergo a Pummerer-like rearrangement via intermediate C. Considering that the alternative path b and c (in the case of disubstituted β-keto sulfides), leading to sulfoxide 4 and α-sulfanyl enone 6 respectively, should be less-favored as a result of both the enhanced α-proton acidity of A, as well as the increased electrophilicity of intermediate C, provided by the carbonyl group, the preferential formation of 3 with high level of selectivity can be reasonably expected. If successful, the proposed method would provide a general, straightforward, and mild synthetic route to O-benzoyloxy β-keto sulfides, avoiding the use of catalysts and saving energy as well as allowing transformation to be carried out at ambient temperature in response to the principle of sustainable chemistry.13
 | ||
| Scheme 1 Uncatalyzed synthesis of α-O-benzoyl substituted β-keto sulfides 3. | ||
α-Acyloxy β-keto sulfides are important molecules due to their applications in different synthetic fields. Investigations into the reactivity of monosubstituted α-acyloxy β-keto sulfides have established them as valuable intermediates for the synthesis of α-acyloxy-and α-amino-thioester derivatives (Scheme 2; figure a) which can be easily transformed into the corresponding sulfur-free biologically active products.14 Moreover, disubstituted α-acyloxy β-keto sulfides (Scheme 2; figure b) play a key role as starting materials for the generation of mono-enolized diketones,15 1,2-diketones,16 dicarboxylic acids17 as well as carbocyclic α-hydroxy carboxylic acids.18 Surprisingly, synthetic strategies for the construction of these latter compounds are scarce and exclusively rely on the use of the Pummerer rearrangement19 and the acetoxylation reaction of phenyl thio ketones with lead tetraacetate.20
 | ||
| Scheme 2 α-O-Acyl-and benzoyl substituted β-keto sulfides in organic synthesis. | ||
Results and discussion
As a proof of concept for our study, the reaction of phenythio acetone 1a and benzoyl peroxide 2 was carried out in CH2Cl2 at room temperature and monitored by GC-MS (Table 1, entry 1).
|
|||
|---|---|---|---|
| Entry | Solvent | 3ab (%) | 4ab (%) |
| a Reaction conditions: 1a (333 μmol), benzoyl peroxide 2 (366 μmol), solvent (0.5 mL).b GC-MS yield.c 23% of 1-ethoxy-1-(phenylthio)propan-2-one was also observed (the formation of this product during the reaction is consistent with our mechanistic proposal).d 0.1 mL of solvent was used.e Isolated yield. | |||
| 1 | CH2Cl2 | 78 | 12 |
| 2 | 1,4-Dioxane | 79 | 12 |
| 3 | Toluene | 82 | 8 |
| 4 | CH3COOEt | 76 | 13 |
| 5 | CH3CN | 71 | 21 |
| 6c | EtOH | 46 | 13 |
| 7 | n-Hexane | 67 | 23 |
| 8 | DMF | 74 | 14 |
| 9 | THF | 75 | 12 |
| 10 | DMSO | 45 | 13 |
| 11 | Acetone | 79 | 11 |
| 12d | Toluene | 87 (78)e | 9 |

The investigated reaction took place extraordinarily smoothly without any kind of catalysts to afford 3a in good conversion (78%) along with only a small amount of the expected by-product sulfoxide 4a (12%, via path b, see: Scheme 1), benzoic acid 6 and benzoic anhydride 5. These preliminary data indicated that the reaction is spontaneous, efficient and relatively rapid at room temperature. To confirm the reaction mechanism, selected control experiments were carried out (Scheme 3). In particular, the reaction was unaffected by the presence of radical scavenger such as butylated hydroxy-toluene (BHT). This result indicate that, as expected, the reaction likely proceeds through a nonradical pathway as illustrated in previously reported studies.11 Furthermore, sulfoxide 7 failed to afford the corresponding O-benzoyloxylation products suggesting the requirement of a nucleophilic sulfur atom other than sufficiently acidic protons in alpha position for the success of this transformation at room temperature.
