
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid crystal growth of bimetallic PdPt nanocrystals with surface atomic Pt cluster decoration provides promising oxygen reduction activity†
Kuan-Wen Wang a,
Zhuang Yub,
Alice Hub,
Yang-Yang Hsuc,
Tian-Lin Chena,
Cheng-Yang Lind,
Chih-Wei Huc,
Ya-Tang Yangd and
Tsan-Yao Chen*ce
a,
Zhuang Yub,
Alice Hub,
Yang-Yang Hsuc,
Tian-Lin Chena,
Cheng-Yang Lind,
Chih-Wei Huc,
Ya-Tang Yangd and
Tsan-Yao Chen*ce
aInstitute of Materials Science and Engineering, National Central University, Taiwan
bDepartment of Mechanical and Biomedical Engineering, City University of Hong Kong, Kowloon, Hong Kong
cDepartment of Engineering and System Science, National Tsing Hua University, Hsinchu 30013, Taiwan. E-mail: chencaeser@gmail.com; Tel: +886-3-5715131 ext. 34271
dDepartment of Electrical Engineering, Institute of Electronic Engineering, National Tsing Hua University, Hsinchu 30013, Taiwan
eInstitute of Nuclear Engineering and Science, National Tsing Hua University, Hsinchu 30013, Taiwan
First published on 5th December 2017
Abstract
Carbon nanotube-supported bimetallic Pd nanoparticles (NPs) with atomic Pt cluster surface decoration (the mole ratio of Pt/Pd is 0.1) are synthesized by a wet chemical method with sodium borohydride as the reducing agent. The core component is controlled by metal ion reduction sequences. The mass activity for the oxygen reduction reaction (ORR) over Pt metal of the presented PdPt NPs is improved by 29.4 times as compared to that of commercial Pt catalysts. Such an enhancement is accounted by an expanded Pt–Pd bond length and heteroatomic intermixing-induced electron relocation.
Introduction
Atomic configuration could be a dominant factor in the oxygen reduction reaction (ORR) activity of bimetallic nanocatalysts. Our previous studies and literature have proved that nanowires and nanorods possess enhanced ORR activity and chemical durability when compared with Pt NPs.1 Such ORR enhancement on Pt-based nanomaterials could be attributed to their low roughness contact (i.e., reduced contact resistance) with the carbon surface, preferential exposure of crystal facets to reaction media, and properly arranged heteroatomic Pt–Pd bond pairs (i.e., facilitated oxygen reduction pathways) on crystal surfaces.2 Apart from composition differences, recent studies have revealed that the electronic and chemical states of NPs can be manipulated via formation of 1-D structures with surface alloying, thus varying the surface oxophilicity and substantially enhancing the ORR performance.3An interesting approach to ORR optimization is to relocate the Pt cluster location on cathode NPs. Because of electron injection effects via local lattice strain and structure configuration, Pd NP with slight atomic Pt clusters decorated in near-surface region shows outstanding ORR activity and thus promoting the development of low temperature fuel cells with low fabrication cost.4 It has been reported that the formation of intra-particle heterogeneous interface (particularly cluster-in-cluster, surface modification, and core–shell structure) in NP can substantially improve their stability and catalytic activity as compared to that of homogeneous alloy. By proper decoration of atomic Pt clusters on Cocore–Pdshell NP, the result from long-term accelerated degradation test (ADT) demonstrated that the numbers of ORR working cycles increase by 10 times and the device efficiency is improved by 55–65% as compared to the commercial Pt catalyst.5 In such a NP, effects of local lattice strain near crystal surface are one of the most influential factors in its electrochemical properties.6
In this work, we demonstrate that ORR activity of PdPt NPs can be substantially improved by ∼29.4 times over Pt metal by decorating Pt clusters with proper atomic structure in the near-surface region as compared to commercial Pt catalyst. Using robust wet chemical fabrication processes, such an NP with Pt metal loading less than 10 wt% could be a potential candidate of low-cost catalyst among fuel cell cathode materials. We also show that the ORR activity of PdPt NPs is optimized by controls of reaction methods. The effect of atomic structure and Pd alloying on ORR performance of PdPt NPs has been elucidated by cross-referencing results of electrochemical analyses (including linear sweep voltammetry (LSV), CO stripping, and CV sweeping curve), X-ray diffraction (XRD), X-ray absorption spectroscopy (XAS), X-ray photoemission spectroscopy (XPS), and high-resolution transmission electron microscopy (HRTEM) characterizations. For clarifying effects of atomic configuration on ORR reactivity, structures and electrochemical properties of control samples (CNT supported Pt and Pd NP prepared by identical method to that of PdPt-s) are compared in this study.
Experimental
Preparation of Pt cluster decorated Pd nanoparticles and electrode for ORR activity test
Atomic Pt clusters decorated Pd NP were prepared by using a wet chemical reduction method with sequential control. Before metallic NPs growth, carbon support (carbon nanotube “CNT from Cnano Technology Ltd.”), was acid treated by immersing in aqua solution of 4.0 M sulphuric acid at 80 °C for 6 h and then washed by distilled water till pH value of washing liquid was 6.0. Processes and schematic representation for growing PdPt-s NPs is shown in Fig. 1. In step 1, 50.0 mg of acid treated CNT powder was immersed in aqueous solution of Pd(IV) (Pd(NO3)4, 99%, Sigma-Aldrich Co.) and stirred at 250 rpm at 25 °C for 4 h resulting in Pd4+/2+ chemisorption on CNT (CNT-Pdads). In this mixture (solution A), Pd/CNT ratio is 30 wt% (i.e., Pd = 15 mg) and distilled water is 30 g. The Pd precursor solution was prepared by dissolving metal powder (Pd, 99%, Sigma-Aldrich Co.) in 1.0 M HCl(aq). After immersion, 3.0 g of solution B containing 10.0 mg (0.252 mmol) NaBH4 (99%, Sigma-Aldrich Co.) was added to solution A and stirred at 250 rpm for 15 s in step 2. In step 3, Pt precursor solution containing 1.0 g distilled water and ∼6.42 mg (∼0.032 mmol) of Pt ions was added in solution A with a molar ratio of Pt/Pd = 0.1. Pt precursor, 17.05 mg of H2PtCl6·6H2O (Sigma-Aldrich, CAS number: 18497-13-7 206083) was dissolved in distilled water at room temperature. In step 3, atomic Pt clusters were decorated on the Pd NP surface via a galvanic replacement of Pd metal with Pt4+ ion simultaneous reduction of residual metal ions (Pd4+ and Pt4+) with H: radicals. To complete metal reduction, additional reduction agent with identical amount of NaBH4 to that in solution B was added to solution A in step 4 subsequent to the addition of Pt4+ precursor and thus resulting in atomic Pt clusters decorated Pd NP (namely PdPt-s). Preparation of Pd-s followed similar processes as those used to prepare PdPt-s, except that only Pt (Pd) precursor was used in reaction system (i.e., only process to step 3 for preparing Pd-s). For preparing Pt-s, Pt4+ precursor was used in step 1 and reaction proceeded to step 3.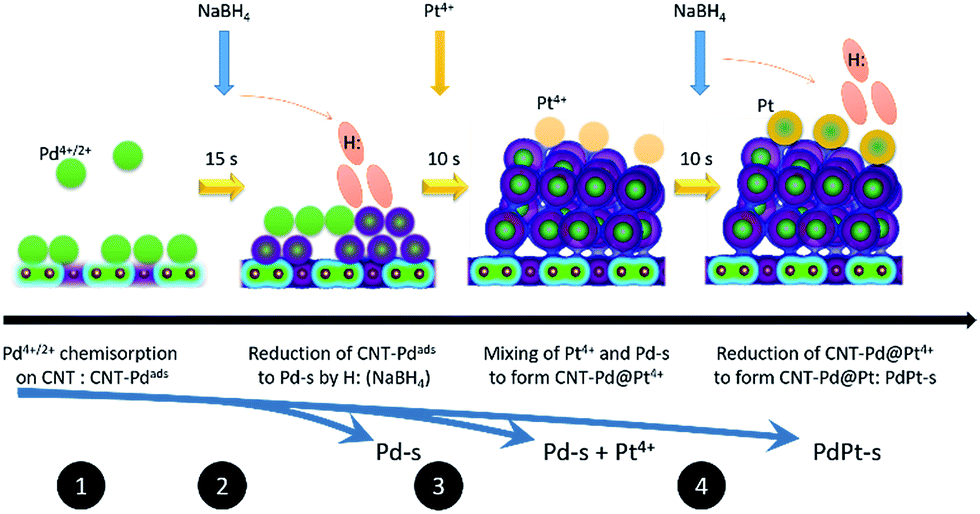 | ||
| Fig. 1 Schematic representation of reaction steps for preparing bimetallic PdPt nanoparticles by wet chemical reduction via interacting with NaBH4. | ||
As for PdPt alloy NPs (namely PdPt-p), polyol reduction method was employed. In reaction system, water solutions of Pt and Pd metal precursor (molar ratio of Pt/Pd = 0.1) were mixed in 30 g of ethylene glycol (EG, 99 wt%, Sigma-Aldrich Co.) in the presence of 10 wt% (vs. EG) PVP polymer stabilizer (PVP-40, MW = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 99%, Sigma-Aldrich Co.). After mixing at 25 °C with a stirring speed of 250 rpm for 1 h, the mixture was heated at 160 °C for 2 h in formation of polymer stabilized PdPt NP in EG (solution C). PdPt NP was extracted by a solvent exchange method to ethanol solution in the previous work.7 The PdPt-p NPs was then mixed with acid treated CNT in ethanol (Pd/CNT ratio is 30 wt%) at a stirring speed of 250 rpm for more than 12 h to form CNT supported PdPt-p. For stabilizing NC in CNT surface, PdPt-p powders were filtered, dried at 75 °C for 3 h, and then annealed in Ar ambient at 270 °C for 1 h.
