
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential one-pot three-step synthesis of polysubstituted 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazole systems†
Helio G. Bonacorso *a,
Gean M. Dal Fornoa,
Carson Wiethana,
Alex Ketzera,
Nilo Zanattaa,
Clarissa P. Frizzoa,
Marcos A. P. Martinsa and
Mark Stradiottob
*a,
Gean M. Dal Fornoa,
Carson Wiethana,
Alex Ketzera,
Nilo Zanattaa,
Clarissa P. Frizzoa,
Marcos A. P. Martinsa and
Mark Stradiottob
aNúcleo de Química de Heterociclos – NUQUIMHE, Departamento de Química, Universidade Federal de Santa Maria, 97105-900 Santa Maria, Brazil. E-mail: helio.bonacorso@ufsm.br
bDepartment of Chemistry, Dalhousie University, Halifax, NS, B3H 4R2 Canada
First published on 11th September 2017
Abstract
This work reports a successful one-pot three-step protocol for the synthesis of a new series of 15 examples of polysubstituted 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazoles, in which sequential Sonogashira cross-coupling, desilylation, and a copper(I)-catalyzed azide–alkyne cycloaddition reaction (CuAAC) with an overall yield of up to 72% were used. The presence of a trifluoromethyl substituent attached to the pyrazole moiety made the Sonogashira cross-coupling reaction challenging. Additionally, the selection of the ancillary ligand XPhos was essential and indispensable for the desired heterocyclic construction.
Introduction
Due to their broad spectrum of biological properties, five-membered nitrogen heterocycles with trifluoromethyl moieties represent sought-after motifs in many fields of medicinal chemistry, biology, materials science, and agriculture.1 Several trifluoromethylated azole cores are present in many of the prescribed and/or profitable drugs commercialized in the U.S. pharmaceutical market. As an example, sitagliptin is an antidiabetic drug that was the fifteenth most prescribed drug overall in 2015, generating $ 2.8 billion; while celecoxib – which is a nonsteroidal anti-inflammatory drug – generated $ 2.2 billion in the U.S. market in the same year (Fig. 1).2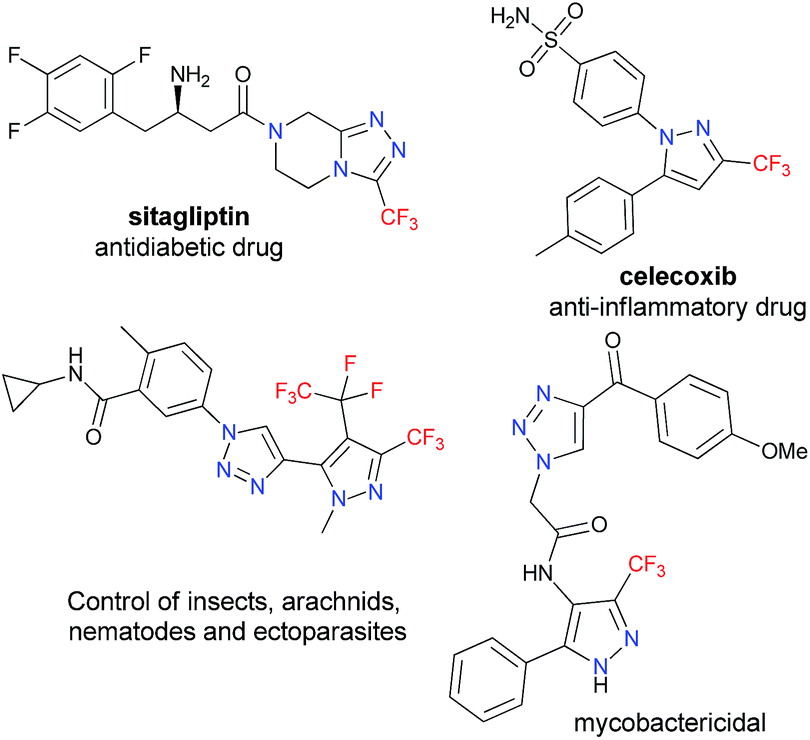 | ||
| Fig. 1 Some fluorinated azoles and pyrazolyl-1,2,3-triazoles with biological activity. | ||
Notably, 20–30% of approved drugs, and about 40% of the new compounds entering phase III trials, possess at least one fluorine atom in their structure.2 This trend is mainly due to the nature of the fluorine atom, which furnishes specific physico chemical properties such as: the introduction of fluorine groups to aromatic compounds, which increases the lipophilicity and metabolic stability of the molecule in vivo; and stronger electrostatic interaction with the receptors that bind the compounds more tightly in the active sites of the enzymes.3 The increasing use of fluorine atoms in active pharmaceutical ingredients parallels advances in understanding of physicochemical attributes, and it gives an enhanced appreciation of how these unique properties can be exploited to address the numerous challenges encountered in pharmaceutical candidate optimization.1
Given our shared interest in the synthesis of fluorinated nitrogenous heterocycles with potential pharmacological application,4 in a recent publication we disclosed cross-coupling reactions involving 4-halo-5-trifluoromethyl-1H-pyrazoles, in order to achieve the related tetrasubstituted structures.5 Despite such success, cross-coupling reactions of this type involving organometallic reagents derived from nitrogen-containing heterocycles continue to represent a significant challenge.
