
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of hierarchical porous carbon derived from carboxyl graphene oxide/phenolic foam for high performance supercapacitors†
Xiaozheng Li a,
Xi Li*a,
Jian Zhoua,
Yulin Donga,
Zhizhong Xiea,
Weiquan Caia and
Chaocan Zhang*b
a,
Xi Li*a,
Jian Zhoua,
Yulin Donga,
Zhizhong Xiea,
Weiquan Caia and
Chaocan Zhang*b
aSchool of Chemistry, Chemical Engineering and Life Science, Wuhan University of Technology, Wuhan 430070, PR China. E-mail: chemlixi@whut.edu.cn; Tel: +86 27 87756662
bSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, PR China. E-mail: polymers@whut.edu.cn
First published on 11th September 2017
Abstract
Hierarchical porous carbon materials were prepared using a facile and low-cost method from phenolic resin and carboxyl graphene oxide by foaming and carbonization. Structural characterization revealed that the prepared materials consisted of macropores, mesopores and micropores from the foaming process, decomposition of the ester groups and carbonization of the polymer, respectively. Due to the advantages of a rational-distribution of porous structures and the excellent electrical conductivity, the best specific capacitance of the porous carbon materials could reach up to 272.6 F g−1 at a current density of 0.1 A g−1 and exhibited an excellent cycle life with 92.31% specific capacitance retained after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in KOH electrolyte. The symmetric supercapacitor based on the prepared material also exhibited a high energy density (15.20 W h kg−1) and power density (10.11 kW kg−1), indicating a promising electrode material for real energy storage in supercapacitors.
000 cycles in KOH electrolyte. The symmetric supercapacitor based on the prepared material also exhibited a high energy density (15.20 W h kg−1) and power density (10.11 kW kg−1), indicating a promising electrode material for real energy storage in supercapacitors.
1. Introduction
Supercapacitors, a new type of advanced energy storage device with the characteristics of high energy and power density, long cycle life and non-polluting to the environment, satisfy many requirements of various fields and have become a hot research area.1–4 The energy storage of supercapacitors depends on the adsorption of electrolyte ions on the electrodes and the charge separation at electrode/electrolyte interfaces. This requires the electrodes to have a large specific surface area for high specific capacitance. Therefore, studies on supercapacitors have mainly concentrated on electrode materials with a large specific surface.5 Activated carbons have been widely employed as the electrode materials due to their advantages of large surface area, good electrical conductivity and an abundant source of raw materials.6–8In theory, the larger the special surface area of the electrode is, the higher the specific capacitance of the supercapacitor is.9 Therefore, the initial research always focused on the micropores structure due to its contribution to the special surface,10 but neglected the blocking effect to electrolyte ions.11 However, many studies showed that the specific capacitance of the supercapacitors did not increase with the special surface area of the electrode and almost all of the experimental specific capacitance values were far below the theoretical values even if the special surface area of the electrode was up to 1000–3000 m2 g−1.12,13 Like the nitrogen-doped activated carbon (NACs) prepared by Li et al., the surface area could reach 2900 m2 g−1, but the specific capacitance was only 184 F g−1 at a current density of 0.4 A g−1.14 This is because most of micropores contributing to the larger specific surface area are non-interconnected or closed pores, which caused the low utilization of the specific surface area and thus the small specific capacitance. In fact, during the charging and discharging process, the electrolyte ions cannot easily get through the micropores structure to reach the electrochemical reaction site on the inner surface of the electrode materials, which results in low both power and energy densities.15 Recently, the hierarchical porous carbon is regarded as a promising electrode material for supercapacitors, which could meet these demands of the fast ion diffusion and high charge storage.16 For the hierarchical porous carbon, the macrospores can be regarded as temporary ion repository to accelerate the flow of electrolyte, the mesoporous afford ion-transport pathways to electrochemical reaction site, and the microporous also increase the specific surface area in favor of charge accommodation.17 It was demonstrated that the hierarchical porous carbon materials with larger macropores, moderate mesopores and abundant micropores exhibited the better capacitance performance than the porous carbon materials with uniform pore size.18
Until now, the research about the carbon materials with hierarchical porous structure has made the considerable progress in terms of both the raw materials and the preparation procedure.19,20 Among these, phenolic resins were usually selected as the carbon precursors for the hierarchical porous carbon owing to the high-yield and economical synthesis.21–23 Phenolic resins also have the thermal and chemical stability which is more important to possess abundant micropores from the crosslinking of macromolecular chains.20 In addition, the hierarchical porous carbon derived from phenolic resins can be designed and fabricated through the template24–26 and activation methods.27–29 However, the methods are usually complicated and prone to the isolated non-interconnected pores which are detrimental to the ion transport. Therefore, a simple and effective method to prepare the hierarchical porous carbon with inner-connective porous structure is highly desired.
Besides, the structural damage of the hierarchical porous carbon due to the collapse is also a problem. The reported work indicated that the incorporation of 1D or 2D carbon materials with the small proportion, such as carbon fibers and graphene, could effectively prevent or alleviate the collapse.30–32 Zou et al. synthesized the three-dimensional (3D) N/O-doped activated carbon by adopting silkworm cocoon and phenolic resin, and the silkworm cocoon as carbon fibers after carbonization was used as a good framework to make the composite materials more hierarchical porosity and flexible.33 Therefore, it was only a lower surface area of 499 m2 g−1 but a higher specific capacitance of 239 F g−1 at current density of 0.57 A g−1.
Graphene, a two-dimensional carbon material with the large theoretical surface area, unique electronic conductivity and good electrochemical stability, is considered as a promising candidate for the electrode materials of the supercapacitors. However, graphene is strongly inclined to restack together because of the strong van der Waals forces between sheets and π–π interaction, which leads to a decrease of the effective specific surface area.34,35 Thus, graphene with planar sheets could not reach the theoretical maximum specific capacitance, 550 F g−1.36 Combining graphene with other materials has been proved to be an effective way to prevent or mitigate the aggregation and these graphene-based hybrid materials have the excellent capacitance performance.37–42 For examples, Zhang et al. fabricated a graphene–RF carbon composite with alkali treated graphene oxide as a base catalyst for the polymerization of formaldehyde and phenol, and the composite exhibited excellent rate performance in supercapacitors.40 Qian et al. synthesized a three-dimensional graphene-based aerogels foam by adopting graphene oxide and resin.43 The as-prepared porous carbon owned a large specific surface area of 1019 m2 g−1 and exhibited a higher specific capacitance of 160 F g−1 at 2 A g−1 in KOH without bio-inspired methods and activation methods. The truth is that the resol molecules were first adsorbed on the surface of graphene oxide sheets so that the graphene oxide could not get together easily. In addition, Qin et al. also designed and prepared 3D flexible O/N Co-doped graphene foams via melamine and graphene oxide.44 The best specific capacitance was up to 375 μF cm−2 at a higher current density of 1 A g−1 due to the unique structure and design. That is, the melamine was used not only as the source of O/N, but also as the template to prevent the agglomeration of graphene.