000, 99%, Sigma-Aldrich Co.). After mixing at 25 °C with a stirring speed of 250 rpm for 1 h, the mixture was heated at 160 °C for 2 h in formation of polymer stabilized PdPt NP in EG (solution C). PdPt NP was extracted by a solvent exchange method to ethanol solution in the previous work.7 The PdPt-p NPs was then mixed with acid treated CNT in ethanol (Pd/CNT ratio is 30 wt%) at a stirring speed of 250 rpm for more than 12 h to form CNT supported PdPt-p. For stabilizing NC in CNT surface, PdPt-p powders were filtered, dried at 75 °C for 3 h, and then annealed in Ar ambient at 270 °C for 1 h.
Methods for preparing slurry samples on electrode in ORR activity test was adopted from previous studies and literature.8 Details of slurry, electrode preparations and electrochemical characterizations are given in (ESI†).
Physical structure characterizations on PdPt nanoparticles
The physical properties of experimental NPs were determined by microscopic (HRTEM) and spectroscopic (X-ray absorption and X-ray photoemission) characterizations at electron microscopy center of National Sun Yat-Sen University and beamlines (BL-17C, 01C1, and BL-24A1) at National Synchrotron Radiation Research Center (NSRRC), Taiwan, respectively. The XRD patterns were collected at beamline of BL-12B2 at Spring-8, Japan.Results and discussion
Crystal structure characterizations
Surface morphology and crystal structure of experimental NPs are determined by TEM analysis (line histogram denoted by yellow double arrow, high resolution image, and low magnification image are shown in Fig. 2 and its inset). Shown in Fig. 2a, a slight angle (Ø 1) between the two (111) lattice fringes (with an interplanar spacing “d(111)” of 2.257 ± 0.03 Å) in an absence of mirror reflection images at two sides of selected axis (yellow double arrows) indicates the stacking of two Pt NPs in Pt-s. Denoted by yellow arrows, Pt-s possesses prevailing defects in NP surface. Such a surface morphology is attributed to the absence of stabilizers upon crystal growth and is revealed by aggregation of Pt NPs (Fig. 2a inset) and the random displacement of interatomic distances between Pt atoms (R1, R2, and R3 shown in line histogram). The distinct inter-particle boundaries and sharp surface morphology indicate the strong self-stabilization of Pt NP that reaches a critical size of heterogeneous crystallite. Above characteristics reveal the fatal issues of high surface free energy defect sites and a lack of heteroatomic bonding for Pt NPs in electrochemical reactions. Those issues cause easy corrosion and atomic restructure of edge atom on surface of Pt NPs (Fig. S1†) in electrochemical characterizations. Therefore, it is expectable to see the low electrochemical stability of Pt-s in an accelerated degradation test (ADT) of oxygen reduction reaction (ORR) (see Fig. 4). Details for structural and electrochemical correlations will be discussed in the latter section. HRTEM image and line histogram of CNT supported Pd NPs (Pd-s) are demonstrated in Fig. 2b. As demonstrated in Fig. 2b inset, Pd NPs are drop-like particles with indistinct inter-particle boundaries, revealing a severe inter-particle collapse as a result of instability (easy oxidation) of Pd NPs (also proved by Pd K-edge XAS analysis). Consequently, it is inevitable for Pd-s to face a similar fate with Pt-s (low electrochemical performances) at both ORR and ADT (Fig. 4). Comparing to that of Pt-s, Pd-s has less interatomic displacement (R1 and R2) in line histogram. Such a feature can be explained by reduction of surface defects via atomic restructure upon interparticle collapse. Fig. 2c shows that PdPt-s NPs are grown in decahedron-like crystal; where d(111) is 2.295 ± 0.03 Å and d(200) is 2.003 ± 0.03 Å. Compared to that of metallic Pt, d(111) and d(200) of PdPt-s are expanded by 1.30% and 2.01%, respectively. Such an uneven lattice displacement explains the preference of Pt allocation at Pd {111} facets by minimization of total surface free energy in a NP system.9 Given that atomic diameter of Pd is smaller than that of Pt, observed lattice expansion in PdPt-s is a combination factor including lattice mismatch, intercalation of Pt atoms at surface hollow sites (proved by bond distances around local coordination shell in EXAFS analysis), and oxidation induced lattice displacement. Those factors are all related to the prevailing surface defect sites on incoherent interface between Pd and Pt crystal upon allocating Pt atoms in Pd NP surface. Among NPs synthesized by NaBH4 reduction (Pt-s, Pd-s, and PtPt-s), PdPt-s possesses the highest extent of uneven atomic distribution (proved by DEX map image in Fig. S2† and line histogram across selected lattice fringe in Fig. 2c) as a result of its largest extent of surface oxide and heteroatomic intermix. HRTEM image, line histogram (Fig. 2d), and EDX map (Fig. S2c†) elucidate the homogeneous distribution of Pd and Pt atoms with ordered atomic displacement in cubic-like NP of PdPt-p.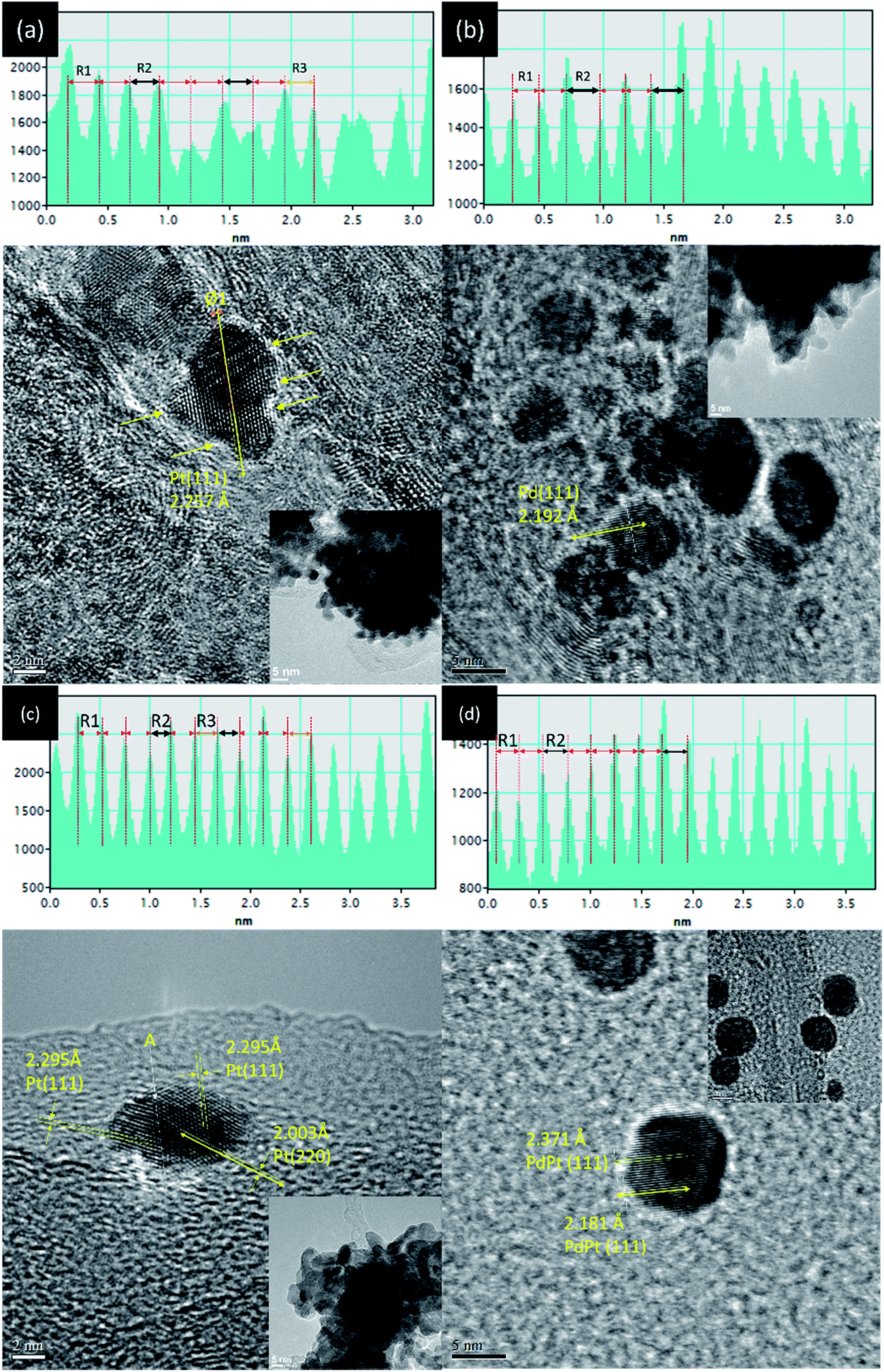 | ||
| Fig. 2 HRTEM images and selected line histogram (across yellow double arrows) of (a) Pt-s, (b) Pd-s, (c) PdPt-s, and (d) PdPt-p NPs. | ||