Given the established anticancer,7 antimycobacterial,8 CB1 receptor ligand,9,10 antibacterial,11 and nematicidal12,13 properties of heterocyclic systems containing pyrazole-1,2,3-triazole scaffolds (Fig. 1), we were encouraged to develop an efficient and useful protocol for obtaining 4-(pyrazol-4-yl)-1H-1,2,3-triazole systems, seeking to avoid some limitations associated with coupling reactions involving trifluoromethyl-substituted pyrazole precursors.5,6
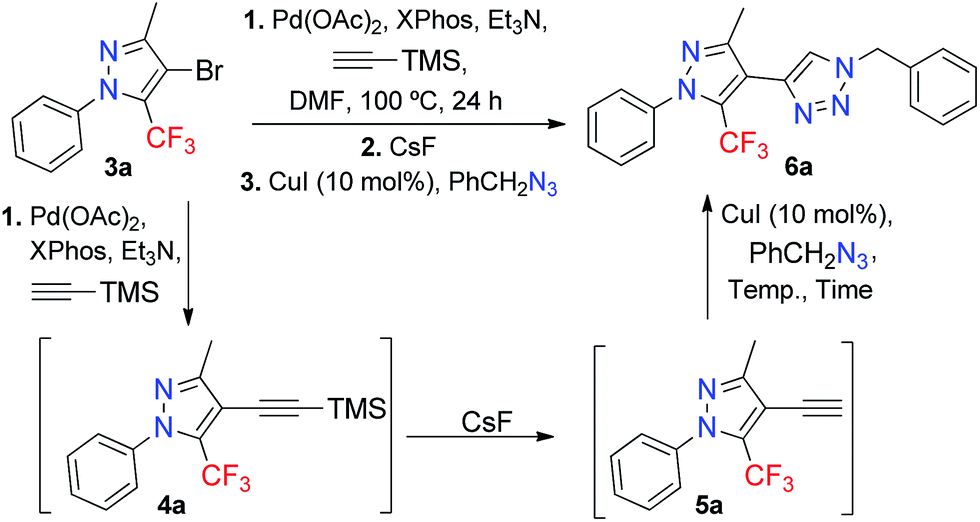
Thus, we envisioned the possibility of performing Sonogashira coupling and copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) in a one-pot fashion. The Pd(0) catalyzed cross-coupling reaction of 4-bromo-5-trifluoromethyl-1H-pyrazole with ethynyltrimethylsilane (TMSA) would furnish the alkyne-containing TMSA-5-trifluoromethyl-1H-pyrazoles, which, after in situ fluorine deprotection, would give terminal alkyne intermediates. The Cu(I)-catalyzed reaction (CuAAC) is a click reaction, in which the reaction involving an azide and the terminal 4-ethynyl-5-trifluoromethyl-1H-pyrazoles would give direct access to 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazoles, in a one-pot sequential metal-catalyzed reaction. Selected examples in the literature detail the synthesis of a 4-(1H-pyrazol-4-yl)-1H-1,2,3-triazole by a similar approach; however, this pyrazolyl-triazole did not have a trifluoromethyl group in the pyrazole core.14 The presence of an electron-withdrawing group like CF3 would make the Sonogashira cross-coupling difficult, and hinder the synthesis of the compounds proposed herein.
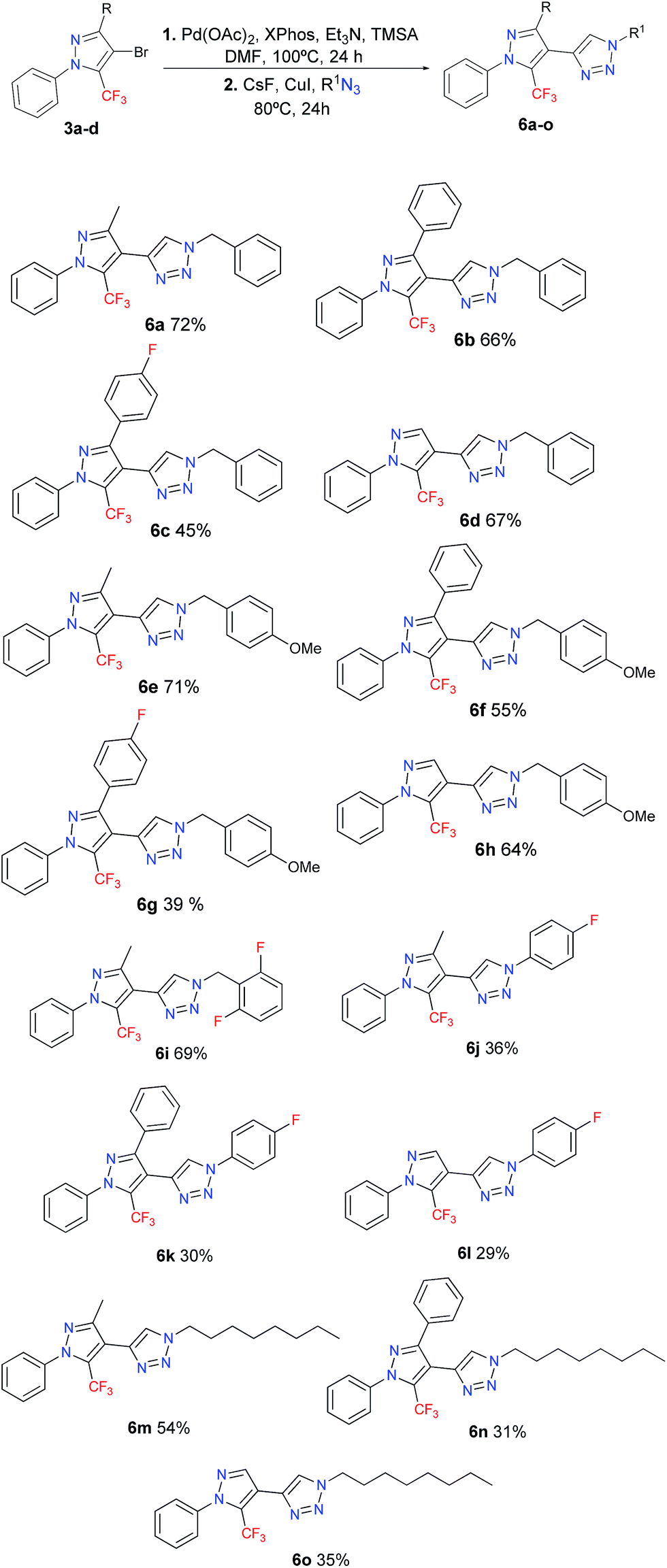
In this work we disclose the Sonogashira cross-coupling, desilylation, and CuAAC in a sequential one-pot three-step reaction – which is summarized in Scheme 1 – for the synthesis of a new series of 15 examples of a polysubstituted 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazole system.
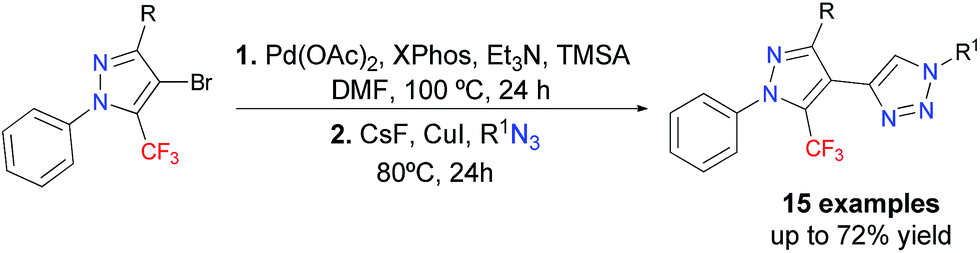 | ||
| Scheme 1 Sequential one-pot, three-step synthesis of 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazoles. | ||
Results and discussion