On the basis of the above statement, we present a convenient and effective template-free approach to prepare the hierarchical porous carbon materials by using carboxyl graphene oxide/phenolic resin composite through the simple foaming method and carbonization. As shown in Scheme 1, the phenolic resin was firstly synthesized through the polymerization of paraformaldehyde and phenol and the carboxyl graphene oxide (CGO) was obtained through chemical modification; then CGO was combined with the phenolic resin to prepare CGO/phenolic resin composite by the esterification reaction and hydrogen bond; thirdly, the CGO/phenolic foam (CGO/PF) was formed by the simple foaming method; finally, the hierarchical porous carbon foam (RGPF) was prepared by the carbonization of CGO/PF. Here, the choice of phenol and CGO as the raw materials is attributed to the four reasons: (1) the synthesis of phenolic resins is high-yield and economical, and the mutual crosslinking could lead to the vast microporous structure during the carbonization process because of the thermal and chemical stability; (2) CGO could easily dispersed in the organic solution to avoid the agglomeration, and have strong interfacial interaction with the phenolic resins; (3) the carboxyl groups from CGO could react with the phenolic hydroxyl groups in phenolic resin to form ester groups, and the ester groups will decompose during the carbonization, which leads to the formation of mesopores; (4) CGO could translate into graphene during the carbonization process and the flexible network of graphene could improve the electron conductivity of electrode materials during the charging and discharging process.
 | ||
| Scheme 1 Schematic illustration of the preparation of RGPF. | ||
On balance, mesopores are mainly produced by the decomposition of the ester groups during the carbonization and all most micropores are formed due to carbonization of polymer. It is worth noting that the formation of macropores comprising thin walls during the foaming process is quite important in supercapacitors because they provide the fast accessibility of the electrolyte ions to the material surface, which accords with the demands of the electrodes for the highly capacitive performance. By taking full advantage of the large specific surface, well-defined pore structure and excellent electrical conductivity, the hierarchical porous carbon material as the working electrode showed a good capacitive behavior. In KOH electrolyte, the specific capacitance can reach up to 272.6 F g−1 at a current density of 0.1 A g−1 when the mass fraction of carboxyl graphene oxide was just only 0.2%.
2. Experimental section
2.1 Materials preparation
Graphene oxide (GO) was prepared from the flake graphite according to the Hummers' method.45Carboxyl graphene oxide (CGO) was obtained through chemical modification.46 In a typical preparation, GO (0.75 g) was first dispersed in the double distilled water by the hyperacoustic. Then 50 mL hydrogen bromide was added in the GO suspension and the mixture was stirred for 12 h at room temperature. Following, oxalic acid (15.0 g) was added and the mixture kept stirring for another 4 h. Finally, CGO was obtained after centrifugation, lavation and vacuum drying at 50 °C. The final CGO suspension was obtained by dispersing CGO into absolute alcohol.
The synthesis of CGO/phenolic foam (CGO/PF) was divided into three steps. The first step is the synthesis of phenolic resin.47,48 Paraformaldehyde (F) and phenol (P) were mixed to obtain a homogeneous mixture. Then sodium hydroxide was added stepwisely in the mixture at 65 °C and the polymerization was kept at 85 °C for 3 h. The second step is the incorporation of CGO to phenolic resin by esterification reaction.49 CGO suspension was added in the above mixture and the mixture reacted for 1 h at 85 °C. The third step is the foaming process according to the literatures.50,51 Phenolic resin (80 g) is mixed homogeneously with surfactants, foaming agents and composite curing agents. Then the mixture was put into a rectangular container at 65 °C for 1 h. CGO/PF with different proportions was obtained by changing the initial mass percent of CGO with 0, 0.05%, 0.10%, 0.20% and 0.30%.
Hierarchical porous carbon foam (RGPF) was prepared by carbonizing CGO/PF in N2 atmosphere at a certain temperature (700, 800, 900 and 1000 °C) for 5 h. The obtained RGPF was denoted as RGPF-m-t, where m stands for initial mass percent of CGO and t for the carbonization temperature. For the comparison, carboxyl graphene oxide and phenolic foam were also carbonized at 800 °C and designated as RCGO-800 and PF-800.
In addition, because the amount of F and P could affect the crosslinking of the resins and then the porous structure of the carbon foam, F and P with different ratios (F/P = 1.6, 1.8, 2.0, 2.2, 2.4) was used in the preparation of the phenolic resins. The porous carbon foams were prepared from the obtained phenolic resins through the same foaming and carbonizing process as above. All the results were shown in ESI materials.† The ratio of F and P played a critical role in the formation of the hierarchical porous structure. On the one hand, the smaller ration of F and P would make the crosslinkage of the phenolic resin lower, then the resin difficult to curing and finally the pore structure of the porous carbon easy to collapse during the carbonization, which caused a little amount of pores and a smaller specific surface area as shown in Table S2.† Obviously, the materials with a smaller specific surface area were inappropriate to the electrode materials. On the other hand, the higher ration of F and P would cause a higher crosslinkage of the resin, which was favorable to the formation of the micropores but unfavorable to the foaming of the resin. Therefore, most of these micropores were closed pores which were not conducive to the ion transport. That was the reason why the porous carbon foam exhibited a larger specific surface area and a poor capacitive performance when the ration of F and P was higher than 2.0. As shown in Fig. S3,† the electrochemical measurements indicated that when the ratio of F and P was 2.0, the porous carbon foam derived from the phenolic resin exhibited the best performance. So, F/P = 2.0 was chosen in the preparation of CGO/PF and RGPF.
2.2 Characterization
The surface morphology and microstructure of the prepared materials were observed by an ULTRA PLUS scanning electron microscopy (SEM, Zeiss, Germany) and a JEM2100F transmission electron microscopy (TEM, JEOL, Japan). Fourier transform infrared (FT-IR) spectra were recorded by a Nexus FT-IR spectrometer (Thermo Nicolet, USA). Raman spectra were obtained from an INVIA Raman spectrometer (Renishaw, UK). X-ray photoelectron spectroscopy (XPS) measurements were carried out by a VG Multilab 2000 spectrometer (Thermo, USA). The nitrogen adsorption/desorption experiments were investigated at the liquid nitrogen temperature (77 K) by an automatic ASAP 2460 sorptometer (Micromeritics, USA). The specific surface areas (SBET, m2 g−1) of the prepared materials were determined by using the Brunauer–Emmett–Teller (BET) method. The micropore surface area (Smicro, m2 g−1) and micropore volume (Vmicro, cm3 g−1) were calculated from the t-plot method. The total pore volume (Vtotal, cm3 g−1) was calculated at a relative pressure P/P0 = 0.995. Average pore width (D, nm) was calculated by 4Vtotal/SBET. The pore size distribution (PSD) was calculated using adsorption isotherms by Barrett–Joiner–Halenda (BJH) model.2.3 Electrochemical measurements
The working electrode was prepared by first grounding RGPF-m-t with acetylene black and polytetrafluoroethylene into a uniform thin slice, then placing the slice between two pieces of nickel foam and finally pressing them together under a pressure of 5 MPa to make the electrode active material adhere to the nickel foam. The weight ratio of RGPF-m-t, acetylene black and polytetrafluoroethylene was 18:1:1. The weight of the prepared working electrode was about 9 mg and the surface area was about 1 cm2.