Fig. 3 compares XRD patterns of experimental NPs (Pt-s, Pd-s, PdPt-s, and PdPt-p), where structural parameters are summarized in Table 1. Accordingly, the three peaks A, B, and C at 39.91°, 46.33°, and 67.74° are diffraction lines of Pt/Pd (111), (200), and (220) planes with d(hkl)_Pt/coherent lengths (D(hkl)_Pt) of 2.256 Å/68.2 Å, 1.957 Å/51.2 Å, and 1.382 Å/47.4 Å, respectively. The varying D(hkl)_Pt at different planes indicates an anisotropic crystal growth between the three facets in Pt-s. For PdPt-s, diffraction lines of the three facets are shifted to lower angles, revealing lattice expansion by 1.52% to 2.15% as compared to that of metallic Pd planes due to formation of Pt to Pd alloy in NP. Uneven lattice strain indicates the preferential positioning of Pt atoms at Pd (111) and (220) facets with substantial displacement between Pt–Pd atoms (also proved by XAS characterization in latter section). As compared to metallic Pt lines, uneven shifts in (111) and (200) lines reveal the preferential local alloying (by galvanic replacement between Pt4+ ions and Pt metal atoms) in (200) planes of PdPt-s. In PdPt-s, Davg is 41.8 Å for (111), 31.5 Å for (200), and 29.4 Å for (220) planes. The largest D(111) again revealing the preferential crystal growth in (111) planes. High local micro-compression strain and Pd–Pt alloy with small Davg together suggest that a considerable extent of local alloying between Pt4+ ion and metallic Pd in (220) planes. Such a phenomenon is expectable considering the nature of surface free energy (γ) minimization by placing Pt atoms on open facets (Pd (220)).10 Given that identical carbon support is employed for all NPs, presence of diffusion scattering (denoted by Q and Q*) at two sides of main diffraction lines implies the high roughness NP surface in PdPt-s by Pt clusters decoration and will be consistently proved by XAS analysis. For XRD pattern of PdPt-p, downshift of diffraction lines to the same extent illustrates a formation of homogeneous PdPt alloy with a similar Davg in the three planes. Meanwhile, PdPt-p shows a strongest preferential growth at (111) facets among experiment NPs. Such a preferential growth is triggered by steric effect of pyrrolidone ligands11 capping on opened facets for minimizing the system energy.9a Size distribution and Davg of experimental NPs are further investigated by TEM analysis with consistent results shown in Fig. S3.†
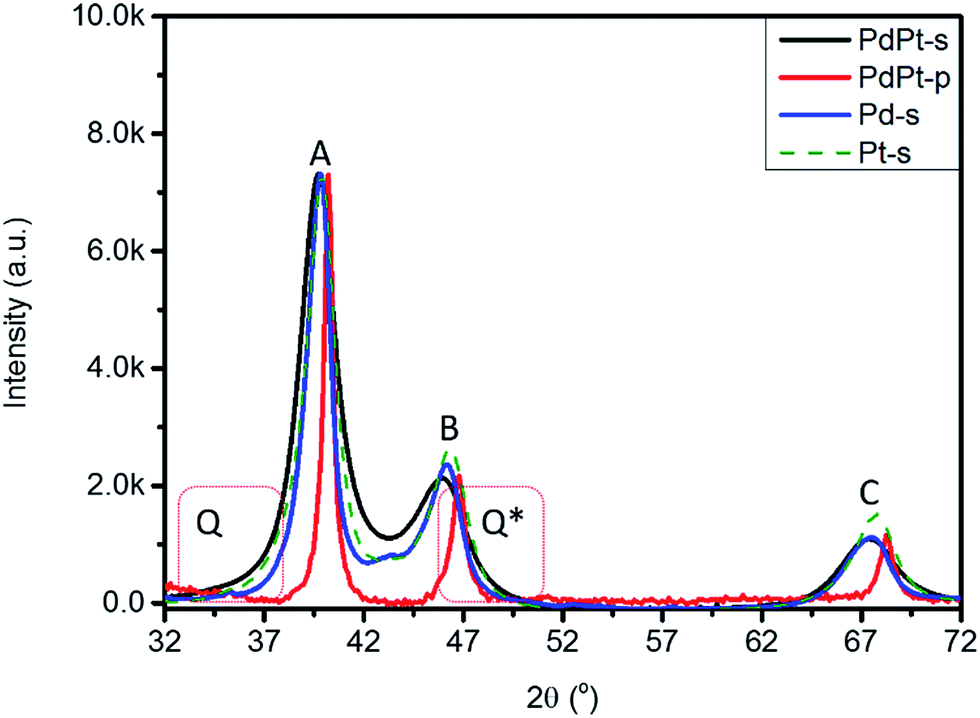 | ||
| Fig. 3 XRD patterns of Pt-s, Pd-s, PdPt-s, and PdPt-p. Q and Q* denote diffusion scattering from NP surface and out-of-phase scattering signals via micro-strain. | ||
| NPs | Facets | 2θ | D (Å) | d (Å) | a (Å) | ε vs. Pta | ε vs. Pda |
|---|---|---|---|---|---|---|---|
| a Inter-planar distance (IPD) of ideal Pt/Pd metals is 2.263/2.233 Å for (111), 1.960/1.934 Å for (200), and 1.386/1.368 Å for (220) planes. | |||||||
| Pt-s | (111) | 39.91 | 68.2 | 2.256 | 3.908 | −0.29 | |
| (200) | 46.33 | 51.2 | 1.957 | 3.915 | −0.12 | ||
| (220) | 67.74 | 47.4 | 1.382 | 3.908 | −0.29 | ||
| Pd-s | (111) | 40.37 | 68.2 | 2.260 | 3.914 | ||
| (200) | 46.96 | 51.1 | 1.966 | 3.932 | |||
| (220) | 67.79 | 39.3 | 1.368 | 3.906 | |||
| PdPt-s | (111) | 39.72 | 41.8 | 2.267 | 3.926 | 0.18 | 1.52 |
| (200) | 45.89 | 31.5 | 1.975 | 3.951 | 0.79 | 2.15 | |
| (220) | 67.32 | 29.4 | 1.389 | 3.930 | 0.26 | 1.58 | |
| PdPt-p | (111) | 40.21 | 75.6 | 2.242 | 3.883 | −0.94 | 0.39 |
| (200) | 46.78 | 71.5 | 1.940 | 3.880 | −1.02 | 0.31 | |
| (220) | 68.24 | 73.4 | 1.373 | 3.883 | −0.93 | 0.37 | |
Position of Pt atoms and the nanostructure of PdPt NPs are further confirmed by XAS characterisation. Fig. 4a compares the normalized Pt L3-edge X-ray absorption near-edge spectra (XANES) of PdPt-s, PdPt-p, and Pt-s. In an L3-edge spectrum, position of inflection point (arrow X) refers to a threshold energy (E0) for 2p to 5d orbital transitions and is proportional to the oxidation state of transition metals. Intensity (ha) and width (Wa) of near-edge absorption peak (peak A) correspond to the extent of empty states and splitting of 5d5/2 orbitals. The width (WB) and intensity (hB) of oscillation hump in post-edge region elucidate the extent of local atomic structure ordering. Shown by similar inflection position, the metallic characteristics of Pt atoms in all NPs are evident while a significantly reduced hB denotes the presence of high density local structure defect (or disordering) around Pt atoms in PdPt-s. Compared to ha of Pt-s and PdPt-p, an increase of intensity by ∼7.7% features the prevailing oxygen chemisorption. Atomic structure of PdPt-s is consistently confirmed by extended X-ray absorption fine structure (EXAFS) spectrum (Fig. 4b and fitting results Table 2). As determined, Pt atoms are stacked as tri-Pt-atom clusters (CNPt–Pt = 2, peak E) atop Pd NP surface. The substantial splitting between peaks E and E* is due to typical destructive interferences between signals from Pt–Pt and Pt–Pd bond pairs with similar amount of coordination numbers (CNPt–Pt = 2.09 and CNPt–Pd = 1.04), where oxygen CN (peak D) per Pt atom is ∼1.5.