We commenced our investigations by targeting an expedient route for the synthesis of 4-bromo-5-(trifluoromethyl)-1H-pyrazoles 3a–d, for use as starting materials for the proposed study (Scheme 2). Compounds 2a–c were prepared by the cyclocondensation reaction of 1,1,1-trifluoro-4-methoxy-alken-2-ones (1a–c) and phenylhydrazine, following procedures found in the literature.15 The starting materials 3a–d were obtained by the subsequent bromination reaction of 2a–c with NBS, following a procedure that we described previously.5 Seeking to increase the scope of this reaction, we also tried to expand this bromination methodology to aryl substituents attached to the 3-position of the pyrazole ring. When we used the same reaction condition for a 3-(furan-2-yl)pyrazole, the formation of a non-separable mixture of dibrominated (both rings) and monobrominated products was observed via GC. For the 1-(4-methoxyphenyl)pyrazole, the formation of another non-separable mixture of monobrominated products (both rings) and the starting pyrazole was observed. However, when we applied the same reaction conditions to the 3-(4-fluorophenyl)pyrazole, the formation of the desired monobrominated pyrazole 3c was observed as a single product in 92% yield. The structurally simple 4-bromo-1-phenylpyrazole 3d was prepared in 38% yield, from the bromination reaction of 4-ethoxy-1,1,1-trifluorobut-3-en-2-one 1d via a cyclocondensation reaction involving the brominated enone 1e and phenylhydrazine, in accordance with a procedure described in the literature.16,17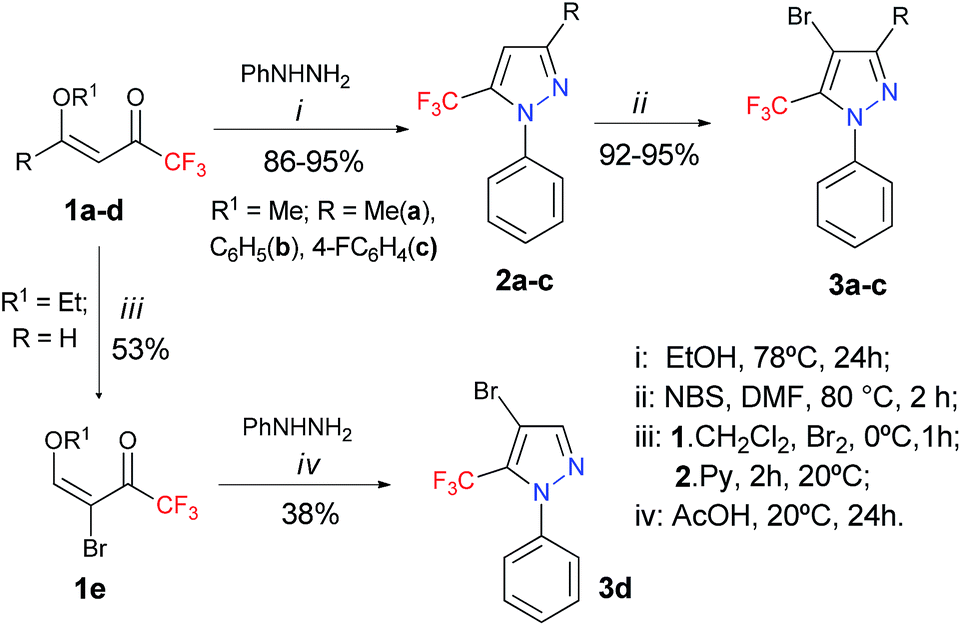 | ||
| Scheme 2 Synthesis of 4-bromo-5-(trifluoromethyl)-1H-pyrazoles 3a–d. | ||
With the starting materials 3a–d in hand, we turned our attention towards targeting an expedient protocol for the Sonogashira cross-coupling reaction between trimethylsilylacetylene (TMSA) and 4-bromo-5-trifluoromethyl-1H-pyrazole 3a – see Table 1. The presence of a trifluoromethyl group makes these substrates less reactive, and, consequently, Pd-catalyzed alkyne coupling transformations become a challenge that has to be overcome. However, according to the literature, for the activation of less reactive substrates, the simple PPh3 or other triarylphosphines have proven to be ineffective.18 In keeping with this trend, poor results were obtained in an initial test involving the reaction between 3a and TMSA, following a procedure similar to that in the literature,19 but using: 3 mol% Pd(OAc)2, 6 mol% PPh3, and 6 mol% CuI as the catalyst; Et3N as the base; and DMF as the solvent, at 100 °C in an argon atmosphere. As expected, this reaction provided a conversion rate of only 18% for 4a.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Ligand | T (°C) | Conversionb [%] |