 | ||
| Scheme 3 Control experiments.a aGC-MS yield. | ||
To further improve the control over the reaction and the conversion, the effect of solvent was investigated. All solvents tested in this study can provide good conversions except EtOH (46%) and DMSO (45%). The best result was obtained with toluene. The expected adduct 3a can be obtained in 82% conversion (Table 1, entry 3). Finally, in more concentrated solution the conversion of 3a could be further increased to 87% (78% isolated yield, entry 12). With the optimal conditions for the α-benzoylation of phenylthioacetone in hand, the scope of the transformation was evaluated with respect to the presence of ring substituents on the phenylthio group (Scheme 4). Various O-benzoyl substituted substrates bearing electron-withdrawing (1f, 1g); electron-donating groups (1b–c) and alogens (1d, 1e) on the phenyl ring were synthesized using this methodology with high efficiency (69–78% yields; 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio 84:16–91:9). Moreover, only a slightly decreased chemical yield (62%; 3:4 ratio 91:9) was observed in the case of para-nitro-substituted β-keto sulfide 1f despite the relatively lower nucleophilicity of the sulfur atom. Notably, the steric hindrance was very well tolerated in the case of ortho-; and meta-substituted substrates such as 1i and 1h that gave the corresponding O-benzoyl substituted compounds 3i (81%; 3:4 ratio 86:14) and 3h (85%; 3:4 ratio 87:13) in high chemical yield.
:4 ratio 84:16–91:9). Moreover, only a slightly decreased chemical yield (62%; 3:4 ratio 91:9) was observed in the case of para-nitro-substituted β-keto sulfide 1f despite the relatively lower nucleophilicity of the sulfur atom. Notably, the steric hindrance was very well tolerated in the case of ortho-; and meta-substituted substrates such as 1i and 1h that gave the corresponding O-benzoyl substituted compounds 3i (81%; 3:4 ratio 86:14) and 3h (85%; 3:4 ratio 87:13) in high chemical yield.
 | ||
| Scheme 4 Substrate scope.a,b,c aReaction conditions: 1 (333 μmol), benzoyl peroxide (366 μmol), toluene (0.1 mL). bIsolated yield. c3:4 ratio determined by GC-MS. d3:4 ratio determined by 1H NMR. eThe reaction was carried out for 5 days. | ||
Furthermore, naphthylthio ketone 1j also reacted well, affording the corresponding product 3j in 84% yield (3:4 ratio 96:4). Under the optimized conditions, phenyl selenide ketone 1k was well tolerated and the product 3k was obtained in 88% yield and excellent selectivity (3:4 ratio > 99:1). Linear (1l) and branched (1m) 3-alkyl-substituted β-keto sulfides, despite the relatively greater steric hindrance, were both effective in the reaction leading to the desired products in good to high yields (79–82%; 3:4 ratio 93:7–97:3). To our delight, a representative aromatic α-acyloxy β-keto sulfide 1n, subjected to the same reaction conditions, was smoothly converted into the corresponding product with a good chemical yield (76%; 3:4 ratio 82:18). On the other hand, 2-(phenylthio)acetonitrile 1p and methyl 2-(phenylthio)acetate 1o also reacted with benzoyl peroxide to yield O-benzoyl substituted derivatives 3p (78%; 3:4 ratio 93:7) and 3o (68%; 3:4 ratio 85:15) respectively, albeit much more slowly than the corresponding ketone derivatives, requiring longer reaction times in order to obtain good yields. This seemed to be due to the higher pKa value of the hydrogens adjacent to the ester and cyano group with respect to the corresponding ketone derivatives which disfavor the formation of the required intermediate B (see: Scheme 1). Next, encouraged by the above-mentioned positive results, the applicability of the reaction protocol to other substituted benzoyl peroxides was then preliminary investigated.
As a result, a decreased yield of 3t (68%), and the formation of a significant amount of sulfoxide 4 (3:4 ratio 74:26) was observed in the case of benzoyl peroxides with a strong electron-withdrawing substituent such as –NO2 on the aromatic ring compared to those with electron-donating group such as 3q (77% yield; 3:4 ratio 90:10), 3r (77%; 3:4 ratio 88:12) and alogens for example 3s (81%; 3:4 ratio 84:16). This behavior may be related to the significantly more electrophilic character of the carbonyl of the benzoyl group (provided by the strong electron-withdrawing group) in intermediate sulfonium A which increases the reaction rate of path b (Scheme 1). β-Keto sulfides 1u and 1v, bearing an aliphatic group on sulfur performed very well, and delivered regioselectively the corresponding α-benzoyloxy β-keto sulfide 3u (80%; 3:4 ratio 94:6) and 3v (66%; 3:4 ratio 91:9) in good to high yields. Similarly, 1w also underwent efficient reaction (75% yield; 3:4 ratio 97:3) to afford only the mono benzoyloxylation product 3w.