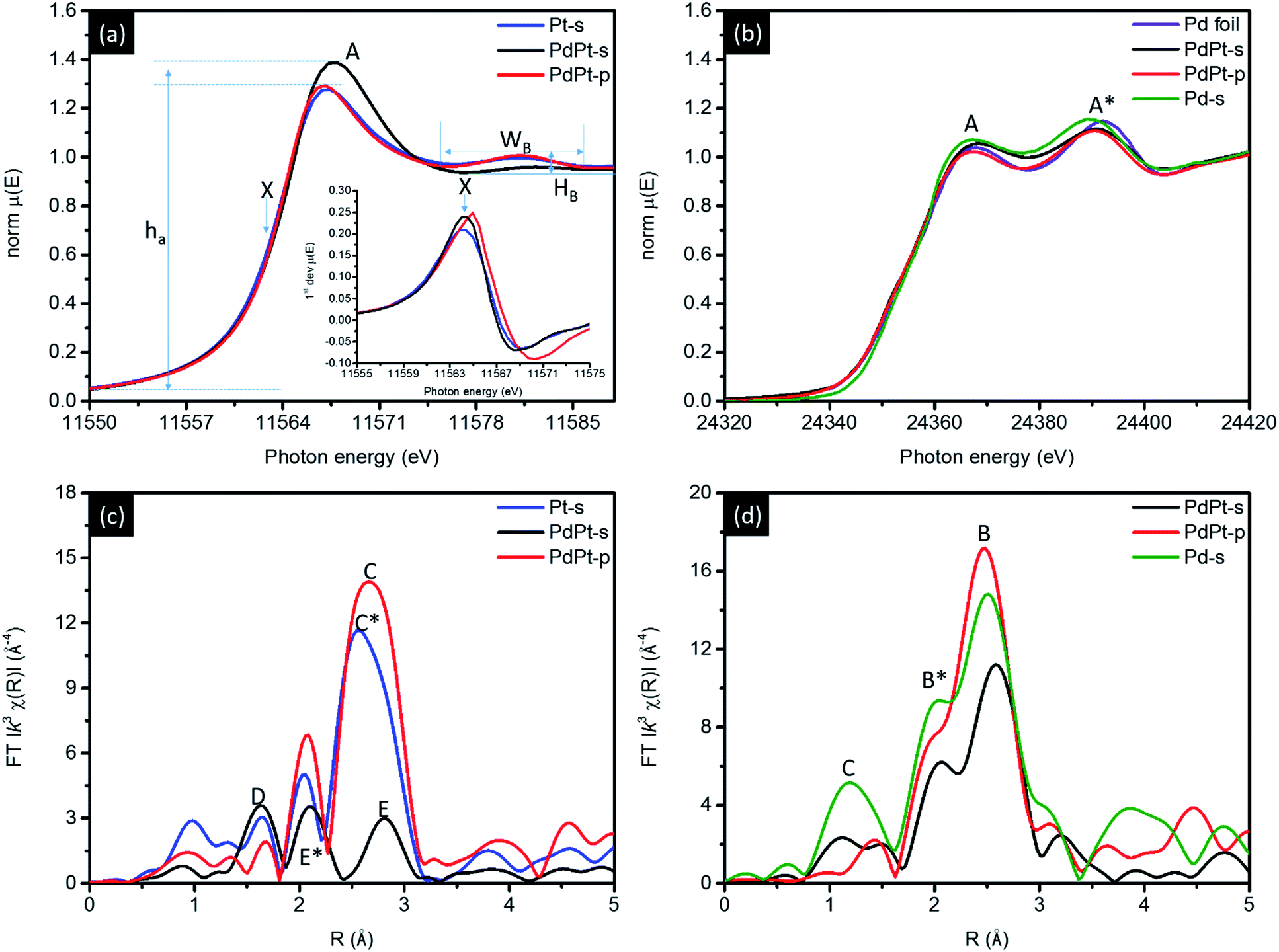 | ||
| Fig. 4 (a) Pt L3-edge, (b) Pd K-edge XANES and EXAFS spectra for CNT supported NPs. Corresponding EXAFS spectra are shown in (c) and (d). | ||
| Edge | EXAFS: local atomic structure | XANES: compositionb | ||||||
|---|---|---|---|---|---|---|---|---|
| NPs | Path | R | CN | χ(Pt–Pt)a | χ(Pd–Pt)a | Pt metal (%) | Pt oxide (%) | |
| a χ(Pt) and χ(Pd) are determined by dividing CNPt–Pt and CNPt–Pd by CNtotal at Pt L3-edge or CNPd–Pt and CNPd–Pd by CNtotal at Pd K-edge. CNtotal is determined by summation of CN for all metallic coordination shells (i.e., CNtotal = CNPt–Pt + CNPt–Pd at Pt L3-edge and CNtotal = CNPd–Pt + CNPd–Pd at Pd K-edge). High error bar of CN suggest the nearly absence of path in analyzed NPs (for example, Pt–O of PdPt-p).b Pt composition is determined by using linear combination fitting (LCF) method. Pd combinations are not determined due to the resolution of LCF method in very limited differences between spectra of NPS at Pd K-edge. | ||||||||
| Pt L3 | Pt-s | Pt–Pt | 2.754 | 6.45 (±0.51) | 100 | 0 | 84.6 | 15.4 |
| PdPt-s | Pt–O | 1.989 | 1.52 (±0.23) | 66.7 | 33.3 | 61.9 | 33.1 | |
| Pt–Pt | 2.747 | 2.09 (±0.29) | ||||||
| Pt–Pd | 2.730 | 1.04 (±0.19) | ||||||
| PdPt-p | Pt–O | 1.989 | 0.09 (±0.09) | 88.1 | 11.9 | 81.7 | 18.3 | |
| Pt–Pt | 2.756 | 7.35 (±0.26) | ||||||
| Pt–Pd | 2.754 | 0.99 (±0.58) | ||||||
| Pd K | NPs | Path | R | CN | χ(Pt–Pd) | χ(Pd–Pd) | Pd metal (%) | Pd oxide (%) |
|---|---|---|---|---|---|---|---|---|
| Pd-s | Pd–O | 2.065 | 2.15 (±0.42) | 0 | 100 | NA | NA | |
| Pd–Pd | 2.762 | 5.12 (±0.35) | ||||||
| PdPt-s | Pd–O | 2.062 | 0.36 (±0.11) | 27.4 | 74.6 | |||
| Pd–Pd | 2.766 | 5.54 (±0.91) | ||||||
| Pd–Pt | 2.787 | 2.09 (±0.52) | ||||||
| PdPt-p | Pd–O | 2.095 | 0.4 (±0.14) | 13.4 | 86.6 | |||
| Pd–Pd | 2.751 | 7.31 (±0.61) | ||||||
| Pd–Pt | 2.778 | 1.13 (±0.28) |
A non-stoichiometry of oxygen CN can be attributed to co-existence of chemisorbed oxygen atoms in atop sites and edge bridge sites between Pt and neighbouring Pd atoms. In the absence of stabilizer agents in crystal growth, a low oxygen CN at Pd atoms (0.36) with a high oxygen CN at metallic Pt atoms (∼1.5) are evidences to formation of island-like-growth Pt clusters as shielding layer capping in PdPt-s surface region. Such an island-like surface Pt-cluster is consistently revealed by the presence of diffusion scattering in XRD patterns and the suppressed post-edge scattering hump in XANES. Results of local atomic structure around Pd atoms complimentary verify such a hypothesis. As indicated in Table 2, CNPd–Pd and CNPd–Pt are 5.54 and 2.09, respectively. Compared with local structure around Pt atoms, similar preference of hetero-atomic intermix (χ) is found both around Pt (χ(Pd–Pt)) and Pd (χ(Pt–Pd)) atoms. Therefore, a greater total CN of Pd than Pt atoms and high CNPt–O elucidate the formation of Pd core crystal with high density of surface Pt cluster decoration. For EXAFS spectrum of PdPt-p, compared to signal of metallic Pt–Pt bond in Pt-s spectrum (peak C*), upshift of atomic bond pair signal (peak C) accounts for a slight out-of-phase interference between X-rays interacting with Pt–Pt (CN = 7.35) and Pt–Pd (CN = 0.99) bond pairs. In the meantime, CN is 7.31 for Pd–Pt and 1.13 for Pd–Pt bond pairs around Pd atoms. Above quantitative structural parameters suggest that Pt and Pd atoms tend to form homo-atomic clusters in PdPt-p. Such a hypothesis is consistently confirmed by results of XRD, EDX map images (Fig. S2†), XPS (Fig. S6† and Table S1†), and CO stripping (Fig. S7†) analyses.
