DOI:
10.1039/C7RA08290H
(Paper)
RSC Adv., 2017,
7, 44401-44409
Effect of borrelidin on hepatocellular carcinoma cells in vitro and in vivo†
Received
27th July 2017
, Accepted 5th September 2017
First published on 15th September 2017
Abstract
Hepatocellular carcinoma (HCC) is the most common type of liver cancer with high prevalence and mortality. Borrelidin, produced by several actinomycete bacteria of Streptomycin sp. exhibited diversiform activities including anti-bacterial, anti-viral, anti-angiogenic, and anti-tumor. However, the effect of borrelidin on HCC cells has not been characterized. The present study demonstrated borrelidin exhibited great potential to inhibit the growth of HCC cells, HepG2 and SMMC7721 in vitro. Western blot and real-time qPCR analysis revealed that borrelidin decreased the expressions of cyclin D1, cyclin D3, cyclin E1, CDK2, CDK4, and CDK6 and increased the expression of p21, thereby inducing G0/G1 cell cycle arrest. Moreover, borrelidin down-regulated expression of Bcl-2, up-regulated expression of Bax and increased cleavages of caspase-9 and caspase-3 to activate caspase-dependent apoptosis in HCC cells. Borrelidin inhibited migration and invasion through suppressing the expression of MMP-2 and MMP-9 in HCC cells. Further investigation indicated that the anti-tumor effect of borrelidin was mediated by MAPKs signaling pathway. In addition, an in vivo experiment revealed that borrelidin suppressed tumor growth in SMMC7721 xenograft model mice with few side effects. Cell cycle arrest and induced apoptosis were also observed in tumor tissues of model mice treated with borrelidin.
1. Introduction
Hepatocellular carcinoma (HCC) is one type of liver cancer with a high incidence and mortality rate worldwide.1 Despite tremendous progress in diagnosis and management, the present therapeutic strategies for treating HCC are still surgical resection or liver transplantation.2 Moreover, HCC is highly resistant to conventional chemotherapeutic drugs such as anthracycline, fluoropyimidine, camptothecin and platinum-based drugs.3,4 As a consequence, the development of new anti-tumor drugs is essential for improving the treatment of hepatic carcinoma.
In the process of our search for new bioactive natural products from actinomycetes associated with animal feces, the secondary metabolites of the fermentation broth of Streptomyces vinaceusdrappus (YIM 100880), which was isolated from Hylobates hoolock feces, were investigated. Borrelidin, a nitrile-containing macrolide, was isolated from the fermentation broth of YIM 100880 by several chromatographic techniques and the structure was elucidated by 1H-NMR, 13C-NMR, and MS spectroscopic data analysis. Borrelidin exhibited a wide spectrum of biological activities, such as anti-bacterial, anti-viral, and anti-tumor.5 It was proved that some of these activities were associated with the inhibition of threonyl-rRNA synthetase (ThrRS).6 Previous study had shown that borrelidin induced cell cycle arrest and apoptosis with activation of the GCN2 kinase pathway in acute lymphoblastic leukemia cells.7 Borrelidin also induced chop-dependent apoptosis through unfolded protein response (UPR) in oral cancer cell.8 In addition, borrelidin was an effective angiogenesis inhibitor through its inhibition of ThrRS and inducing caspases cascade in endothelial cells.9 The remarkable bioactivities have prompted the development of borrelidin as a potential anti-tumor candidate.
Currently, the effect of borrelidin on HCC remains unknown. In the present study, our investigation was designed to explore the effect of borrelidin on proliferation, cell cycle arrest, apoptosis, migration and invasion of two HCC cells, HepG2 and SMMC7721 in vitro and xenograft growth inhibition in vivo. Real-time qPCR and western blot analysis of molecular markers associated with cell cycle arrest and apoptosis as well as MAPKs cell signaling cascade were performed to identify the underlying mechanisms.
2. Results and discussion
2.1. Borrelidin inhibited proliferation of HCC cells
Borrelidin was isolated by our co-author and provided with a purity of >98.0% determined by high-pressure liquid chromatography.