The cyclic voltammetry (CV), galvanostatic charge/discharge (CP) and electrochemical impedance spectroscopy (EIS) were carried out on CHI660D electrochemical workstation (Shanghai CH Instruments Co, China) in KOH solution (6 M). The counter and reference electrodes were platinum column and Hg/HgO electrode, respectively.
The specific capacitance (C, F g−1) was calculated from the CV curves by using eqn (1) as well as from the CP curves according to the eqn (2).
 | (1) |
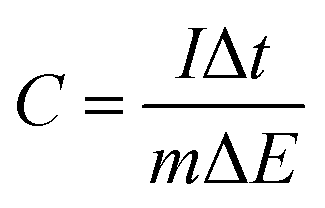 | (2) |
In this paper, the value of the specific capacitance was the average of six samples. From the average specific capacitance, the energy density (E, W h kg−1) and power density (P, W kg−1) were calculated from CP curves of two-electrode system according to the eqn (3) and (4), respectively.
 | (3) |
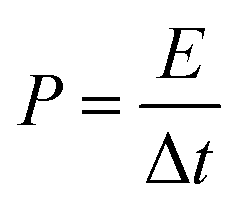 | (4) |
3. Results and discussion
3.1 Materials characterization
FT-IR was employed to investigate the chemical characteristics of the prepared samples. The typical FTIR spectra of RCGO-800, PF-800 and RGPF-0.20%-800 were showed in Fig. 1(a). The three FTIR spectra were similar and in accord with the reported papers.20 In each FTIR spectrum, there was a wide and strong absorption peak at 3440 cm−1, which could be considered as the contribution of the –OH groups residual in phenol and graphene.20,54 The characteristic peak at 1326 cm−1 was mainly contributed by C–O vibration, and the one at 1630 cm−1 corresponded to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration.55,56 In the FTIR spectra of RGPF-0.20%-800, a weak absorption peak was observed at 2340 cm−1, which could be attributed to the residual O–CO groups formed by the esterification reaction between carboxyl graphene oxide and phenolic resin. In addition, compared with CGO and phenolic resin,57,58 the peaks owing to the oxygen groups such as CO (1732 cm−1) and C–O–C (1048 cm−1) disappeared, indicating these oxygen groups were decomposed during the carbonization, which is helpful for the pore formation.54
C vibration.55,56 In the FTIR spectra of RGPF-0.20%-800, a weak absorption peak was observed at 2340 cm−1, which could be attributed to the residual O–CO groups formed by the esterification reaction between carboxyl graphene oxide and phenolic resin. In addition, compared with CGO and phenolic resin,57,58 the peaks owing to the oxygen groups such as CO (1732 cm−1) and C–O–C (1048 cm−1) disappeared, indicating these oxygen groups were decomposed during the carbonization, which is helpful for the pore formation.54
 | ||
| Fig. 1 (a) FT-IR spectra and (b) Raman spectra of RCGO-800, PF-800 and RGPF-0.20%-800. XPS spectra for C 1s region of (c) RCGO-800, (d) PF-800 and (e) RGPF-0.20%-800. | ||
Raman spectroscopy is an effective tool for the characterization of carbon materials. As shown in Fig. 1(b), all the Raman spectra of the three carbon material (RGPF-0.20%-800, PF-800 and RCGO-800) had two obvious peaks located at about 1325 cm−1 and 1586 cm−1. The peak loaded at 1325 cm−1 is regarded as D bands corresponding to sp3 carbon atoms, and it also represents irregular model concerned with structure defects and content. Meanwhile, G bands loaded at 1586 cm−1 is derived from stretching vibration of sp2 carbon atoms, and it certainly indicates order-induced mode.59,60 The intensity ratio I(D)/I(G) is usually served as a standard to evaluate the quality of the graphene structures.61 For RCGO-800, I(D)/I(G) was the highest up to 1.27, meaning the reduction of CGO at 800 in N2 atmosphere. This also could be verified by the obvious 2D bands at 2700 cm−1.62 I(D)/I(G) values of RGPF-0.20%-800 and PF-800 were similar, indicating the incorporation of CGO with small amount had little influence on the Raman spectra.
X-ray photoelectron spectroscopy (XPS) is a powerful method to analysis the elemental composition on the material surface. Fig. 1(c–e) is the C 1s spectra of RCGO-800, PF-800 and RGPF-0.20%-800. Five typical peaks locating at 284.4, 285.1, 286.1, 288.2 and 289.6 eV corresponded to CC, C–C, C–O, CO and O–CC, respectively.52,63 In the three spectra, the two wide and high peaks at 284.4 and 285.1 eV indicated that most of the carbons in the prepared porous materials were aromatic because they owned sp2 carbon atoms and delocalized-bonds.64 The percentages of the groups on the surface were summarized in Table 1. Compared to CGO,58 the percentage distribution of CO and O–CO in RCGO-800 reduced, indicating the reduction of CGO to RCGO during the carbonization. The carboxyl groups were only derived from the CGO. So there was no O–CO in the PF-800. The content of oxygen groups on the surface of RGPF-m-800 was changed with the initial content of CGO. In CGO/PF, the statues of oxygen groups on the surface of RGPF-m-800 was changed with the initial content of CGO. In CGO/PF, the statues of oxygen groups mainly originated from –OH on the phenyl rings, –COOH on CGO and –O–CO from the esterification reaction. Among these oxygen groups, –O–CO was the easiest to thermally decompose.20 When the content of CGO increased from 0.05% to 0.20%, the number of –O–CO in CGO/PF increased and the content of –OH decreased, which caused the content of oxygen decreased after carbonization. However, when the content of CGO increased from 0.2% to 0.30%, the content of oxygen increased because the number of –O–CO in CGO/PF was up to the maximum and extra –COOH was introduced. This also was verified by the contents of C–O, CO and O–CC in the RGPF-m-800. RGPF-0.05%-800 had the largest number of C–O which could come from –OH. In the meantime, RGPF-0.30%-800 had the maximum of CO and O–CC due to –COOH in CGO. The thermolysis of O–CC benefited formation of the micropores during the carbonization. The carbonization temperature also affected the chemical structure of the carbon materials. With the rise of carbonization temperature, the structure of C–O, CO and O–CC would decompose, leading to the oxygen content became less.