| a Reaction conditions: pyrazole 3a (0.5 mmol), TMSA (0.75 mmol), Et3N (1.5 mmol), and solvent (2 mL).b Conversion determined by GC.c 1.5 mol% of Pd(OAc)2 and 3 mol% of ligand were used.d Isolated yield after column chromatography. | ||||
| 1 | MeCN | CyJohnPhos (L1) | 110 | 28 |
| 2 | MeCN | JohnPhos (L2) | 110 | 33 |
| 3 | MeCN | tBuXPhos (L5) | 110 | 19 |
| 4 | MeCN | SPhos (L3) | 110 | 77 |
| 5 | MeCN | XPhos (L4) | 110 | 94 |
| 6 | THF | XPhos (L4) | 65 | 31 |
| 7 | DMF | XPhos (L4) | 100 | 98 (83)d |
| 8 | DMF | XPhos (L4) | 25 | 32 |
| 9 | DMF | XPhos (L4) | 50 | 81 |
| 10c | DMF | XPhos (L4) | 100 | 89 |
 |
||||
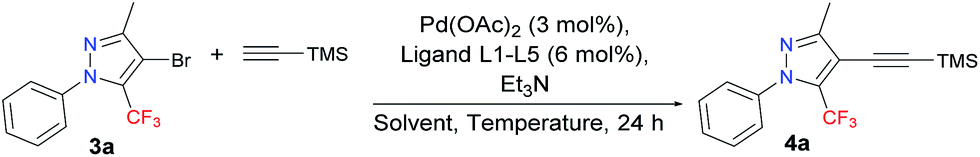
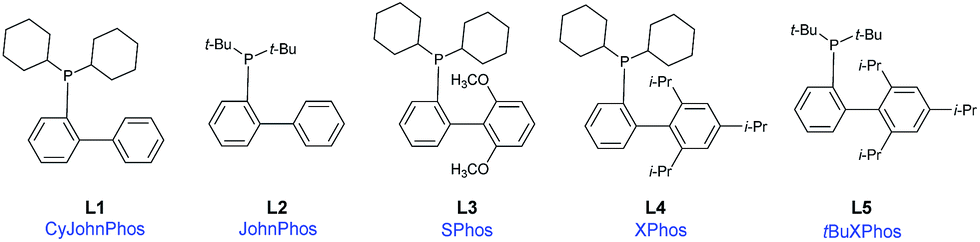
To address this difficulty, a screening involving several highly effective, bulky electron-rich dialkylbiarylphosphine ligands (L1–L5) – with Pd(OAc)2 as the catalyst, Et3N as the base, and MeCN as the solvent, at 110 °C in an argon atmosphere (Table 1, entries 1–5) – was performed. These ligands have been used in a variety of transformations especially in Pd-catalyzed carbon–carbon, carbon–nitrogen, and carbon–oxygen bond-forming processes – for the synthesis of pharmaceuticals, natural products, polymers, and new materials.20
Among the five different phosphine ligands tested, the best conversion was found with 3 mol% of Pd(OAc)2 and 6 mol% of XPhos (L4) – see Table 1, entry 5. Xphos was previously reported like an active ligand in copper-free Sonogashira coupling reaction with various aryl chlorides and tosylates in the presence of PdCl2(CH3CN)2;18 or aryl(heteroaryl) halides in combination with precatalyst in continuous-flow21 or by copper-free Sonogashira coupling of aryl chlorides on charcoal,22 or by Negishi cross-coupling reaction of trifluoromethylated 1H-pyrazoles.5
In light of a previous report stating that copper suppresses the desired transformation,18 we chose a copper-free version, which, under such conditions, is also known as the Heck alkynylation.