Finally, the substrate scope was extended to include the synthesis of some challenging α-branched O-benzoyloxy β-keto sulfides. As summarized in Scheme 5, gratifyingly acyclic disubstituted β-keto sulfides 1aa–1ad performed well and delivered the corresponding products 3aa–3ad in moderate to good yields (63–77%; 3:4 ratio 80:20–94:6).
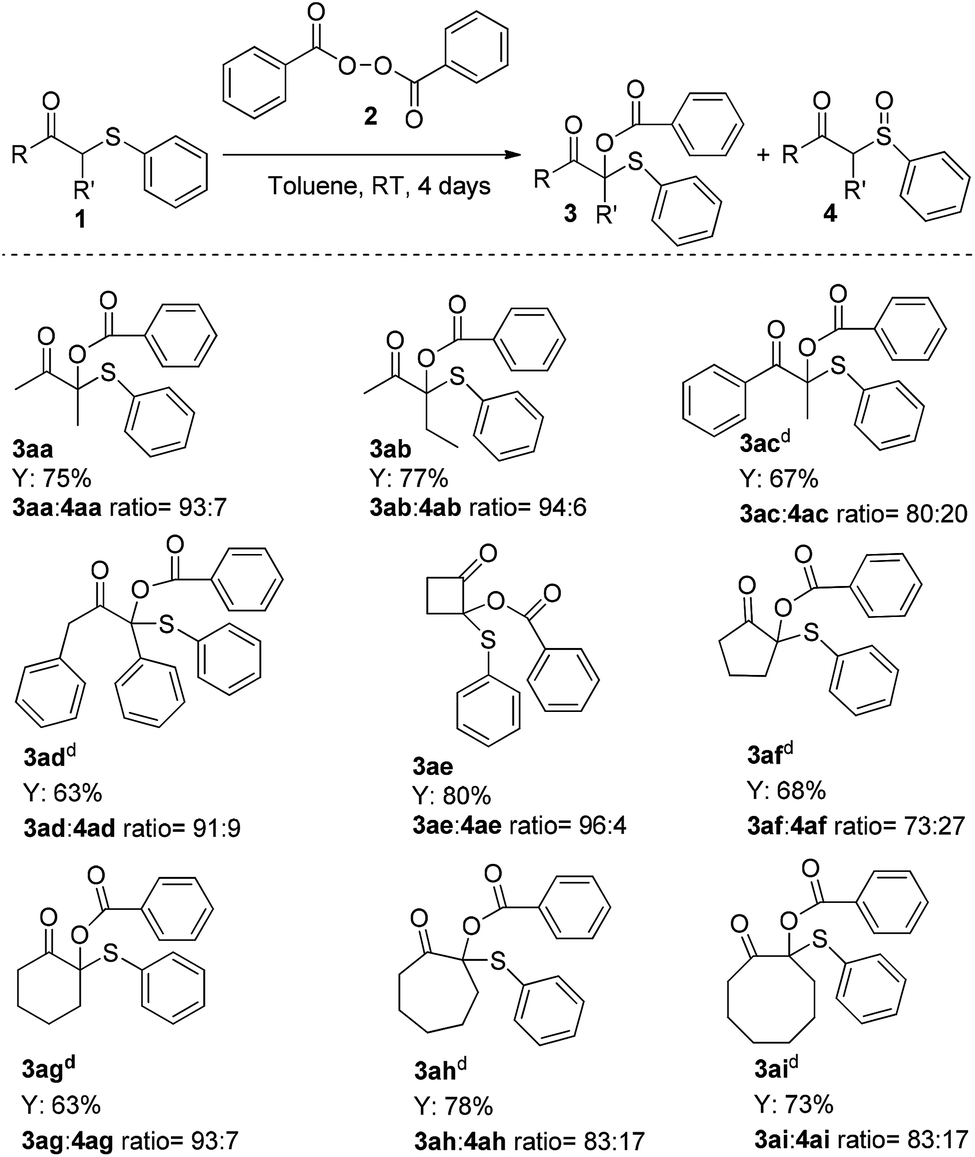 | ||
| Scheme 5 Substrate scope for the formation of quaternary α-branched O-benzoyloxy β-keto sulfides.a,b,c aFollowing the general procedure. bIsolated yield. c3:4 ratio determined by GC-MS. d3:4 ratio determined by 1H NMR. | ||
The reaction proved to be rather general; in addition to acyclic β-keto sulfides, cyclic β-keto sulfides 1ae–1ai also could be used giving the corresponding quaternary adducts21 in moderate to high yields (63–80%; 3:4 ratio 73:27–96:4). Interestingly, under these very mild reaction conditions, in all the cases examined only trace amount of the corresponding α,β-unsaturated sulfide by-product (Scheme 1, path c), which is the major drawback of the Pummerer reaction,19d,22 was detected.