| Samples | Element content (at%) | O/C (at%) | C 1s (at%) | |||||
|---|---|---|---|---|---|---|---|---|
| C | O | CC |
C–C | C–O | CO |
O–CO |
||
| RCGO-800 | 93.64 | 6.36 | 6.79 | 45.83 | 42.82 | 5.62 | 3.70 | 2.03 |
| PF-800 | 94.54 | 5.46 | 5.78 | 49.34 | 39.73 | 10.22 | 0.71 | |
| RGPF-0.05%-800 | 92.58 | 7.42 | 8.01 | 45.54 | 40.00 | 8.27 | 3.36 | 2.83 |
| RGPF-0.10%-800 | 95.27 | 4.73 | 4.96 | 46.86 | 38.49 | 7.98 | 3.29 | 3.38 |
| RGPF-0.20%-800 | 95.88 | 4.12 | 4.30 | 47.25 | 38.08 | 7.70 | 3.39 | 3.58 |
| RGPF-0.30%-800 | 91.56 | 8.44 | 9.22 | 48.41 | 36.26 | 6.62 | 4.75 | 3.97 |
| RGPF-0.20%-700 | 92.04 | 7.96 | 8.65 | 48.29 | 35.2 | 9.62 | 3.67 | 3.21 |
| RGPF-0.20%-900 | 94.26 | 5.74 | 6.09 | 38.77 | 47.31 | 5.89 | 4.83 | 3.20 |
| RGPF-0.20%-1000 | 95.30 | 4.70 | 4.93 | 37.10 | 49.86 | 4.99 | 5.54 | 2.51 |
To understand the surface properties and pore distribution of the prepared porous materials, N2 adsorption/desorption isotherms of RCGO-800, PF-800 and RGPF-m-t (Fig. 2) were analyzed. The isotherms of RCGO-800 (Fig. 2(c)) is the typical I adsorption isotherms, indicating the presences of micropores (<2 nm).65 The isotherms of PF-800 (Fig. 2(c)) exhibited a mixture adsorption isotherm of type I at the lower pressure and type IV with a H2 hysteresis in the higher relative pressure of 0.450 to 0.995. This demonstrated the presence of micropores and mesopores (2–50 nm), which in agreement with the pore size distribution calculated by the BJH model shown in Fig. 2(d).66 For RGPF-m-t (Fig. 2(a) and (b)), the isotherm showed a type I at the lower pressure attributed to the micropores. But at the higher pressure, a slight hysteresis existed, implying a small quantity of mesopores with the smaller diameter. The pore size distribution in Fig. 2(d) was consistent with this. Table 2 presented the specific surface area, total pore volume and micropore volume of the prepared porous carbon materials.
 | ||
| Fig. 2 (a–c) N2 adsorption/desorption isotherms and (d) pore size distribution of prepared materials. | ||
| Samples | SBET (m2 g−1) | Smicro (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (cm3 g−1) | D (nm) |
|---|---|---|---|---|---|
| RCGO-800 | 281.90 | 222.36 | 0.13 | 0.09 | 1.78 |
| PF-800 | 141.62 | 80.56 | 0.11 | 0.03 | 3.22 |
| RGPF-0.05%-800 | 609.54 | 547.49 | 0.25 | 0.21 | 1.60 |
| RGPF-0.10%-800 | 679.13 | 599.83 | 0.27 | 0.23 | 1.60 |
| RGPF-0.20%-800 | 837.55 | 744.36 | 0.33 | 0.28 | 1.58 |
| RGPF-0.30%-800 | 616.74 | 561.64 | 0.24 | 0.21 | 1.56 |
| RGPF-0.20%-700 | 224.11 | 198.22 | 0.13 | 0.075 | 2.34 |
| RGPF-0.20%-900 | 672.12 | 599.11 | 0.27 | 0.23 | 1.61 |
| RGPF-0.20%-1000 | 397.11 | 335.31 | 0.16 | 0.13 | 1.65 |
PF-800 had the smallest specific surface area because of the fewest micropores. It is noteworthy that all the specific surface area of RGPF-m-t except RGPF-0.20%-700 was much larger than that of PF-800 and RCGO-800, and RGPF-0.20%-800 had the largest one, which further demonstrated the micropores mainly formed by the decomposition of O–CC.
However, the carbon materials only with the micropore structure were unfavorable to the capacitive performance because it is difficult for inner pores to be fully accessed by the ions. It also needs a certain amount of macropores to ensure the fast and accessible ion diffusion.17,67 In this work, the macropores formed by the foaming of CGO/PF, which were obviously observed from the SEM and TEM images shown in Fig. 3. In the SEM pictures of PF and RGPF, there were mass of the honeycomb-like structure with external open pores in the diameter range from nanometer to micron. It could derive from the above representation that the prepared carbon materials had the hierarchical porous structure with macropores, mesopores and micropores.
 | ||
| Fig. 3 (a–i) SEM images of the prepared materials, the insets are SEM images under high magnification; (j) TEM image of RGPF-0.20%-800. | ||
3.2 Electrochemical tests
The above discussion about the surface properties of the prepared materials revealed that most of RGPF-m-t had a hierarchical porous structure and a large specific surface area, illustrating they were the ideal candidate electrode materials for the supercapacitors. The material characterization also demonstrated that the content of CGO as well as the carbonization temperature could affect the surface structure, and resultantly would affect their electrochemical properties. The electrochemical performance of these materials was evaluated by CV, CP and EIS in a 6 M KOH solution by using a three-electrode system.Firstly, the optimization of the potential window was carried out using RGPF-0.02%-800 by CV at 5 mV s−1. The higher cut-off potential varied from 0.0 to 0.5 V vs. Hg/HgO with respect to −1.0 V vs. Hg/HgO. As shown in Fig. 4(a), the oxygen evolution reaction was observed clearly when the higher cut-off potential was higher than 0.3 V vs. Hg/HgO, which would affect the measurement of the double-layer capacitance and the stability of the electrode. Hence, we selected the potential window of −1.0 to 0.3 V vs. Hg/HgO for the following double-layer-capacitance investigation.