The decoration of Pt clusters on NP surface is further confirmed by cross-referencing results of local structure inspection (Fig. 4c and d and Table 2), geometrical correlation (Fig. S4†), and electrochemical analyses (Fig. 5 and S5†). Fig. 4c compares the Pd K-edge XANES spectra of experimental NPs, where two sorption peaks A and A* are typical characteristics of metallic Pd atom. As indicated, PdPt-p shows similar spectra features to that of Pd foil which confirms majority of Pd atoms are in metallic state. Such a result is consistently revealed by its radial distribution profile in EXAFS spectra (Fig. 4d), where peaks B and B* are respectively the X-ray interferences from Pd–Pd and Pd–Pt bond pairs. For PdPt-s, the two radial peaks are shifted to high radial site to different extents elucidating the extension of interatomic bond length and the local distortion between Pd and Pt atoms. Those characteristics are complimentary disclosed by uneven micro-strain and local alloying on different planes of PdPt-s (Table 1). It is possible to have mild solid solution of Pd–Pt in PdPt-s surficial region. Given that the reaction rate of metal ion reduction by H: radicals (nano second) is far higher than that of ion–metal exchange rate in galvanic replacement, surface decoration of Pt clusters could be the main structure in PdPt-s. This statement is consistently explained by high Pt–O CN and low Pt–Pt CN of PdPt-s. The surface decoration of Pt clusters is further revealed by CV, XAS, XPS (Fig. S6†), and CO stripping (Fig. S7†) analysis. More evidences are revealed by comparing RSF curves of PdPt-s and Pd-s NP at Pd K-edge. Shown in Fig. 4c, radial peak C centered at 1.3 Å originates from interference of X-ray between Pd and O atoms chemisorbed in carbon supported Pd NP. Given that Pd-s is synthesized by using identical method to PdPt-s except the presence of Pt atoms, the strong Pd–O peak can be attributed to the surface oxidation by O chemisorption in Pd-s. Consequently, two important structural indexes prove that Pt atoms are preferentially located at defect sites as protection layer in NP surface. (1) In the absence of polymer stabilizer, Pd–O CN in PdPt-s NPs substantially decreases by reducing Pt atoms on Pd core surface by NaBH4. (2) A dramatic suppression of peak B (Pd–Pd and Pd–Pt) intensity because of destructive interference of backscattering waves as a result of Pd–Pt local alloy in NP surface. Taking structural information, geometric correlation, and intrinsic properties of selected metals into considerations, nanostructure of experimental NPs can be confirmed by cross-referencing the local structure and oxidation states of Pt and Pd atoms (Fig. S4†).
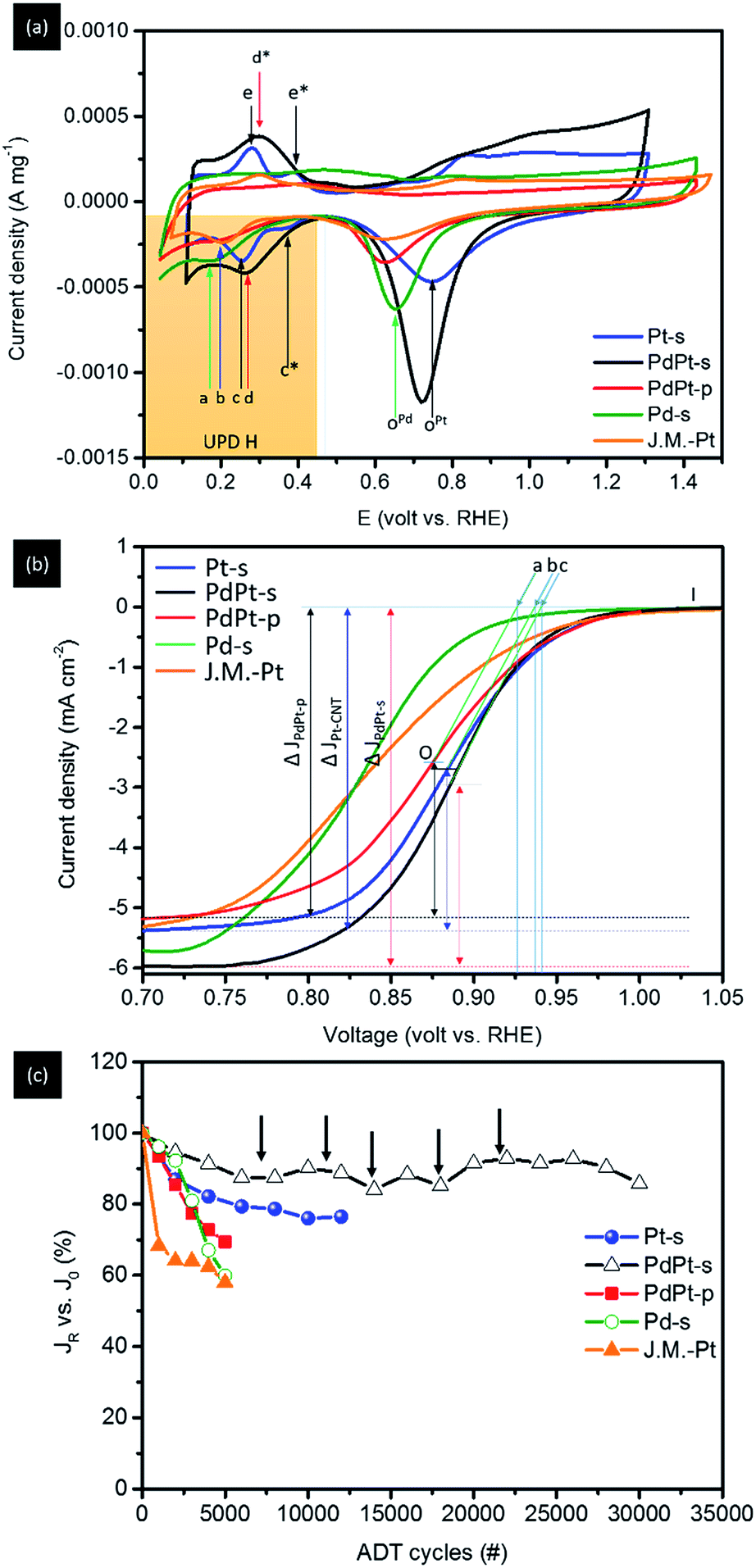 | ||
| Fig. 5 (a) CV, (b) LSV, and (c) ADT curves of experimental Pt-s, PdPt-s, PdPt-p, Pd-s NPs, and commercial Pt catalysts (J.M.-Pt). In CV curve of Pt-s, current peaks c and c* in negative sweep curve refer to adsorption responses of H+ at (111) and (200) facets of metallic Pt. The significant higher current density of peak c reveals the preferential grow of (111) facets in Pt-s. The counterpart responses of H+ desorption appears at peaks e and e* in positive sweep curve. | ||
Hypothetical mechanisms for crystal growth in self aligned wet chemical method (Pt-s and PdPt-s) and polyol reduction method (PdPt-p) are proposed by cross-referencing experimental results, solid (liquid) state ions diffusion, and chemical reaction kinetics. As demonstrated in Scheme 1, NP in both the two methods are grown in heterogeneous nucleation – crystal pathways in three stages including (a) metallic ion incubation and embryo formation in substrate, (b) crystal nucleation, and (c) heterogeneous crystal growth followed by atomic cluster decoration in crystal surface (in bimetallic PdPt-s NP). In Pt-s NP, the three stages are (a) chelation between Pt4+ ions and dangled (C5:) or thiolated defect sites (C5–C2H6SO) to form embryos on carbon support, (b) reduction of Pt4+ ions on embryos surface by H: radicals (provided by sodium borohydride) to form Pt nuclei, and (c) Pt NP formation with preferential growth at (111) facets. In stage (c), strong bonding between thiol groups and Pt atoms forms a steric barrier to protect NP from oxidation. Nucleation rate in stage (b) is dominated by kinetics of three reaction pathways and result in the final atomic configurations of NP. The reaction kinetics of three pathways are (1) Kads-Pt: chemisorption of Pt4+ atop embryo surface 〈eqn (1)〉 followed by its reduction by electrons [e] donated by H: radicals 〈eqn (2)〉,12 (2) Kads-C: chemisorption 〈eqn (1)〉 and reduction of chemisorbed Pt4+ (Ptads-C4+) into Pt0 at C5: or C5–C2H6SO sites by [e] 〈eqn (2)〉,13 (3) diffusion 〈eqn (3)〉 and allocation of Pt0 atoms or Ptads-C4+ from carbon to embryos surface.