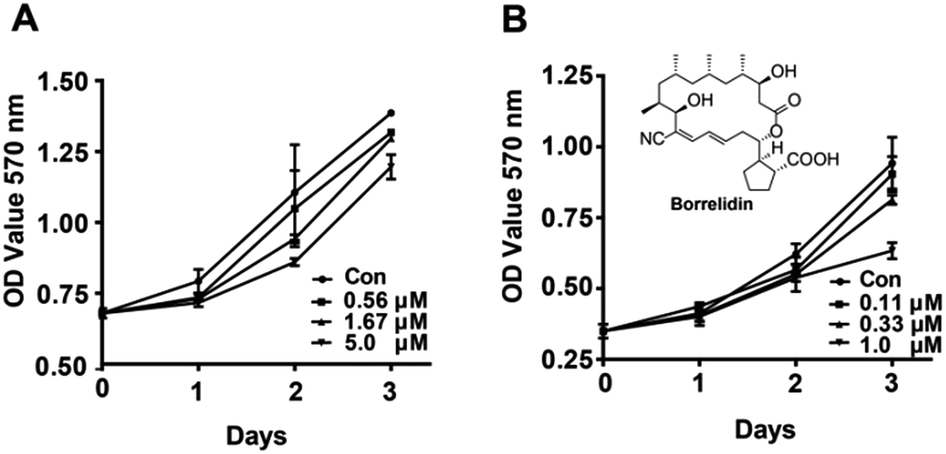
The effects of borrelidin on the growth inhibition of two HCC cells, HepG2 and SMMC7721 were assessed by MTT assay. The results showed that borrelidin time- and dose-dependently inhibited the growth and proliferation of the two HCC cells (Fig. 1). Half inhibitory concentrations (IC50) of borrelidin in HepG2 and SMMC7721 cells were 6.7 ± 0.3 and 1.5 ± 0.2 μM, respectively. According to the results, the concentrations below IC50 were used to conduct following-up studies.
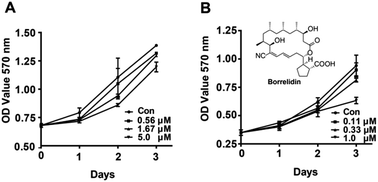 |
| | Fig. 1 Effect of borrelidin on growth and proliferation of HCC cells. The growth curve of HCC cells was determined by MTT assay (A) HepG2 cells were treated with borrelidin (0, 0.56, 1.67 and 5.0 μM) for 0 h, 24 h, 48 h and 72 h. (B) SMMC7721 cells were treated with borrelidin (0, 0.11, 0.33 and 1.0 μM) for 0 h, 24 h, 48 h and 72 h. The control (Con) group was treated with vehicle. Data were represented as means ± SD (n = 3). | |
2.2. Borrelidin caused HCC cells G0/G1 phase arrest
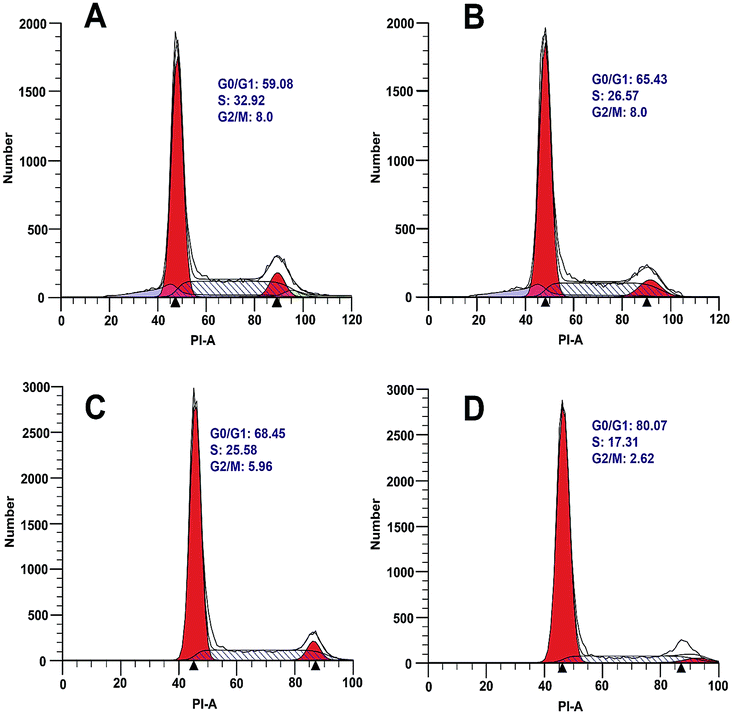
Oncogenic transformation or loss of tumor suppressor function drives abnormal growth and proliferation in cancer cells, which often results from uncontrolled cell cycle.10 In order to explore the effect of borrelidin on the cell cycle progression of HepG2 and SMMC7721 cells, the cells were incubated with borrelidin