 | ||
| Fig. 4 (a) CV curves of the RGPF-0.02%-800 at 5 mV s−1 in different potential windows; (b) CV curves at 20 mV s−1; (c) specific capacitance from CV curves as a function of scan rate; (d) CP curves at 1 A g−1; (e) specific capacitance from CP curves as a function of current density; (f) Nyquist plots from 10 mHz to 100 kHz for PF-800 and RGPF-m-800. | ||
The impact of the content of CGO on electrochemical behaviors of RGPF-m-800 was investigated and the results were shown in Fig. 4(b)–(f). As shown in Fig. 4(b), all the CV curves of the RGPF-m-800 samples presented quasi-rectangular shapes with a wider potential range of −1.0 to 0.3 V, showing the typical double-layer capacitive behavior.52,53 The currents changed with the content of CGO. Fig. 4(c) showed the specific capacitance of all the prepared RGPF-m-800 calculated by the eqn (1) at different scan rates. All the capacitance of RGPF-m-800 is higher than that of pure PF-800 and the composite materials owning 0.20% CGO reach the highest capacitance at a scan rate of 5 mV s−1. In addition, Fig. 4(c) also showed the capacitance decreased with the increase of the scan rates, which was regarded corresponding to the diffusion of electrolyte ions. At a low scan rate, electrolyte ions have sufficient time to diffuse on the electrode surface and finish the charge propagation at the interfaces of electrode/electrolyte. With the increase of scanning ratio, the charge propagation would be affected by decrease of the ion effective diffusion, resulting in a lower capacitance.
The CP experiments of RGPF-m-800 with different current densities in Fig. 4(d) and (e) also exhibited the same results. As showed in the Fig. 4(d), the profile of these charge/discharge curves was like an isosceles triangle, indicating a reversible adsorption/desorption of ions which is the characteristic of the electric double-layer capacitors.14,68–70 It was clearly noted from Fig. 4(e) that RGPF-0.20%-800 owns the highest specific capacitance at the same current density calculated from the CP curves, and the changing rules of the specific capacitance with the content of CGO was consistent with that in the CV measurement.
RGPF-0.20%-800 exhibits a higher capacitance up to 272.6 F g−1 at a current density of 0.1 A g−1, and the capacitance still sustained 153.1 F g−1 at 20 A g−1. Compared with the specific capacitance of PF-800 (158.9 F g−1 at 0.1 A g−1), there was a huge and obvious improvement for RGPF-0.20%-800 after adding about 18 μg CGO in each working electrode. Meanwhile, compared with the reported porous carbon derived from phenolic resin summarized in Table S3,† RGPF-0.20%-800 also exhibited an excellent capacitive performance. The first reason for the excellent electrochemical performance of RGPF-0.20%-800 is that it had the largest special surface area and the well-balanced pore distribution shown in the above surface structure analysis for the fast diffusion and storage of ions. A porous carbon material with the excellent capacitive performance should have an appropriate proportion of macropores, mesopores and micropores.71,72 The macropores as a bulk-buffering reservoir for the electrolyte made the ion diffusion distance shorten, mesopores provided a large, accessible surface area which enables the rapid and reversible ion transport during charging storage, and micropores could be helpful for the charge accommodation. Moreover, the incorporation of RCGO was also helpful for the electron conduction.73 In addition, a foaming method was applied to forming hierarchical porous structure instead of the conventional method to make pores, such as template method and acid or alkali activation. Hard-to-control process and complicated post-treatment limited the large scale application of these conventional methods. In contrast, the foaming method was simple, easy to control and able to form a large amount of open pores, which was more suitable for the extensive preparation of the porous carbon materials.
EIS was performed to evaluate the conductive ability of the electrode materials. Fig. 4(f) showed the Nyquist plots of RGPF-m-800. All the Nyquist plots exhibited a straight line in the low frequency region and a semicircle in the high frequency region. The straight line related to the ion diffusion. The larger the value of slope was, the faster the ion diffusion was, and the better the capacitive performance of the electrode was. It was obvious that the straight line in the Nyquist plot of RGPF-0.20%-800 was nearly vertical, indicating a closely ideal capacitive performance due to the hierarchical porous structure. The radius of the semicircle represented charge transfer resistance (Rct) at the electrode/electrolyte interface.74 Obviously, RGPF-0.20%-800 showed the smallest Rct compared to other RGPF. In the high-frequency region, the intercept of the semicircle with the real axis corresponded to the internal resistance (Rs) coming from the ionic resistance of the electrolyte, the electrical resistance of the electrode, and interfacial resistance between the electrode and electrolyte.75 All the Rs values of RGPF were about 0.4 Ω. This was also due to the hierarchical porous structure which could shorten the ion diffusion distance and prompt ion transport fast.
The carbonization temperature was another pivotal factor in determining the structures of the prepared materials as well as the resultant supercapacitive performance. Fig. 5(a–e) showed the CV, CP and EIS curves of RGPF-0.20%-t. The results indicated the appropriate carbonization temperature was at 800 °C because of the larger special surface area, the appropriate pore distribution and the good electron conduction.
 | ||
| Fig. 5 (a) CV curves at 20 mV s−1; (b) specific capacitance from CV curves of as a function of scan rate; (c) CP curves at 1 A g−1; (d) specific capacitance from CP curves as a function of current density; (e) Nyquist plot from 10 mHz to 100 kHz for RGPF-0.20%-t; (f) capacitance retention of RGPF-0.20%-800 over 10000 cycles at 1 A g−1, the inset is the typical measured CP curves. | ||
In addition, the cyclic stability, an important criterion for the supercapacitor applications, was investigated by the charge/discharge curves of RGPF-0.20%-800 at the current density of 1 A g−1. During the measurement process, all the curves kept a symmetric triangular shape and owned the same transformation. The specific capacitance retention ratio shown in Fig. 5(f) was calculated based on the eqn (2). After 10000 cycles, RGPF-0.20%-800 can retain 92.31% of its initial specific capacitance. In a word, this hierarchical porous carbon presented a good cycle life and capacitance value, implying a promising application for the supercapacitors.
To further explore the capacitance performance of the RGPF and PF electrode for the actual application, RGPF-0.20%-800 or PF-800 was characterized as electrodes in a two-electrode symmetric supercapacitor by CV and CP using 6 M KOH solution. The performance of the symmetric supercapacitor was shown in Fig. 6. The CV curves with various potential windows from 0–1.0 to 0–1.5 V at 5 mV s−1 were presented in Fig. 6(a). It was noted that the CV curves hold the rectangular shapes with a mirror image feature at the potential window up to 1.3 V, demonstrating a double-layer capacitive behavior and the maximum working potential window of 1.3 V. With the increase of scan rate from 5 mV s−1 to 100 mV s−1, the profile of the CV curves of RGPF-0.20%-800 had no significant change as shown in Fig. 6(b), which was due to the hierarchical porous structure. Fig. 6(c) showed the CP curves of the RGPF-0.20%-800 with a regular isosceles triangle shapes, also indicating an ideal double-layer capacitive behavior. The specific capacitance could be calculated from CP curves by the eqn (2). The capacitance at 0.1 A g−1 was 64.9 F g−1, and still retain 31.55 F g−1 at a high current density of 10 A g−1. According to the specific capacitance, the energy density and power density of the symmetric supercapacitor were calculated by eqn (3) and (4) respectively, and their relationship was depicted by Ragone plots shown in Fig. 6(d). The maximum energy density of the RGPF-0.20%-800 could reach 15.20 W h kg−1 corresponding to the power density of 90.90 W kg−1 at a current density of 0.1 A g−1. More remarkably, even when the power density of devices was 10.11 kW kg−1, the energy density still remained 4.54 W h kg−1. Taking into account of the high-yield, economical, environmentally friendly of the raw materials and simple synthetic method, this hierarchical porous carbon derived from carboxyl graphene oxide/phenolic foam are worthy of further investigation to be applied actual production for the supercapacitors.