|
Kads-Pt = d(Pt4+)/dt = aexp(−αq)
| (1) |
| Ka = [e]n[Ptads-C4+]m | (2) |
|
D = D0exp(−Ed/kBT)
| (3) |
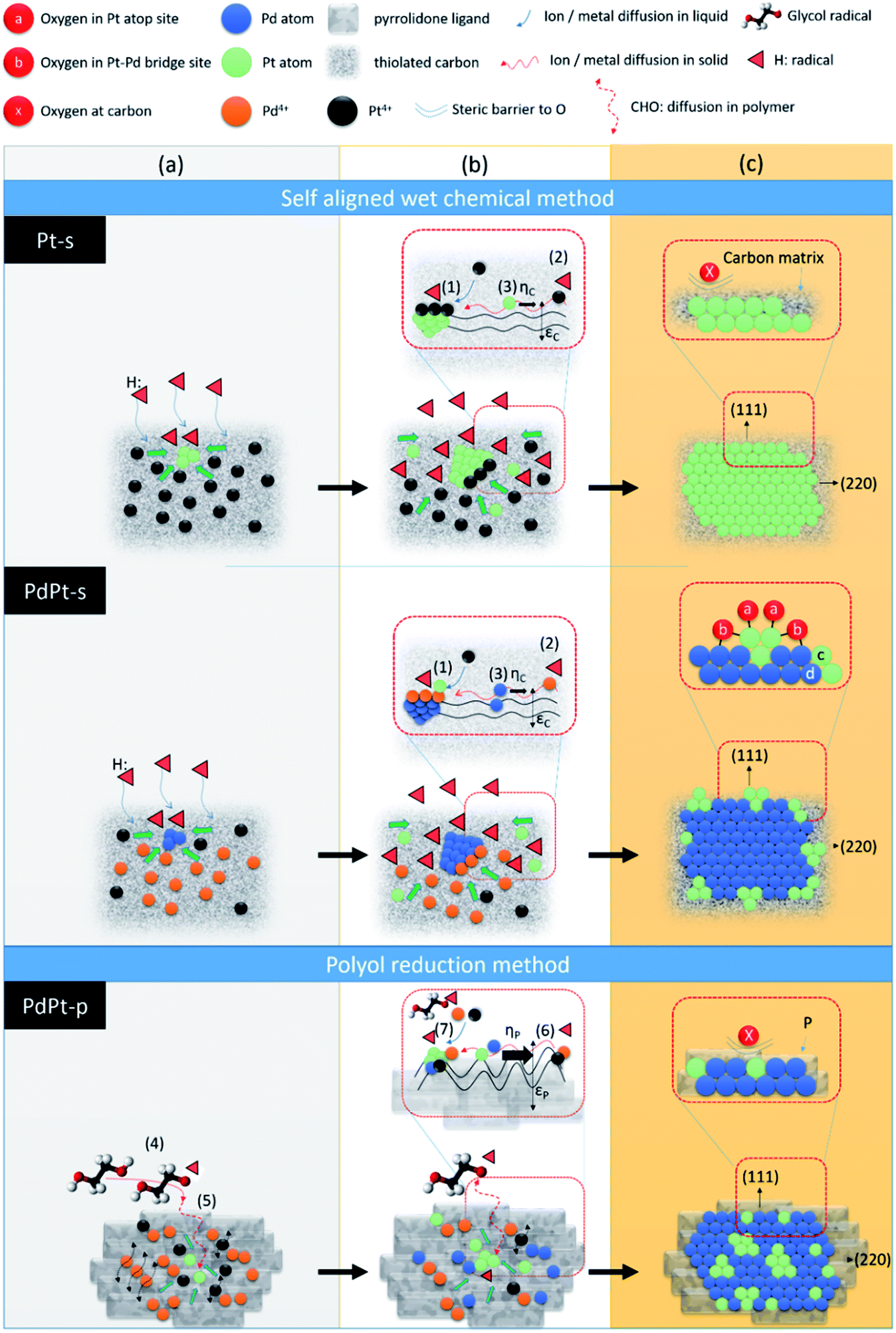 | ||
| Scheme 1 Schematic representation for atomic structure relocation in reaction pathways of experimental NCs upon crystal growth. In self-aligned wet chemical process, three steps correspond to (a) metal–ligand (carbon) complex formation, (b) core crystal nucleation, (c) sorption of Pt metal ion followed formation of atomic cluster in core crystal surface. In polyol process, three steps correspond to (a) PdPt nucleation (PdPtni) by success reactions of chelation of Pd and Pt ions (PVP–Pd or PVP–Pt) by pyrrolidone ligands and their interaction with CHO:, (b) heterogeneous crystal growth by subsequent sorption of PVP–Pt/PVP–Pd and reduction in PdPtni surface, (c) atomic relaxation formation of PdPt crystal. Reaction pathways refer to (1) Pt4+ diffusion, sorption, and interaction with H:/e− in Ptni surface, (2) reduction of Pt4+ or Pd4+ ion by H:/e− followed by their accommodation in CNT surface, (3) metal/metal ion diffusion in CNT surface, where ηc and εc denote the chemical resistances of defect sites parallel and perpendicular to metal/metal ion diffusion vectors, diffusion of C2H6O2: in (4) ethylene glycol and in (5) PVP molecules followed by reduction of PVP–Pt4+/Pd4+, (6) diffusion of metal/metal ions in PVP molecules where ηp and εp denote the resistances of friction coefficient and chemical strength of pyrrolidone ligands parallel and perpendicular to metal/metal ion diffusion vectors in PVP molecule, (7) reduction of Pd4+/Pt4+ into metal nuclei by thermal activated CHO:. | ||
Crystal growth of PdPt-s goes on the same pathways as Pt-s except that Pd4+ and Pt4+ ions are coexisting in the second pathway. Therefore, Ka of second step is affected by concentrations of H: radical [e], Ptads-C4+ and Pdads-C4+ in reaction system (eqn (4))
| Ka = [e]n[Pdads-C4+][Ptads-C4+] | (4) |
In polyol reduction method, PdPt-p NP is grown through pathways including (4) decomposition of ethylene glycol into metastable radical pairs of HOCH2–CHO:–H:, (5) diffusion of radicals to solid surface in polymer matrix, (6) diffusion of reduced metal atom to NP surface in polymer matrix, and (7) reduction of metal ions to solid phases surface by CHO: radicals. Kinetics of pathway (4), is controlled by reaction temperature in Arrhenius correlation. Kinetics of pathways (5) is controlled by a slow diffusion of CHO: radicals in polymer phases. Kinetics of pathway (6) shows the similar form but suffered from higher value of Ed as compared to that of pathway (3) due to the diffusion of Pt4+ and Pd4+ ions in highly flexible carbon matrix with substantial vibration amplitude in all directions. Reaction kinetics of pathway (7) is controlled by thermal reduction of polymer chelated metal ions. In this pathway, metal ion reduction can be described by a reaction of “aPt(L)4+R + bPd(L)4+D + cCHO:(L) + dPd(h,k,l)(S) ↔ ePd01−xPt0x@Pd(h,k,l)(S)”; where Pt4+R, Pd4+D, and Pd(h,k,l)(S) refer to the residual Pt4+ and dissolved Pd4+ ions after nucleation reaction, and the molar volume of the solid material (i.e., number density of reaction sites (area) at Pd crystallites, [Pd(S)]). Hereby, kinetics of NP growth with an approximation of reagent concentrations can be shown by eqn (5):
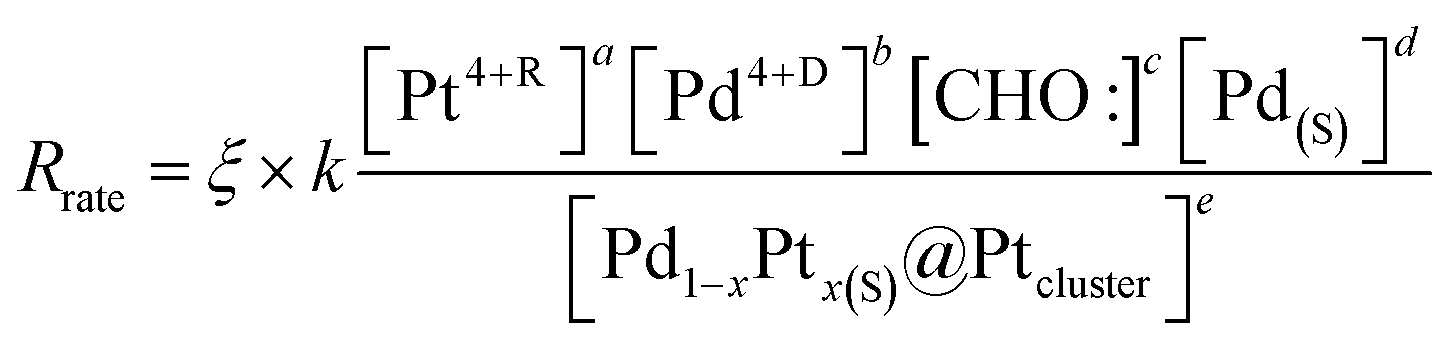 | (5) |
Surface chemical composition of experimental NPs is analysed by using CV analysis (Fig. 5a). For Pt-s, the two redox reaction peaks c (0.23 V vs. RHE) and c* (0.38 V vs. RHE) at a potential more negative to approximately 0.46 V vs. RHE (hydrogen underpotential deposition region, i.e., UPD H) refer to current responses of protons (H+) adsorption at (111) and (200) facets of Pt metal during the negative sweep, and the coupling signals of H+ desorption at peaks e (0.235 V vs. RHE) and e* (0.405 V vs. RHE) during the positive sweep, respectively. Accordingly, both adsorption and desorption profiles clearly reveal the substantially weakened H+ interaction on (200) facets, therefore proving the preferential grown of {111} facets for Pt/C. To clarify the extent of local alloying between Pt and Pd atoms in NP surface, CV curve of Pd/C is compared as reference. In this curve, H+ adsorption peak during negative sweep at 0.17 V (vs. RHE) together with the absence of desorption peak in positive sweep show the typical characteristics of strong H affinity of Pd metal. Consequently, H+ desorption peak should be located between peaks a and c for cases of NP with considerable extents of surface alloy between Pt and Pd atoms. For PdPt-s, preferential (111) growth is evidently shown by substantially enhanced (111) current peak (peak d) with the absence of (200) current. The broadened redox peaks (d at 0.238 V vs. RHE and d* at 0.415 V vs. RHE) indicate the various chemical states (i.e., a slight extent of local surface PdPt alloy and diffused inter-faceted regions) on its surface. Given that the potential of H+ desorption peak (peak d) is higher than that of Pt-s (peak c), surface local alloy between Pt and Pd atoms should be a minor factor in the electrochemical properties of PdPt-s. Therefore, complexity of chemical states can be rationalized by heteroatomic intermixing between atomic Pt clusters and Pd regions in NP surface; where sorption sites of H+ including atop, bridge, and hollow sites in Pt clusters, Pd crystal, and on Pd–Pt interfaces. In the CV curve of PtPd-p, potential of H+ sorption peak (peak b) is 0.2 V vs. RHE. It locates between those of Pd-s and Pt-s, therefore, indicating the formation of Pd–Pt alloy. The indistinct difference between current density of (111) and (200) facets in H UPD reveals the isotropic crystal growth at (111) and (200) facets. Therefore, by cross-referencing results of XRD and CV analyses, one can note that PdPt-s is grown with a metallic Pd core decorated by Pt clusters in NP surface. The shift of diffraction peaks A and B in opposite sides suggests that Pt atoms tend to alloy on opened (200) facets. In PdPt-p, homogeneous Pd–Pt alloy occurs with a structure of Pt clusters on Pd crystal NP. Such a structure is revealed by presence of homoatomic Pt–Pt bond pairs in EXAFS and upshift of A and B peaks from that of metallic Pd (111) and (200) in diffraction pattern of PtPd-p. Comparison between surface to bulk ratio and extent of oxidation further reveals location of Pt atoms in NPs; where correlations of spherical NPs can be estimated by geometric calculations and are discussed as simplified interpretations. As shown in Fig. S4,† surface to bulk ratios (η) are 24.7% for PdPt-s (4.2 nm), 16.1% for Pt-s (5.5 nm), and 20.1% for PdPd-p (7.3 nm). Considering that all PdPt NPs are grown in homogeneous alloy, the oxidation extent of Pt (determined by fitting corresponding XANES spectra using linear combination fitting method) should be determined to a similar value to their η (PdPt-p). A slightly lower Pt oxidation to η (also found in Pt-s) could be attributed to the protection of Pt from oxidation by carbon support at interface. However, extent of Pt oxidation is determined to be 38.2% for PdPt-s. This value is far higher than that is expected in homogeneous alloy and can only be rationalized by exposing Pt clusters to NP surface (i.e., decoration of Pt atoms in NP surface).