Conclusions
In this work a practical and versatile α-benzoyloxylation of β-keto sulfides under mild conditions is demonstrated. Through this procedure, a series of α-benzoyloxy-β-keto sulfides were obtained in moderate to high yields. Besides, α-branched cyclic and acyclic β-keto sulfides were also compatible with the current protocol and provide the corresponding O-benzoyloxy β-keto sulfides in moderate to high yields. The wide functional group tolerance highlights the potential of this transformation as an effective method for the operationally simple installation of a benzoyloxy group in the α-position of a sulfur atom in the presence of a carbonyl group. We prospect that the impact of our findings will be beyond the research on sustainable organic synthetic approaches, due to the role of sulfur-bearing compounds in bioactive and medicinally relevant compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the MIUR, by the University of Cagliari (FIR 2016-FIR 2017) and from RAS and Fondazione Banco di Sardegna is acknowledged. Sardinia Scientific Park Polaris is thanked for free access to IR spectrophotometer. We thank Dr Danilo Loche for aid in analyses and for useful discussions.Notes and references
- (a) P. Metzner and A. Thuillier, Sulfur Reagents in Organic Synthesis, Academic Press, San Diego, 1994 Search PubMed; (b) R. J. Cremlyn, An Introduction to Organosulfur Chemistry, John Wiley & Sons, Chichester, 1996 Search PubMed; (c) V. K. Aggarwal and C. L. Winn, Acc. Chem. Res., 2004, 37, 611–620 CrossRef CAS PubMed; (d) T. Toru and C. Bolm, Organosulfur Chemistry in Asymmetric synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed; (e) X. Huang, S. Klimczyk and N. Maulide, Synthesis, 2012, 175–183 Search PubMed; (f) L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC; (g) F. Dénès, C. H. Schiesser and R. Renaud, Chem. Soc. Rev., 2013, 42, 7900–7942 RSC; (h) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed; (i) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Andy Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (j) B. M. Trost and M. Rao, Angew. Chem., Int. Ed., 2015, 54, 5026–5043 CrossRef CAS PubMed; (k) L. A. Damani, Sulphur-Containing Drugs and Related Organic Compounds, Wiley, New York, 1989 Search PubMed; (l) H. Liu and X. Jiang, Chem.–Asian J., 2013, 8, 2546–2563 CrossRef CAS PubMed.
- R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed.
- (a) S. Brand, M. F. Jones and C. M. Rayner, Tetrahedron Lett., 1995, 36, 8493–8496 CrossRef CAS; (b) P. Magnus, M. Giles, R. Bonnert, G. Johnson, L. McQuire, M. Deluca, A. Merritt, C. S. Kim and N. Vickert, J. Am. Chem. Soc., 1993, 115, 8116–8129 CrossRef CAS; (c) S. L. Schreiber and K. Satake, J. Am. Chem. Soc., 1984, 106, 4186–4188 CrossRef CAS.