and then followed by flow cytometric analysis to examine the DNA distribution. The results suggested that borrelidin caused a G0/G1 phase cell cycle arrest in HCC cells (Fig. 2). Cell cycle progression was known to be tightly regulated by different cyclins, cyclin dependent kinases (CDKs) and cyclin dependent kinase inhibitors (CKIs) in different phase of cell cycle.11 Among of cyclins, cyclin D and cyclin E, played important roles in the G1/S phase progression of cells. Cyclin D interacted with CDK4 and CDK6 to allow expression of cyclin E which bound and activated CDK2 to form CDK2/cyclin E complex, thus promoting cell cycle progression from G1 to S phase.
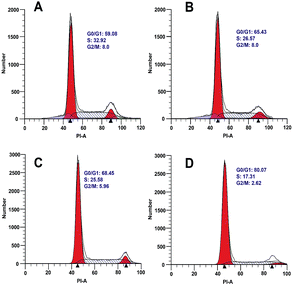 |
| | Fig. 2 Measurement of the cell cycle in HCC cells treated with borrelidin. Flow cytometric analysis on cell cycle arrest of HepG2 cells treated with (A) vehicle, (B) 5.0 μM borrelidin for 24 h, respectively. SMMC7721 cells treated with (C) vehicle, (D) 1.0 μM borrelidin for 24 h, respectively. The vehicle-treated group was the control (Con). | |
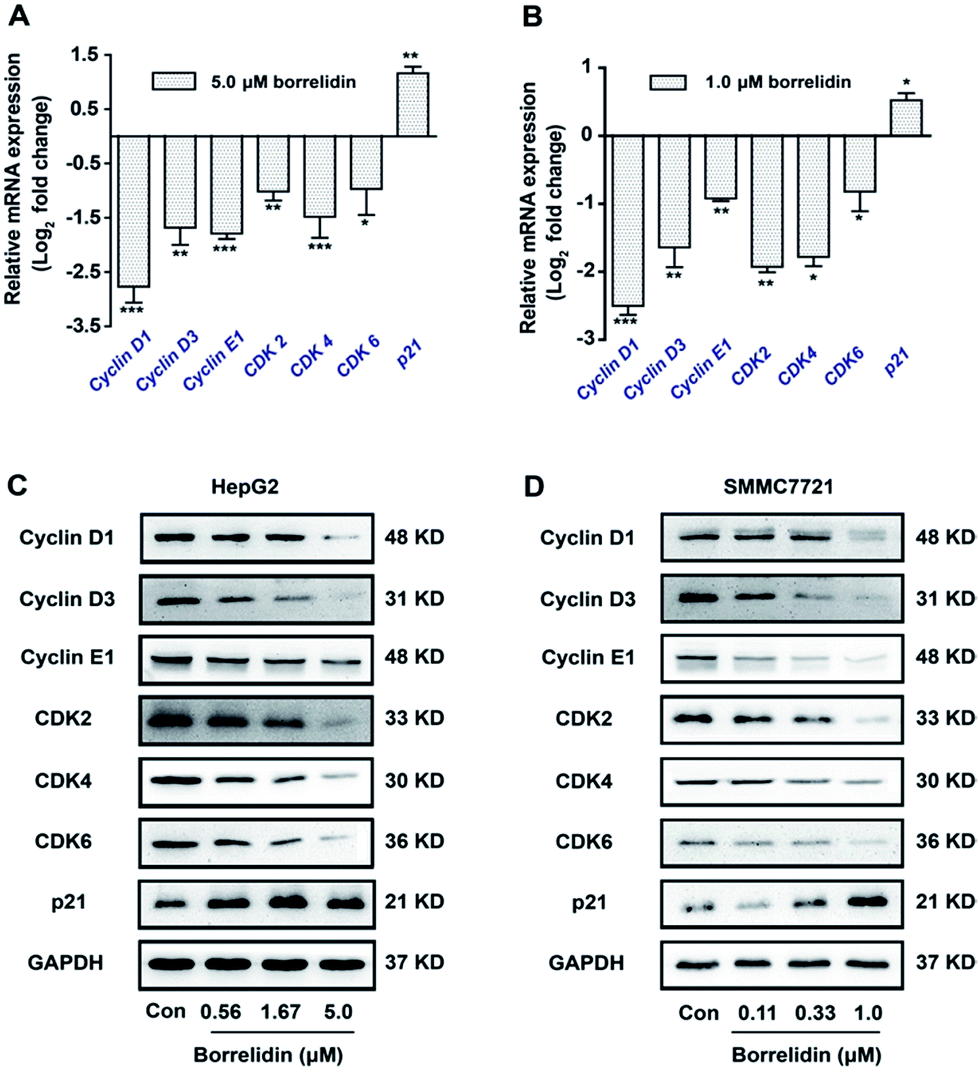
On the other hand, the activities of cyclins/CDKs complex were negatively regulated by CKIs, especially p21.11,12 The disorders of these biomarkers would result in the G0/G1 phase cell cycle arrest in cells.13 To disclose the underlying mechanism, we focused on the genes related to cell cycle progression