We next focused on studying the effect of solvent on the reaction, including transformations in THF at 65 °C and DMF at 100 °C (Table 1, entries 6–7). The best conversion was observed when the reaction was conducted in DMF at 100 °C. The effects of lowering the reaction temperature were also investigated. We observed that a reduction in reaction temperature from 100 °C to 25 °C and 50 °C significantly decreased the conversion observed, from 98% to 32% and 81%, respectively (Table 1, entries 8–9), which indicates that 100 °C was probably close to the optimal temperature for this reaction. Finally, the effects of lowering the catalyst concentration to 3 mol% of XPhos and 1.5 mol% of Pd(OAc)2 resulted in low conversion (Table 1, entry 10).
After establishing the best reaction condition for Sonogashira coupling (Table 1, entry 7), we next turned our attention to optimizing the sequential desilylation-CuAAC cycloaddition in a one-pot fashion. It has previously been reported that TMS acetylenes can be deprotected by using several reagents – such as tetrabutylammonium fluoride (TBAF), fluorine salts,23 or silver salts24 – to give terminal alkynes. We chose CsF as the reagent for deprotecting TMSA-5-trifluoromethyl-1H-pyrazole 4a in order to generate the terminal alkyne 5a, which subsequently reacted with 1.5 equivalents of benzyl azide and 10 mol% of CuI to furnish the 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazole 6a (Table 2).
|
||||
|---|---|---|---|---|
| Entry | CsF (eq.) | T (°C) | t (h) | Yieldb [%] |
| a Reaction conditions: pyrazole 3a (0.5 mmol), Pd(OAc)2 (3 mol%), XPhos (6 mol%), TMSA (0.75 mmol), Et3N (1.5 mmol), DMF (2 mL), CuI (10 mol%), and PhCH2N3 (0.75 mmol).b Isolated yields after column chromatography.c Benzyl chloride (0.75 mmol) and NaN3 (0.75 mmol) were used instead of benzyl azide.d 5a was isolated. | ||||
| 1 | 2 | 25 | 24 | 0d |
| 2 | 2 | 50 | 24 | 40 |
| 3 | 2 | 80 | 24 | 58 |
| 4 | 2 | 100 | 24 | 45 |
| 5 | 3 | 80 | 24 | 72 |
| 6c | 3 | 80 | 24 | 64 |
| 7 | 3 | 100 | 24 | 61 |
| 8 | 3 | 80 | 16 | 64 |
| 9 | 3 | 60 | 24 | 63 |
In optimizing the temperature we observed that when the reaction was conducted at 25 °C, only the 4-ethynyl-5-trifluoromethyl-1H-pyrazole 5a was isolated (Table 2, entry 1). However, with an increase in temperature, the formation of the desired product was observed (Table 2, entries 2–4). When the reaction was conducted at 100 °C, decomposition of the product and a lower yield than that obtained at 80 °C was observed. The use of 3 equiv. of CsF was necessary to obtain an optimal yield (Table 2, entry 5). The worst yield resulted when we tried to substitute the use of pre-formed benzyl azide with the in situ formation of benzyl azide, by adding equimolar quantities of benzyl chloride and NaN3 to the reaction (Table 2, entry 6).