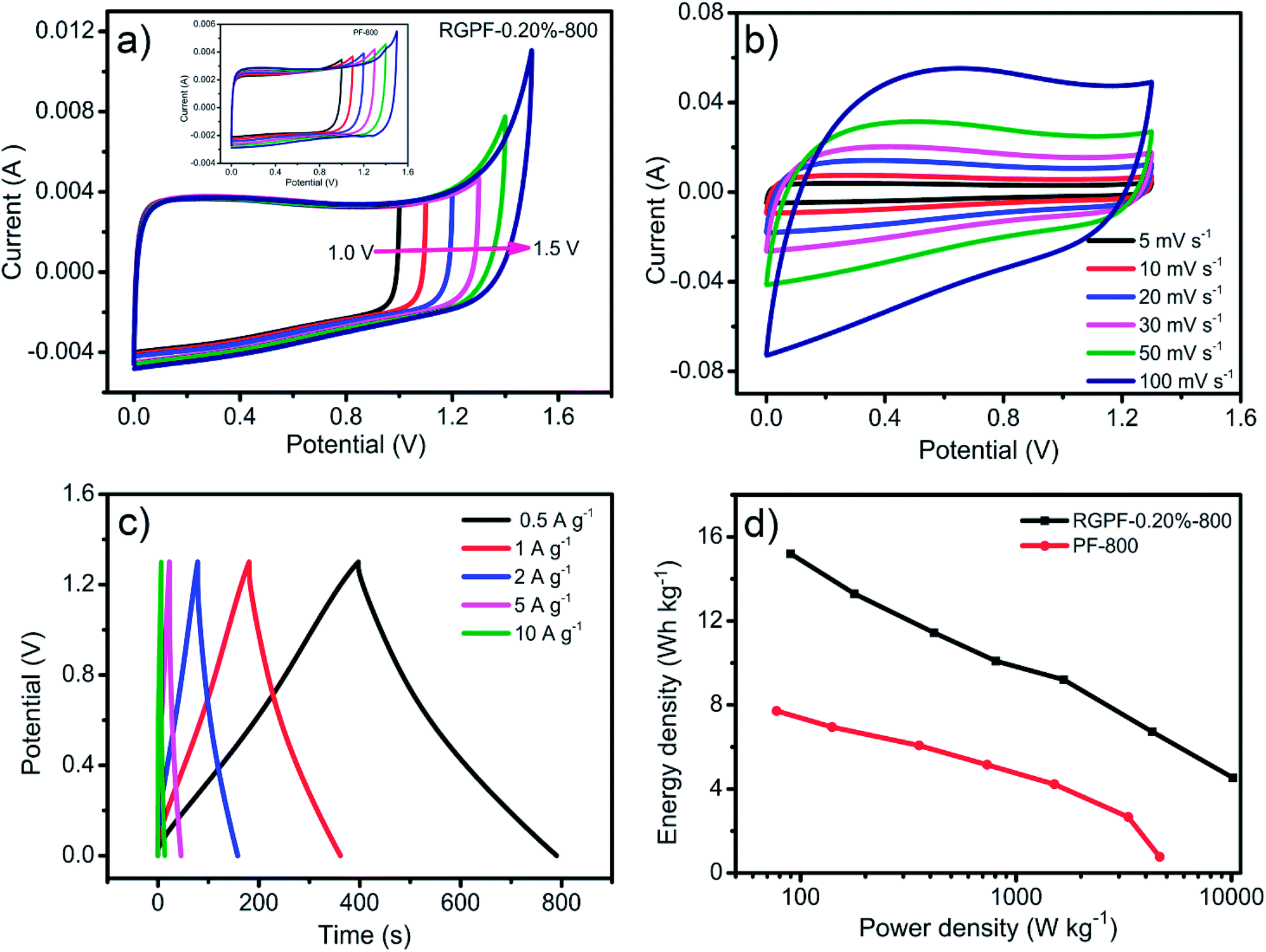 | ||
| Fig. 6 (a) CV curves with different potential windows at 5 mV s−1, (b) CV curves at different scan rates, (c) CP curves at different current density and (d) Ragone plots for RGPF-0.20%-800 and PF-800 in two-electrode system in 6 M KOH. | ||
4. Conclusions
In summary, the hierarchical porous carbon materials were prepared with a facile and low-cost method from phenolic resin and CGO by foaming and carbonization. Structure characterizations revealed that the hierarchical porous carbon materials with a large special surface area consisted of macropores, mesopores and micropores from the foaming process, decomposition of the ester groups and carbonization of the polymer, respectively. The rational-distribution hierarchical porous structures could shorten the ion transport distance and increase the charge accommodation, whereas the excellent electrical conductivity could be conducive to the electron transport. Because of the above advantages, RGPF-0.20%-800 exhibited a high specific capacitance of 272.6 F g−1 at a current density of 0.1 A g−1 in KOH electrolyte, which is 113.7 F g−1 higher than PF-800. Meanwhile, the electrode materials RGPF-0.20%-800 also showed an excellent cycle life with 92.31% specific capacitance retained after 10000 cycles. Moreover, symmetric supercapacitor based on RGPF-0.20%-800 also exhibited a high energy density (15.20 W h kg−1) and power density (10.11 kW kg−1). All the excellent capacitance performances indicate the synthesized hierarchical porous carbon material is a promising candidate for real energy storage in supercapacitors. The approach proposed in this work also provides a novel strategy for the mass preparation of the advanced capacitance materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Nation Natural Science Foundation of China (Grant No. 51273155 and 21476179) and the Fundamental Research Funds for the Central Universities (No. 2016IB005 and 2017IB007). We also thank the professors in Materials Research and Testing Center of Wuhan University of Technology for their kind help with the SEM, TEM and Raman measurements.References
- Z. Li, K. K. Guo and X. L. Chen, RSC Adv., 2017, 7, 30521–30532 RSC.
- X. F. Bing, X. Jiang, M. Tian, J. T. Wang, W. M. Qiao and L. C. Ling, RSC Adv., 2017, 7, 26650–26657 RSC.