- (a) R. Pummerer, Chem. Ber., 1909, 42, 2282–2291 CrossRef CAS; (b) A. Padwa, D. E. Gunn Jr and M. H. Osterhout, Synthesis, 1997, 1353–1377 CrossRef CAS; (c) S. K. Bur and A. Padwa, Chem. Rev., 2004, 104, 2401–2432 CrossRef CAS PubMed; (d) K. S. Feldman, Tetrahedron, 2006, 62, 5003–5034 CrossRef CAS; (e) S. Akai and Y. Kita, Top. Curr. Chem., 2007, 274, 35–76 CrossRef CAS; (f) L. H. S. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832–5844 CrossRef CAS PubMed; (g) O. de Lucchi, U. Miotti and G. Modena, The Pummerer Reaction of Sulfinyl Compounds, Org. React., 2004, 40(3), 157–405 Search PubMed.
- (a) T. Benneche, P. Strande and U. Wiggen, Acta Chem. Scand., 1988, 43, 74–77 CrossRef; (b) S. Avolio, C. Malan and P. M. Knochel, Synlett, 1999, 1820–1822 CrossRef CAS; (c) G. A. Kraus and H. Maeda, Tetrahedron Lett., 1995, 36, 2599–2602 CrossRef CAS; (d) S. Brand, M. F. Jones and C. M. Rayner, Tetrahedron Lett., 1995, 36, 8493–8496 CrossRef CAS; (e) T. Koizumi, T. Fuchigami and T. Nonaka, Chem. Lett., 1987, 1095–1096 CrossRef CAS; (f) T. Koizumi, T. Fuchigami and T. Nonaka, Bull. Chem. Soc. Jpn., 1989, 62, 219–225 CrossRef CAS; (g) D. Uguen, Tetrahedron Lett., 1984, 25, 541–542 CrossRef CAS.
- R.-S. Lee, J. Chin. Chem. Soc., 1997, 44, 77–80 CrossRef CAS.
- R. Blanc, K. Groll, S. Bernhardt, P. N. Stockmann and P. Knochel, Synthesis, 2014, 1052–1058 Search PubMed.
- (a) J. Nokami, M. Hatate, S. Wakabayashi and R. Okawara, Tetrahedron Lett., 1980, 21, 2557–2558 CrossRef CAS; (b) T. Fuchigami, Y. Nakagawa and T. Nonaka, Tetrahedron Lett., 1986, 27, 3869–3872 CrossRef CAS; (c) T. Fuchigami, K. Yamamoto and Y. Nakagawa, J. Org. Chem., 1991, 56, 137–142 CrossRef CAS; (d) T. Fuchigami, K. Yamamoto and H. Yano, J. Org. Chem., 1992, 57, 2946–2950 CrossRef CAS.
- D. Zhu, D. Chang, S. Gan and L. Shi, RSC Adv., 2016, 6, 27983–27987 RSC.
- S.-R. Guo, P. S. Kumar, Y.-Q. Yuan and M.-H. Yang, Eur. J. Org. Chem., 2016, 4260–4264 CrossRef CAS.
- (a) W. A. Pryor and H. T. Bickley, J. Org. Chem., 1972, 37, 2885–2893 CrossRef CAS; (b) D. I. Davies, D. H. Hey and B. Summers, J. Chem. Soc. C, 1970, 2653–2660 RSC; (c) H. B. Henbest, J. A. W. Reid and C. J. M. Stirling, J. Chem. Soc., 1964, 1220–1223 RSC; (d) L. Horner and E. Jürgen, Liebigs Ann. Chem., 1957, 602, 135–153 CrossRef CAS; (e) W. A. Pryor and W. H. Hendrickson Jr, J. Am. Chem. Soc., 1983, 105, 7114–7122 CrossRef CAS; (f) G. Sosnovsky, Tetrahedron, 1962, 18, 15–19 CrossRef CAS.
- (a) K. Gollmer and H. Ringsdorf, Makromol. Chem., 1969, 121, 227–257 CrossRef CAS; (b) Yu. I. Puzin, G. V. Leplyanin, E. A. Kozheparova and G. A. Tolstikov, Dokl. Akad. Nauk SSSR, 1991, 320, 349–352 CAS; (c) M. Hojo, R. Masuda, T. Ichi, K. Yoshinaga, S. Munehira and M. Yamada, Synthesis, 1982, 424–426 CrossRef CAS; (d) N. E. Allen, D. B. Boyd, J. B. Campbell, J. B. Deeter, T. K. Elzey, B. J. Foster, L. D. Hatfield, J. N. Hobbs Jr, W. J. Hornback, D. C. Hunden, N. D. Jones, M. D. Kinnick, J. M. Morin Jr, J. E. Munme, J. K. Swartzendruber and D. G. Vogt, Tetrahedron, 1989, 45, 1905–1928 CrossRef CAS; (e) J. E. Baldwin, C. Lowe and C. J. Schofield, Tetrahedron Lett., 1986, 27, 3461–3464 CrossRef CAS; (f) J. E. Baldwin, M. A. Christie, S. B. Haber and L. I. Kruse, J. Am. Chem. Soc., 1976, 98, 3045–3047 CrossRef CAS PubMed.