Given the optimized reaction conditions (Table 2, entry 5), the scope for the synthesis of 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazoles was explored – the results are shown in Table 3. We investigated the scope of the azide in this reaction by exploring the reactivity of different benzyl, alkyl, and aryl azides. The best results were obtained for the benzyl azides, while the worst results were noted for the alkyl and aryl azides. We also utilized fluorinated azides, such as 1-azido-4-fluorobenzene and 2-(azidomethyl)-1,3-difluorobenzene, given the biological potential that these compounds may have. Compound 6i feature the (2,6-difluorobenzyl)-1,2,3-triazole fragment, that is present in the anticonvulsant Rufinamide, a drug which is used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders.25 All the new products were fully characterized on the basis of GC-MS and 1H/13C NMR spectroscopic data, as well as elemental analysis or high-resolution mass spectrometric data. For the final compounds 6a–o, the 13C{1H} NMR spectra recorded in CDCl3 displayed a through-space 13C–19F spin–spin coupling between the fluorine atoms of the CF3 group and the C-5 carbon of the triazole. The signal for the hydrogenated C-5 carbon of the triazole was observed in the range of 121.1–123.8 ppm, as a broad singlet or a quartet with a JCF between 2–4 Hz. The 19F NMR spectra were recorded for representative compounds. The CF3 signals appeared as a singlet, in average at −55.3 ppm because are very little influenced by both electronic and steric effects. Notably, the average yield obtained for 6a–o (51%) corresponds to an 80% yield across each of the three synthetic steps involved in our one-pot protocol (Sonogashira cross-coupling, desilylation, and copper(I)-catalyzed azide–alkyne cycloaddition).
Conclusions
In summary, we successfully developed a new one-pot, three-step protocol for the synthesis of a series of 15 examples of polysubstituted 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazoles, by employing a sequential Sonogashira cross-coupling, desilylation, and copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC). The presence of a trifluoromethyl group in the pyrazole core made Sonogashira cross-coupling a challenge, and the selection of ancillary ligand XPhos was indispensable for the desired heterocyclic transformation in search of new bioactive molecules. The new useful method applied successful to pyrazoles in this work creates the possibility also for other azoles containing electron-withdrawing groups.Experimental section
General considerations
Unless otherwise indicated, all reagents and solvents used were obtained from commercial suppliers and were used without further purification. The THF was purified by refluxing over Na/benzophenone, followed by distillation. The DMF and MeCN were purified and stored over activated 4 Å molecular sieves. Column chromatography was performed on silica gel (230–400 mesh). The melting points were determined using glass capillary tubes in the Electrothermal MEL-TEMP 3.0 apparatus, with uncorrected values. 1H and 13C spectra were acquired on a Bruker DPX 400 (1H at 400 MHz and 13C at 100 MHz) or a Bruker Avance III HD 600 (1H at 600 MHz and 13C at 150 MHz), with 5 mm sample tubes, 298 K, and digital resolution of 0.01 ppm, in CDCl3 and using TMS as an internal reference. 19F NMR spectra of selected compounds were registered on a Bruker Avance III 600 spectrometer (at 565 MHz), in CDCl3 and using fluorobenzene as external reference with the chemical shifts reported according to the CFCl3 standard. The CHN elemental analyses were performed on a Perkin-Elmer 2400 CHN elemental analyzer (University of São Paulo, SP, Brazil). Gas chromatography was done using an Agilent Technologies 6890N Network GC system, autosampler 7683 series, injector 7683B series, detector 5975 B inert XL EI/CI MSD. High-resolution mass spectra (HRMS) were obtained for all compounds using an LTQ Orbitrap Discovery mass spectrometer (Thermo Fisher Scientific).General procedure for the synthesis of 3-substituted-1-phenyl-5-(trifluoromethyl)-1H-pyrazoles (2a–c)
The pyrazoles were prepared by following the procedures described in the literature.5,15 A solution of phenylhydrazine (5.5 mmol, 0.59 g) in ethanol (20 mL) was added dropwise – via a pipette over the course of 5 min – into a 100 mL round bottom flask containing a magnetic stir bar and a solution of the corresponding enone 1a–c (5 mmol) in ethanol (50 mL), which was kept at 0 °C under magnetic stirring. The system was subsequently left to warm to room temperature over a 1 h period. A reflux condenser was then affixed to the reaction flask, and the flask was submerged in an oil bath, set to 78 °C, that was continually stirred for 24 h. The solvent was removed in a rotary evaporator under reduced pressure, and the residual crude pyrazoles 2a–c were purified via column chromatography, by using 5% ethyl acetate in hexane solution as eluent.General procedure for the synthesis of 4-bromo-1-phenyl-5-(trifluoromethyl)-1H-pyrazoles (3a–c)