by using real-time qPCR. As shown, borrelidin decreased the mRNA expressions of cyclin D1, cyclin D3, cyclin E1, CDK2, CDK4 and CDK6 and increased p21 (Fig. 3A and B). We further analyzed the protein expressions of these molecules by western blot assay and the up-regulations and down-regulations of them in a dose-dependent manner were consistent with their transcriptional target genes (Fig. 3C and D). In summary, borrelidin caused G0/G1 phase cell cycle arrest in HCC cells through down-regulating cyclins and CDKs and enhancing p21 expression in cell cycle progression.
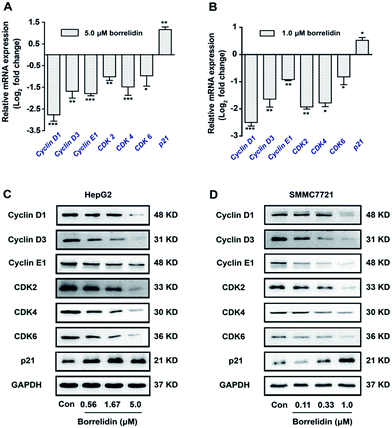 |
| | Fig. 3 Regulation of borrelidin on G0/G1 phase related molecules. The mRNA levels of cyclin D1, cyclin D3, cyclin E1, CDK2, CDK4, CDK6 and p21 of HepG2 cells (A) and SMMC7721 cells (B) treated with borrelidin were quantified by real-time qPCR. The protein levels of cyclin D1, cyclin D3, cyclin E1, CDK2, CDK4, CDK6 and p21 of HepG2 cells (C) and SMMC7721 cells (D) treated with borrelidin were assessed by western blot. The control (Con) group was treated with vehicle. Data were represented as means ± SD (n = 3). *p < 0.05 vs. control; **p < 0.01 vs. control; ***p < 0.001 vs. control. | |
2.3. Borrelidin induced HCC cells apoptosis
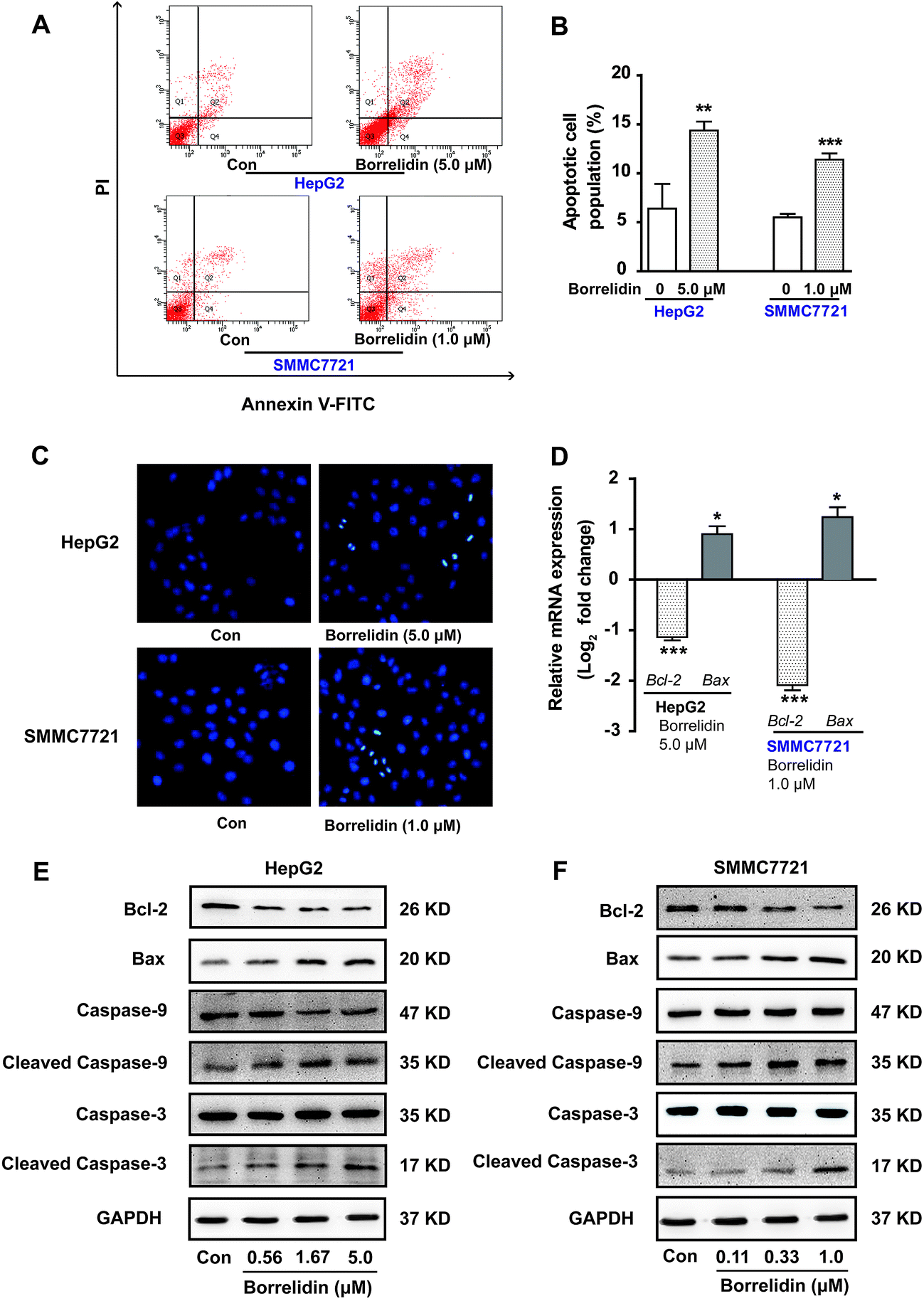