Fig. 5b and Table 3 show the linear sweep voltammetry (LSV) spectra and corresponding parameters of Pt-s, PdPt-s, and PdPt-p NPs measured in a O2-saturated KOH electrolyte (0.1 M) on a rotational working electrode at 1600 rpm. As shown, the onset potential (Voc vs. RHE) of NP follows the trend: PdPt-s (c = 0.941 V) > Pt/C (b = 0.935 V) > PdPt-p (a = 0.928 V) and is inversely proportional to their Pd–Pt intermix “χ(Pd–Pt) at Pt L3-edge” (Table 2). Moreover, their mass activity (MA, current densities vs. Pt metal loading) possesses the same sequence at 0.85 V (Table 3). With a 4-electron pathway (n ∼ 4.0 ± 0.05), results mentioned above demonstrate the facilitation of oxygen reduction by a superior reaction kinetic of PdPt-s (Jk = 20.08 mA cm−2 at 0.85 V vs. RHE) among NPs under investigation. Such a promotion on reaction kinetics can be attributed to a substantial electron relocation to Pt sites with a local distortion in their neighbouring coordination environment. The former factor facilitates oxygen splitting step and the latter provides high density of low activation energy open sites (high surface free energy with wide open space) for allocating oxygen radicals in subsequent steps. With the participation of the two factors, an impressive ORR MA at 2040.6 mA mgPt−1 (at 0.85 V) is achieved in PdPt-s. In this event, PdPt-s truly demonstrates its merits in ORR with a ∼29.4-fold MA of commercial Pt catalyst (J.M.-Pt) in the optimum case. On the other hand, PdPt-p forms a homogeneous alloy between Pd and Pt atoms in NP. It possesses smaller heteroatomic intermix (χ(Pd–Pt) and χ(Pt–Pd)) and local distortion comparing to that of PdPt-s. Consequently, a substantially reduced MA to 734.0 mA mgPt−1 is expectable due to the presence of low density Pt active sites (also indicated by a reduction of n to 3.7–3.8) and slight local distortion between Pt and Pd atoms in NP surface exposed to reaction media. For clarifying the effects of Pd–Pt intermix and surface configuration to NP ORR activity, electrochemical and structural characterization results of Pt-s are compared as reference. As clearly indicated in Table 3, Jk and MA of Pt-s are improved by 3–4 times as compared to that of J.M.-Pt. In Pt-s, the lower extent of local lattice distortion (Tables 1 and 2) and absence of Pd–Pt intermix explains the reduction of Jk and the substantially suppression of MA compared with that of PdPt-s.
| NPs | V on set (1600 rpm) | N (0.5 V) | ECSA_H (m2 g−1) | Jk (0.85 V) (mA cm−2) | MAPt (0.85 V) (mA mg−1) | MAtotal (0.85 V) (mA mg−1) | J0 (mA cm−2) | JR final (mA cm−2) | ΔJ (JR − J0/J0) vs. J0 (%) | ADT cycles |
|---|---|---|---|---|---|---|---|---|---|---|
| a Current densities (J0, JR final) are determined at 0.85 V of a LSV curve. | ||||||||||
| J.M.-Pt | 0.922 | 4.0 | 270.3 | 5.34 | 67.1 | 67.1 | 2.33 | 1.35 | 42.0 | 5000 |
| Pt-s | 0.935 | 4.1 | 35.4 | 15.22 | 271.9 | 271.9 | 4.28 | 3.28 | 24.0 | 12000 |
| Pd-s | 0.891 | 5.5 | 51.7 | 14.9 | NA | 32.7 | 1.91 | 1.15 | 40.1 | 5000 |
| PdPt-s | 0.941 | 4.1 | 909.4 | 20.08 | 2040.6 | 329.9 | 4.87 | 4.18 | 14.0 | 30000 |
| PdPt-p | 0.928 | 3.8 | 584.5 | 9.75 | 734.0 | 118.1 | 3.52 | 2.43 | 31.5 | 5000 |
Electrochemical durability of NPs in ORR is illustrated by results of ADT (Fig. 5c) complementary with CV analysis (Fig. S5†) at corresponding test cycles. Retention current (JR) vs. initial current (J0) of NPs (JR vs. J0) with increasing ADT cycles is shown in Fig. 5c. As revealed, JR vs. J0 is progressively decreased by 14.0% for PdPt-s at 30000 cycles, 31.5% for PdPt-p at 5000 cycles, and 24.0% for Pt-s at 12 000 cycles (Table 3). Those results show that PdPt-s has the highest structural and electrochemical durability among experimental NPs. It could be accounted to a structural reinforcement by high ratio of heteroatomic contact.15 Current fluctuation of PdPt-s (denoted by black arrows) can be attributed to simultaneous dissolution and re-deposition of Pt/Pd atoms in NP during potential sweeping cycles. Such a statement is revealed by fluctuation of peak potential at ∼0.727 to 0.750 V vs. RHE (shown by red solid arrow) with uneven changes of redox peak intensity as affected by the changes of chemisorption states for oxygen adsorption in PtPd-s surface. For Pt-s, intensity of CV peaks is progressively faded away without significant changes in shape with ADT cycles. The fading of CV profile suggests a dissolution induced atomic restructure between facets of Pt NP. Considering that Pd is a noble metal, total MA (MAtotal) is compared to verify the industrial value of experimental NPs. As shown in Table 3, MAtotal of NPs follows the trend of PdPt-s (329.9 mA mg−1) > Pt-s (271.9 mA mg−1) > PdPt-p (118.1 mA mg−1) > J.M.-Pt (67.1 mA mg−1). Compared to Pt-s, a substantially reduced MAtotal of PdPt-p can be rationalized by the surface coverage of high oxygen/metal affinity ligands (pyrrolidone in our case). Such a capping layer hinders the diffusion kinetics of oxygen molecules and increases the interface resistance for charge exchange in NP surface. Both the two factors reduce the Jk and Voc as compared to that of Pt-s. Meanwhile, pyrrolidone ligand possesses a high metal chelation affinity. It forms a metastable metal-complex and thus reduces the re-deposition rate of dissolved metal ions in NP surface during potential cycles. Such characteristics explain the highest JR degradation among experimental NPs in ADT. Charge transfer number (n) is another important index for NPs in ORR application. For cases of NPs with n ∼ 4.0 (i.e., complete charge transition), all electrons are contributed from the reduction of chemisorption oxygen in surface reaction sites. In those cases, current degradation is caused by reduction of surface reaction site which is because of inter-particle sintering during ADT. For Pd-s, n is determined to be 5.5. This value is substantially higher than 4.0 indicating the presence of exceed electrons from Pd oxidation. For PdPt-p, n is determined to be 3.8 implying the formation of intermediate hydrogen peroxide. Such a feature could be attributed to the strong chelation effect of PVP-ligands that shielding (suppressing) the surface Pt sites from ORR.