Pyrazoles 3a–c were prepared by following the procedures described in the literature.5 An oven-dried 25 mL round bottom flask, equipped with a rubber septum and a stirrer bar, was charged with the pyrazole (2a–c, 5 mmol) and NBS (12.5 mmol, 2.22 g for 2a; 15 mmol, 2.67 g for 2b–c). The system was evacuated and backfilled with argon (operation performed three times in total). Anhydrous DMF (15 mL) was added, and the mixture was stirred at 80 °C for 2 h. Upon cooling to room temperature, the crude reaction mixture was purified via column chromatography (without prior removal of the DMF), by using 2% ethyl acetate in hexane solution as eluent.General procedure for the synthesis of 4-bromo-1-phenyl-5-(trifluoromethyl)-1H-pyrazole (3d)
This procedure was adapted from the literature.16 A phenylhydrazine (5.2 mmol, 0.56 g) was slowly added to a stirred solution of the enone 1e (5.0 mmol, 1.23 g) in AcOH (15 mL), and this was then stirred at 25 °C for 24 h. The reaction was then poured into water and extracted with AcOEt (3 × 20 mL). The combined organic extracts were washed with a solution of NaHCO3 and water, then dried with Na2SO4 and filtered, and the solvent was then removed in a rotary evaporator under reduced pressure. The residual crude pyrazole was purified via column chromatography by using 5% ethyl acetate in hexane solution as eluent.General procedure for the synthesis of azides
The alkyl azides and 1-azido-4-fluorobenzene were prepared by following the procedures described in the literature.27General procedure for the synthesis of 3-methyl-1-phenyl-5-(trifluoromethyl)-4-((trimethylsilyl)ethynyl)-1H-pyrazole (4a)
An oven-dried 5 mL round bottom flask, equipped with a rubber septum and a stirrer bar, was charged with the 4-bromo-3-methyl-1-phenyl-5-(trifluoromethyl)-1H-pyrazole 3a (0.5 mmol, 152.5 mg), Pd(OAc)2 (3 mol%, 3.35 mg), and ligand (L1–L5, 6 mol%). The system was evacuated and backfilled with argon (operation performed three times), then Et3N (1.5 mmol, 0.21 mL), TMSA (0.75 mmol, 0.11 mL), and dry solvent (2 mL) were added successively in an argon atmosphere. The round bottom flask was sealed and the mixture was stirred at the indicated temperature for 24 h. The crude reaction mixture was then cooled to room temperature, filtered through a silica pad with ethyl acetate (60 mL), and the filtrate was concentrated under reduced pressure and the pyrazole 4a was purified via column chromatography, using 2% ethyl acetate in hexane solution as eluent.General procedure for the synthesis of 4-(5-(trifluoromethyl)-1H-pyrazol-4-yl)-1H-1,2,3-triazoles (6a–o)
An oven-dried 5 mL round bottom flask, equipped with a rubber septum and a stirrer bar, was charged with the 4-bromopyrazoles (3a–d, 0.5 mmol), Pd(OAc)2 (3 mol%, 3.35 mg), and XPhos (6 mol%, 14.3 mg). The system was evacuated and backfilled with argon (operation performed three times), and then Et3N (1.5 mmol, 0.21 mL), TMSA (0.75 mmol, 0.11 mL), and DMF (2 mL) were added successively in an argon atmosphere. The round bottom flask was sealed, and the mixture was stirred at 100 °C for 24 h. After cooling to room temperature, the system was opened, and CsF (1.5 mmol, 0.227 g), CuI (10 mol%, 10.0 mg), and organic azide (0.75 mmol) were added. The mixture was then stirred at 80 °C for 24 h. The crude reaction mixture was then cooled to room temperature, filtered through a silica pad with ethyl acetate (80 mL), and the filtrate was concentrated under reduced pressure. Purification by flash column chromatography furnished the desired product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank: the Coordination for Improvement of Higher Education Personnel (CAPES) for the fellowships, and the National Council for Scientific and Technological Development (CNPq) for the financial support (Process numbers 306.883/2015-5 and 402.075/2016-1-CNPq/Universal).Notes and references
- V. Nenajdenko, Fluorine in Heterocyclic Chemistry Vol. 1:5-Membered Heterocycles and Macrocycles, Springer-Verlag, Heidelberg, 2014 Search PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- C. D. Murphy and G. Sandford, Expert Opin. Drug Metab. Toxicol., 2015, 11, 589 CrossRef CAS PubMed.
- (a) H. G. Bonacorso, W. C. Rosa, S. M. Oliveira, I. Brusco, E. S. Brum, M. B. Rodrigues, C. P. Frizzo and N. Zanatta, Bioorg. Med. Chem. Lett., 2017, 27, 1551 CrossRef CAS PubMed; (b) M. M. Lobo, S. M. Oliveira, I. Brusco, P. Machado, L. F. S. M. Timmers, O. N. de Souza, M. A. P. Martins, H. G. Bonacorso, J. M. dos Santos, B. Canova, T. V. F. da Silva and N. Zanatta, Eur. J. Med. Chem., 2015, 102, 143 CrossRef CAS PubMed; (c) S. M. Oliveira, C. R. Silva, A. P. Wentz, G. R. Paim, M. S. Correa, H. G. Bonacorso, A. S. Prudente, M. F. Otuki and J. Ferreira, Pharmacol., Biochem. Behav., 2014, 124, 396 CrossRef CAS PubMed; (d) G. Trevisan, M. F. Rossato, C. I. B. Walker, S. M. Oliveira, F. Rosa, R. Tonello, C. R. Silva, P. Machado, A. A. Boligon, M. A. P. Martins, N. Zanatta, H. G. Bonacorso, M. L. Athayde, M. A. Rubin, J. B. Calixto and J. Ferreira, Neuropharmacology, 2013, 73, 261 CrossRef CAS PubMed; (e) J. Milano, M. F. Rossato, S. M. Oliveira, C. Drewes, P. Machado, P. Beck, N. Zanatta, M. A. P. Martins, C. F. Mello, M. A. Rubin, J. Ferreira and H. G. Bonacorso, Life Sci., 2008, 83, 739 CrossRef CAS PubMed; (f) P. D. Sauzem, P. Machado, M. A. Rubin, G. da S. Sant'Anna, H. B. Faber, A. H. de Souza, C. F. Mello, P. Beck, R. A. Burrow, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Eur. J. Med. Chem., 2008, 43, 1247 CrossRef PubMed; (g) W. Cunico, C. A. Cechinel, H. G. Bonacorso, M. A. P. Martins, N. Zanatta, M. V. N. de Souza, I. O. Freitas, R. P. P. Soares and A. U. Krettlia, Bioorg. Med. Chem. Lett., 2006, 16, 649 CrossRef CAS PubMed.