Precious studies had shown that borrelidin could induce apoptosis in various cancer cells, including breast cancer, oral cancer and acute lymphoblastic leukemia cells.7,8,14 To observe whether borrelidin could induce apoptosis of HCC cells, Hoechst 33258 staining was conducted in HepG2 and SMMC7721 cells with borrelidin treatment. As shown in Fig. 4C, borrelidin-treated group exhibited typical morphological features of apoptosis including shrinking nuclei and dense fluorescence emission in contrast with control group. Besides, flow cytometric analysis of annexin V/PI showed that significant increase in population of apoptotic cells were detected in borrelidin-treated HepG2 and SMMC7721 cells (Fig. 4A and B). The anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax, were the well-characterized members of the Bcl-2 family that regulated cell apoptosis.15 Moreover, the activation of caspases cascade played a dominant role in the induction of cell apoptosis.16 Caspase-9, an initiator caspase, was cleaved into the active form and then activated caspases-3 by proteolytic cleavage, subsequently induced the digestion of structural proteins, DNA degradation and ultimately phagocytosis.16–18 To further explore the effects of borrelidin on the apoptosis-related molecules, the mRNA and protein expression of Bcl-2, Bax, caspase-3 and caspase-9 cleavages in two HCC cells were investigated by real-time qPCR or western blot assay. As shown, the expression of Bcl-2 was significantly down-regulated, while