Conclusions
A robust method on growing bimetallic PdPt NP with high extent of surface alloy, high density of Pt cluster on Pd surface, and low Pt metal loading (∼10 wt%) is developed based on self-aligned wet chemical reduction method with precise control on reaction sequences and time. Structural characterization results combining heterogeneous nucleation crystal growth theory suggest the nanostructure of bimetallic PdPt NP is controlled by reaction time and precursor inlet sequence. We demonstrate that such PdPt NP performs a 29.4-times the value of MA as compared to that of commercial Pt catalyst. The structural and electrochemical characterizations show that such impressive ORR performances are attributed to the local electron relocation and atomic structure distortion in the presence of high activity tr-Pt-atom clusters capping atop Pd NP surface. In such a unique surface configuration, Pt clusters with high Pd intermix perform high oxygen splitting kinetics and the neighbouring edge sharing Pd CN with high local distortion provide low activation energy open sites for conducting subsequent oxygen radical reduction steps. By combining the two factors, PdPt-s gains an impressive MA of 2040.6 mA mgPt−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the help by staffs of National Synchrotron Radiation Research Center (NSRRC), Hsinchu, Taiwan for helping in various synchrotron-based measurements. T.-Y. Chen acknowledges the funding support from the National Tsing Hua University, Taiwan (N103K30211 and 103N1200K3) and the Ministry of Science and Technology, Taiwan (MOST 103-2112-M-007-022-MY3 and MOST 105-3113-E-006-019-CC2).Notes and references
- (a) C.-W. Liu, Y.-C. Wei, C.-C. Liu and K.-W. Wang, J. Mater. Chem., 2012, 22, 4641 RSC; (b) Y.-T. Liang, S.-P. Lin, C.-W. Liu, S.-R. Chung, T.-Y. Chen, J.-H. Wang and K.-W. Wang, Chem. Commun., 2015, 51, 6605 RSC; (c) Y. Liu, Y. Jiang and W. Chen, Nano Energy, 2013, 2, 836 CrossRef.
- S. Sun, F. Jaouen and J.-P. Dodelet, Adv. Mater., 2008, 20, 3900 CrossRef CAS.
- (a) J.-N. Zheng, L.-L. He, F.-Y. Chen, A.-J. Wang, M.-W. Xue and J.-J. Feng, Electrochim. Acta, 2014, 137, 431 CrossRef CAS; (b) Q. Zhang, L. Li, B. Jiang, K. Wang, D. Tang and B. Dou, Int. J. Hydrogen Energy, 2017, 42, 17102 CrossRef CAS.
- Y. Zhang, L. Zhao, J. Walton, Z. Liu and Z. Tang, Int. J. Hydrogen Energy, 2017, 42, 17112 CrossRef CAS.
- (a) K. Sasaki, H. Naohara, Y. Cai, Y. M. Chio, P. Liu, M. B. Vukmirovic, J. X. Wang and R. R. Adzic, Angew. Chem., Int. Ed., 2010, 49, 8602 CrossRef CAS PubMed; (b) S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333 CrossRef CAS PubMed; (c) P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454 CrossRef CAS PubMed; (d) L. Mi, W. Wei, S. Huang, S. Cui, W. Zhang, H. Hou and W. Chen, J. Mater. Chem. A, 2015, 3, 20973 RSC.
- (a) W. Wei, L. Mi, Y. Gao, Z. Zheng, W. Chen and X. Guan, Chem. Mater., 2014, 26, 3418 CrossRef CAS; (b) X. Wang, M. Vara, M. Luo, H. Huang, A. Ruditskiy, J. Park, S. Bao, J. Liu, J. Howe, M. Chi, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2015, 137, 15036 CrossRef CAS PubMed; (c) W. Wei, L. Mi, S. Cui, B. Wang and W. Chen, ACS Sustainable Chem. Eng., 2015, 3, 2777 CrossRef CAS.
- T.-Y. Chen, T.-L. Lin, C.-C. Chen, C.-M. Chen and C.-F. Chen, J. Chin. Chem. Soc., 2009, 56, 1236 CrossRef CAS.
- (a) T.-Y. Chen, S.-T. Chang, C. W. Hu, Y.-F. Liao, Y. J. Sue, Y.-Y. Hsu, K.-W. Wang and Y.-T. Liu, CrystEngComm, 2016, 18, 5860 RSC; (b) Y.-T. Liang, S.-P. Lin, C.-W. Liu, S.-R. Chung, T.-Y. Chen, J.-H. Wang and K.-W. Wang, Chem. Commun., 2015, 51, 6605 RSC.
- (a) T. Bligaard and J. K. Nørskov, Electrochim. Acta, 2007, 52, 5512 CrossRef CAS; (b) J.-M. Lin, T.-L. Lin, U.-S. Jeng, Y.-J. Zhong, C.-T. Yeh and T.-Y. Chen, J. Appl. Crystallogr., 2007, 40, s540 CrossRef CAS.
- (a) J. Zhang, F. H. B. Lima, M. H. Shao, K. Sasaki, J. X. Wang, J. Hanson and R. R. Adzic, J. Phys. Chem. B, 2005, 109, 22701 CrossRef CAS PubMed; (b) M. Wang and Q. Chen, Chem.–Eur. J., 2010, 16, 12088 CrossRef CAS PubMed; (c) A. Navrotsky, C. Ma, K. Lilova and N. Birkner, Science, 2010, 330, 199 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Adv. Mater., 2002, 14, 833 CrossRef CAS.
- (a) S. Z. Roginsky and J. Zeldovich, Acta Physicochim. URSS, 1934, 1, 554 Search PubMed; (b) M. J. D. Low, Chem. Rev., 1960, 60, 267 CrossRef CAS; (c) H. A. Taylor and N. Thon, J. Am. Chem. Soc., 1952, 74, 4169 CrossRef CAS; (d) I. S. Mclintock, Nature, 1967, 216, 1204 CrossRef CAS.
- M. E. Davis and R. J. Davis, in Fundamentals of Chemical Reaction Engineering, ed. E. D. Glandt, M. T. Klein and T. F. Edgar, McGraw-Hill Higher Education, New York, 2003, pp. 1–24 Search PubMed.
- (a) T.-Y. Chen, I.-L. Chen, Y.-T. Liu, T.-L. Lin, P.-W. Yang, C.-Y. Wu, C.-C. Hu, T.-J. M. Luo and C.-H. Lee, CrystEngComm, 2013, 15, 982 RSC; (b) V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847 CrossRef CAS; (c) J.-P. Jolivet, Metal Oxide Chemistry and Synthesis – From Solution to Solid State, John Wiley & Sons, Chichester, 2003 Search PubMed.
- (a) M. Li, Z. Zhao, T. Cheng, A. Fortunelli, C.-Y. Chen, R. Yu, Q. Zhang, L. Gu, B. Merinov, Z. Lin, E. Zhu, T. Yu, Q. Jia, J. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. Duan, Science, 2016 Search PubMed; (b) N. Deogratias, M. Ji, Y. Zhang, J. Liu, J. Zhang and H. Zhu, Nano Res., 2015, 8, 271 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of CNT supported Pt catalysts before and after ADT, HRTEM and EDX map images of PdPt nanoparticle synthesized by using wet chemical reduction and polyol method, geometrical calculation results are enclosed. See DOI: 10.1039/c7ra08405f |
| This journal is © The Royal Society of Chemistry 2017 |