- C. Wiethan, W. C. Rosa, H. G. Bonacorso and M. Stradiotto, Org. Biomol. Chem., 2016, 14, 2352 CAS.
- K. M. Clapham, A. S. Batsanov, M. R. Bryce and B. Tarbitb, Org. Biomol. Chem., 2009, 7, 2155 CAS.
- T. S. Reddy, H. Kulhari, V. G. Reddy, A. V. S. Rao, V. Bansal, A. Kamal and R. Shukla, Org. Biomol. Chem., 2015, 13, 10136 Search PubMed.
- N. R. Emmadi, C. Bingi, S. S. Kotapalli, R. Ummanni, J. B. Nanubolu and K. Atmakur, Bioorg. Med. Chem. Lett., 2015, 25, 2918 CrossRef CAS PubMed.
- R. Distinto, C. Zanato, S. Montanari, M. G. Cascio, P. Lazzari, R. Pertwee and M. Zanda, J. Fluorine Chem., 2014, 167, 184 CrossRef CAS.
- C. Zanato, M. G. Cascio, P. Lazzari, R. Pertwee, A. Testa and M. Zanda, Synthesis, 2015, 47, 817 CrossRef CAS.
- N. Nayak, J. Ramprasad, U. Dalimba, P. Yogeeswari, D. Sriram, H. S. S. Kumar, S. K. Peethambar and R. Achur, Res. Chem. Intermed., 2016, 42, 3721 CrossRef CAS.
- X. Chen, Y. Xiao, G. Wang, Z. Li and X. Xu, Res. Chem. Intermed., 2016, 42, 5495 CrossRef CAS.
- M. Maue, A. Decor, J. J. Hahn, W. Hallenbach, R. Fischer, H. G. Schwarz, U. Görgens, K. Ilg, K. Raming and J. Köbberling, Int. Pat., WO 2015193218A1, 2015.
- E. Merkul, F. Klukas, D. Dorsch, U. Gradler, H. E. Greiner and T. J. J. Muller, Org. Biomol. Chem., 2011, 9, 5129 CAS.
- (a) M. Hojo, R. Masuda, Y. Kokuryo, H. Shioda and S. Matsuo, Chem. Lett., 1976, 5, 499 CrossRef; (b) M. Hojo, R. Masuda and E. Okada, Synthesis, 1986, 1013 CrossRef CAS; (c) G. M. Siqueira, A. F. C. Flores, G. Clar, N. Zanatta and M. A. P. Martins, Quim. Nova, 1994, 17, 24 CAS; (d) H. G. Bonacorso, M. A. P. Martins, S. R. T. Bittencourt, R. V. Lourega, N. Zanatta and A. F. C. Flores, J. Fluorine Chem., 1999, 99, 177 CrossRef CAS.
- V. G. Nenajdenko, A. L. Reznichenko and E. S. Balenkova, Russ. Chem. Bull., 2006, 55, 172 CrossRef CAS.
- I. I. Gerus, L. M. Kacharova and S. I. Vdovenko, Synthesis, 2001, 431 CrossRef CAS.
- D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993 CrossRef CAS PubMed.
- R. W. Burli, W. R. K. Esmieu, C. J. Lock, K. F. Malagu, A. P. Owens and W. E. Harte, Int. Pat., WO2014066836A1, 2014.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- W. Shu and S. L. Buchwald, Chem. Sci., 2011, 2, 2321 RSC.
- A. Komaromi and Z. Novak, Chem. Commun., 2008, 4968 RSC.
- F. Friscourt and G.-J. Boons, Org. Lett., 2010, 12, 4936 CrossRef CAS PubMed.
- V. Aucagne and D. A. Leigh, Org. Lett., 2006, 8, 4505 CrossRef CAS PubMed.
- S. Hakimian, A. Cheng-Hakimian, G. D. Anderson and J. W. Miller, Expert Opin. Pharmacother., 2007, 8, 1931 CrossRef CAS PubMed.
- T. N. Ngo, S. A. Ejaz, T. Q. Hung, T. T. Dang, J. Iqbal, J. Lecka, J. Sevigny and P. Langer, Org. Biomol. Chem., 2015, 13, 8277 CAS.
- K. Yamamoto, T. Bruun, J. Y. Kim, L. Zhang and M. Lautens, Org. Lett., 2016, 18, 2644 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra. See DOI: 10.1039/c7ra07960e |
| This journal is © The Royal Society of Chemistry 2017 |