- H. Su, H. T. Zhang, F. Y. Liu, F. J. Chun, B. B. Zhang, X. Chu, H. C. Huang, W. L. Deng, B. N. Gu, H. P. Zhang, X. T. Zheng, M. H. Zhu and W. Q. Yang, Chem. Eng. J., 2017, 322, 73–81 CrossRef CAS.
- B. Hu, L. B. Kong, L. Kang, K. Yan, T. Zhang, K. Li and Y. C. Luo, RSC Adv., 2017, 7, 14516–14527 RSC.
- Q. Shi, R. Y. Zhang, Y. Y. Lu, Y. H. Deng, A. A. Elzatahrya and D. Y. Zhao, Carbon, 2015, 84, 335–346 CrossRef CAS.
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS PubMed.
- T. Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- Q. Wang, J. Yan, Y. B. Wang, T. Wei, M. L. Zhang, X. Y. Jing and Z. J. Fan, Carbon, 2014, 67, 119–127 CrossRef CAS.
- J. Zhou, Z. S. Zhang, Z. H. Li, T. T. Zhu and S. P. Zhuo, RSC Adv., 2015, 5, 46947–46954 RSC.
- P. Sennu, V. Aravindan and Y. S. Lee, J. Power Sources, 2016, 306, 248–257 CrossRef CAS.
- X. J. He, N. Zhang, X. L. Shao, M. B. Wu, M. X. Yu and J. S. Qiu, Chem. Eng. J., 2016, 297, 121–127 CrossRef CAS.
- W. X. Wang, H. Y. Quan, W. M. Gao, R. Zou, D. Z. Chen, Y. H. Dong and L. Guo, RSC Adv., 2017, 7, 16678–16687 RSC.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- B. Li, F. Dai, Q. F. Xiao, L. Yang, J. M. Shen, C. M. Zhang and M. Cai, Energy Environ. Sci., 2016, 9, 102–106 CAS.
- X. L. Yu, J. M. Lu, C. Z. Zhan, R. T. Lv, Q. H. Liang, Z. H. Huang, W. C. Shen and F. Y. Kang, Electrochim. Acta, 2015, 182, 908–916 CrossRef CAS.
- Y. S. Lim, C. W. Lai and S. B. Abd Hamid, RSC Adv., 2017, 7, 23030–23040 RSC.
- L. Yao, G. Z. Yang, P. Han, Z. H. Tang and J. H. Yang, J. Power Sources, 2016, 315, 209–217 CrossRef CAS.
- J. Zhou, Z. S. Zhang, W. Xing, J. Yu, G. X. Han, W. J. Si and S. P. Zhuo, Electrochim. Acta, 2015, 153, 68–75 CrossRef CAS.
- P. Cheng, S. Y. Gao, P. Y. Zang, X. F. Yang, Y. L. Bai, H. Xu, Z. H. Liu and Z. B. Lei, Carbon, 2015, 93, 315–324 CrossRef CAS.
- X. J. Li, W. Xing, J. Zhou, G. Q. Wang, S. P. Zhuo, Z. F. Yan, Q. Z. Xue and S. Z. Qiao, Chem.–Eur. J., 2014, 20, 13314–13320 CrossRef CAS PubMed.
- J. Y. Tian, Z. M. Liu, Z. H. Li, W. G. Wang and H. Y. Zhang, RSC Adv., 2017, 7, 12089–12097 RSC.
- L. X. Jiang, Q. L. Cheng, Y. M. Xia, V. Pavlinek, P. Saha and C. Z. Li, J. Mater. Sci., 2014, 49, 7489–7496 CrossRef CAS.
- C. Ma, Y. Yu, Y. J. Li, J. L. Shi, Y. Song and L. Liu, J. Electrochem. Soc., 2014, 161, A1330–A1337 CrossRef CAS.
- F. Liu, X. Yan, F. T. Fan, C. C. Zhao, R. T. Liu, Y. Gao and Y. Q. Wang, Micro Nano Lett., 2016, 11, 372–377 Search PubMed.
- C. Ma, Y. J. Li, J. L. Shi, Y. Song and L. Liu, Chem. Eng. J., 2014, 249, 216–225 CrossRef CAS.
- L. N. Sui, Y. T. Wang, L. Y. Yu, H. Z. Dong and L. F. Dong, J. Electrochem. Soc., 2016, 163, E179–E184 CrossRef CAS.
- J. Choma, K. Jedynak, W. Fahrenholz, J. Ludwinowicz and M. Jaroniec, Appl. Surf. Sci., 2014, 289, 592–600 CrossRef CAS.
- L. K. C. de Souza, N. P. Wickramaratne, A. S. Ello, M. J. F. Costa, C. E. F. da Costa and M. Jaroniec, Carbon, 2013, 65, 334–340 CrossRef CAS.
- X. H. Xia, X. F. Zhang, S. Q. Yi, H. Chen and H. B. Liu, J. Mater. Res., 2016, 31, 1665–1673 CrossRef CAS.
- V. Drach, M. Wiener, G. Reichenauer, H. P. Ebert and J. Fricke, Int. J. Thermophys., 2007, 28, 1542–1562 CrossRef CAS.
- J. Z. Feng, C. R. Zhang, J. Feng, Y. G. Jiang and N. Zhao, ACS Appl. Mater. Interfaces, 2011, 3, 4796–4803 CAS.
- R. W. Fu, B. Zheng, J. Liu, S. Weiss, J. Y. Ying, M. S. Dresselhaus, G. Dresselhaus, J. H. Satcher and T. F. Baumann, J. Mater. Res., 2003, 18, 2765–2773 CrossRef CAS.
- B. X. Zou, Y. Gao, B. Liu, Y. P. Yu and Y. H. Lu, RSC Adv., 2016, 6, 4483–4489 RSC.
- Y. Fang, B. Luo, Y. Y. Jia, X. L. Li, B. Wang, Q. Song, F. Y. Kang and L. J. Zhi, Adv. Mater., 2012, 24, 6348–6355 CrossRef CAS PubMed.
- X. W. Yang, J. W. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS PubMed.
- C. L. Bao, Y. Q. Guo, L. Song, Y. C. Kan, X. D. Qian and Y. Hu, J. Mater. Chem., 2011, 21, 13290–13298 RSC.
- C. X. Guo and C. M. Li, Energy Environ. Sci., 2011, 4, 4504–4507 CAS.
- Y. J. Lee, H. W. Park, G. P. Kim, J. Yi and I. K. Song, Curr. Appl. Phys., 2013, 13, 945–949 CrossRef.
- Z. B. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 CAS.