- (a) M. B. Gawande, V. D. B. Bonifàcio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24–44 CrossRef CAS PubMed; (b) G. Brahmachari and B. Banerjee, Curr. Green Chem., 2015, 2, 274–305 CrossRef CAS.
- (a) T. Suzuki, Y. Honda, K. Izawa and R. M. Williams, J. Org. Chem., 2005, 70, 7317–7323 CrossRef CAS PubMed; (b) F. Capitta, A. Frongia, P. P. Piras, P. Pitzanti and F. Secci, Org. Biomol. Chem., 2012, 10, 490–494 RSC; (c) F. Capitta, A. Frongia, P. P. Piras, P. Pitzanti and F. Secci, Adv. Synth. Catal., 2010, 352, 2955–2960 CrossRef CAS; (d) F. Capitta, N. Melis, F. Secci, G. Romanazzi and A. Frongia, J. Sulfur Chem., 2014, 35, 649–660 CrossRef CAS.
- Y. Sugihara, A. Yamato and I. Murata, Tetrahedron Lett., 1981, 22, 3257–3260 CrossRef CAS.
- B. M. Trost, C. Pissot-Soldermann and I. Chen, Chem.–Eur. J., 2005, 11, 951–959 CrossRef CAS PubMed.
- B. M. Trost, Chem. Rev., 1978, 78, 363–382 CrossRef CAS.
- B. M. Trost and H. Hiemstra, Tetrahedron, 1986, 42, 3323–3332 CrossRef CAS.
- (a) M. Naora, T. Murae, T. Tsuyuki and T. Takahashi, Bull. Chem. Soc. Jpn., 1986, 59, 1767–1776 CrossRef CAS; (b) T. Patonay, E. Patonay-Péli and G. Litkei, Tetrahedron, 1987, 43, 1827–1834 CrossRef CAS; (c) F. Eiden and F. Meinel, Arch. Pharm., 1979, 312, 302–312 CrossRef CAS; (d) S. Ito, Y. Kubota and M. Asami, Chem. Lett., 2016, 45, 16–18 CrossRef CAS; (e) B. M. Trost and A. J. Bridges, J. Am. Chem. Soc., 1976, 98, 5017–5019 CrossRef CAS; (f) G. H. Posner, E. Asirvatham and S. F. Ali, J. Chem. Soc., Chem. Commun., 1985, 542–543 RSC.
- (a) M. D. Lawlor, T. W. Lee and R. L. Danheiser, J. Org. Chem., 2000, 65, 4375–4384 CrossRef CAS PubMed; (b) B. M. Trost and A. J. Bridges, J. Am. Chem. Soc., 1976, 4, 5017–5019 CrossRef; (c) D. G. McCarthy, C. C. Collins, J. P. O'Driscoll and S. E. Lawrence, J. Chem. Soc., Perkin Trans. 1, 1999, 3667–3675 RSC; (d) P. A. Wender, H. Kogen, H. Y. Lee, J. D. Munger Jr, R. S. Wilhelm and P. D. Williams, J. Am. Chem. Soc., 1989, 111, 8957–8958 CrossRef CAS; (e) D. M. X. Donnelly, B. M. Fitzpatrick, B. A. O'Reilly and J.-P. Finet, Tetrahedron, 1993, 49, 7967–7976 CrossRef CAS.
- J.-S. Yu, H. M. Huang, P.-G. Ding, X.-S. Hu, F. Zhou and J. Zhou, ACS Catal., 2016, 6, 5319–5344 CrossRef CAS.
- J. Durman, J. I. Grayson, P. J. Hunt and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1986, 1939–1945 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterization of new compounds. See DOI: 10.1039/c7ra10888e |
| This journal is © The Royal Society of Chemistry 2017 |