- X. Li, X. B. Zang, Z. Li, X. M. Li, P. X. Li, P. Z. Sun, X. Lee, R. J. Zhang, Z. H. Huang, K. L. Wang, D. H. Wu, F. Y. Kang and H. W. Zhu, Adv. Funct. Mater., 2013, 23, 4862–4869 CAS.
- M. X. Wang, Q. Liu, H. F. Sun, E. A. Stach, H. Y. Zhang, L. Stanciu and J. Xie, Carbon, 2012, 50, 3845–3853 CrossRef CAS.
- M. H. Yeh, L. Y. Lin, C. P. Lee, H. Y. Wei, C. Y. Chen, C. G. Wu, R. Vittal and K. C. Ho, J. Mater. Chem., 2011, 21, 19021–19029 RSC.
- Y. Q. Qian, I. M. Ismail and A. Stein, Carbon, 2014, 68, 221–231 CrossRef CAS.
- T. F. Qin, Z. Y. Wan, Z. L. Wang, Y. X. Wen, M. T. Liu, S. L. Peng, D. Y. He, J. Hou, F. Huang and G. Z. Cao, J. Power Sources, 2016, 336, 455–464 CrossRef CAS.
- J. Chen, B. W. Yao, C. Li and G. Q. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- S. G. Peng, C. Y. Liu and X. J. Fan, Integr. Ferroelectr., 2015, 163, 42–53 CrossRef CAS.
- X. M. Hu, Y. Y. Zhao and W. M. Cheng, Polym. Compos., 2015, 36, 1531–1540 CrossRef CAS.
- Y. F. Ma, W. Zhang, C. P. Wang, Y. Z. Xu and F. X. Chu, J. Appl. Polym. Sci., 2013, 129, 3096–3103 CrossRef CAS.
- Y. Lu, B. K. Li, X. Wang, J. Zheng, J. X. Pu, Q. Ding, L. P. Li, K. Xu, P. Gao and L. Chen, Cryobiology, 2012, 64, 27–32 CrossRef CAS PubMed.
- W. Zhang, Y. F. Ma, F. X. Chu and C. P. Wang, Adv. Mater. Res., 2011, 250–253, 523–527 CAS.
- X. W. Zhuang, S. H. Li, Y. F. Ma, W. Zhang, Y. Z. Xu, C. P. Wang and F. X. Chu, Adv. Mater. Res., 2011, 250–253, 450–454 CrossRef CAS.
- X. J. Li, J. Zhou, W. Xing, F. Subhan, Y. Zhang, P. Bai, B. J. Xu, S. P. Zhuo, Q. Z. Xue and Z. F. Yan, Electrochim. Acta, 2016, 190, 923–931 CrossRef CAS.
- D. Z. Zhu, Y. W. Wang, W. J. Lu, H. Zhang, Z. Y. Song, D. Luo, L. H. Gan, M. X. Liu and D. M. Sun, Carbon, 2017, 111, 667–674 CrossRef CAS.
- F. Zhang, Z. P. Wang, D. L. Wang, S. L. Wang and X. G. Xu, IEEE J. Sel. Top. Quantum Electron., 2017, 23, 200–205 CrossRef.
- C. Nethravathi, B. Viswanath, C. Shivakumara, N. Mahadevaiah and M. Rajamathi, Carbon, 2008, 46, 1773–1781 CrossRef CAS.
- S. H. Park, S. M. Bak, K. H. Kim, J. P. Jegal, S. I. Lee, J. Lee and K. B. Kim, J. Mater. Chem., 2011, 21, 680–686 RSC.
- S. H. Feng, Z. S. Yuan, M. Leitch and C. B. Xu, J. Anal. Appl. Pyrolysis, 2015, 115, 184–193 CrossRef CAS.
- L. L. Yang, X. Li, S. L. Yan, M. M. Wang, P. Liu, Y. L. Dong and C. C. Zhang, Anal. Methods, 2015, 7, 5303–5310 RSC.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- L. L. Wang, Y. G. Liu, C. B. Chong, J. Wang, Z. Q. Shi and J. Pan, Mater. Lett., 2016, 170, 217–220 CrossRef CAS.
- L. Ye, Q. H. Liang, Z. H. Huang, Y. Lei, C. Z. Zhan, Y. Bai, H. Li, F. Y. Kang and Q. H. Yang, J. Mater. Chem. A, 2015, 3, 18860–18866 CAS.
- Y. H. Liu, L. L. Yuan, Y. Yue, M. L. Hu and H. L. Wei, Electrochim. Acta, 2016, 196, 153–161 CrossRef CAS.
- R. S. Zhong, Y. H. Qin, D. F. Niu, J. W. Tian, X. S. Zhang, X. G. Zhou, S. G. Sun and W. K. Yuan, J. Power Sources, 2013, 225, 192–199 CrossRef CAS.
- J. Z. Chen, J. L. Xu, S. Zhou, N. Zhao and C. P. Wong, Nano Energy, 2016, 25, 193–202 CrossRef.
- G. H. An, H. J. Ahn and W. K. Hong, J. Power Sources, 2015, 274, 536–541 CrossRef CAS.
- G. H. An, B. R. Koo and H. J. Ahn, Phys. Chem. Chem. Phys., 2016, 18, 6587–6594 RSC.
- Y. R. Liu, B. P. Lin, D. Li, T. Xu, X. Q. Zhang, Y. Sun and H. Yang, Microporous Mesoporous Mater., 2014, 200, 245–252 CrossRef CAS.
- A. Bello, F. Barzegar, D. Momodu, J. Dangbegnon, F. Taghizadeh, M. Fabiane and N. Manyala, J. Power Sources, 2015, 273, 305–311 CrossRef CAS.
- X. F. Bing, Y. J. Wei, M. Wang, S. Xu, D. H. Long, J. T. Wang, W. M. Qiao and L. C. Ling, J. Colloid Interface Sci., 2017, 488, 207–217 CrossRef CAS PubMed.
- S. J. He and W. Chen, J. Power Sources, 2014, 262, 391–400 CrossRef CAS.
- Z. S. Wu, Y. Sun, Y. Z. Tan, S. B. Yang, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2012, 134, 19532–19535 CrossRef CAS PubMed.
- M. J. Zhi, F. Yang, F. K. Meng, M. Q. Li, A. Manivannan and N. Q. Wu, ACS Sustainable Chem. Eng., 2014, 2, 1592–1598 CrossRef CAS.
- M. Yu, J. Li and L. J. Wang, Chem. Eng. J., 2017, 310, 300–306 CrossRef CAS.
- L. J. Zhang, Y. Z. Jiang, L. W. Wang, C. Zhang and S. X. Liu, Electrochim. Acta, 2016, 196, 189–196 CrossRef CAS.
- Z. J. Fan, J. Yan, T. Wei, L. J. Zhi, G. Q. Ning, T. Y. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366–2375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08602d |
| This journal is © The Royal Society of Chemistry 2017 |