Bax up-regulated in HepG2 and SMMC7721 cells after borrelidin treatment (Fig. 4D–F). Western blot analysis demonstrated that the protein levels of cleaved caspase-9 and cleaved caspase-3 were significantly increased in a dose-dependent manner in the borrelidin treated group (Fig. 4E and F). We concluded that borrelidin could induce apoptosis through unbalance of Bcl-2 family and activation of caspase family in HCC cells.
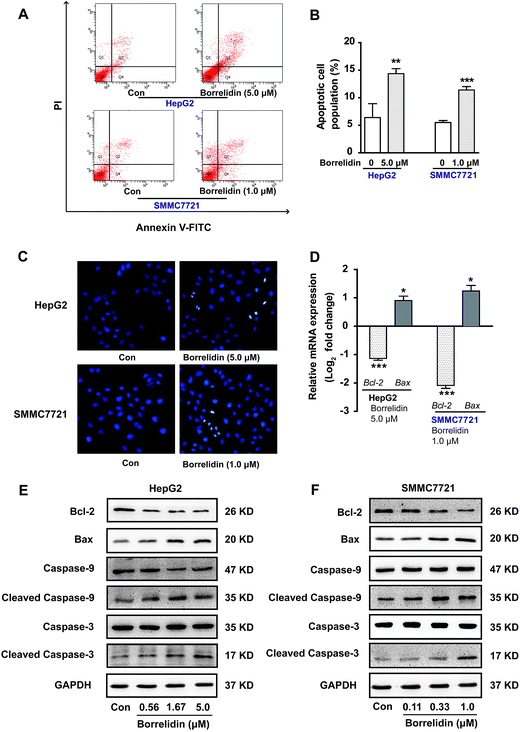 |
| | Fig. 4 Effect of borrelidin on inducing apoptosis of HCC cells. (A) Flow cytometric analysis on apoptosis of HepG2 and SMMC7721 cells treated with borrelidin. (B) The percentages of annexin V-positive cells of HepG2 and SMMC7721 cells treated with borrelidin. (C) Morphological alterations of HepG2 and SMMC7721 cells treated with borrelidin stained with Hoechst 33258. (D) The mRNA levels of Bcl-2, Bax of borrelidin treated HepG2 cells and SMMC7721 cells were quantified by real-time qPCR. The protein levels of Bcl-2, Bax, caspase-3, cleaved caspase-3, caspase-9 and cleaved caspase-9 of HepG2 cells (E) and SMMC7721 cells (F) treated with borrelidin were assessed by western blot. The vehicle-treated group was the control (Con). Data were represented as means ± SD (n = 3). *p < 0.05 vs. control; **p < 0.01 vs. control; ***p < 0.001 vs. control. | |
2.4. Borrelidin inhibited the migratory and invasive ability of HCC cells
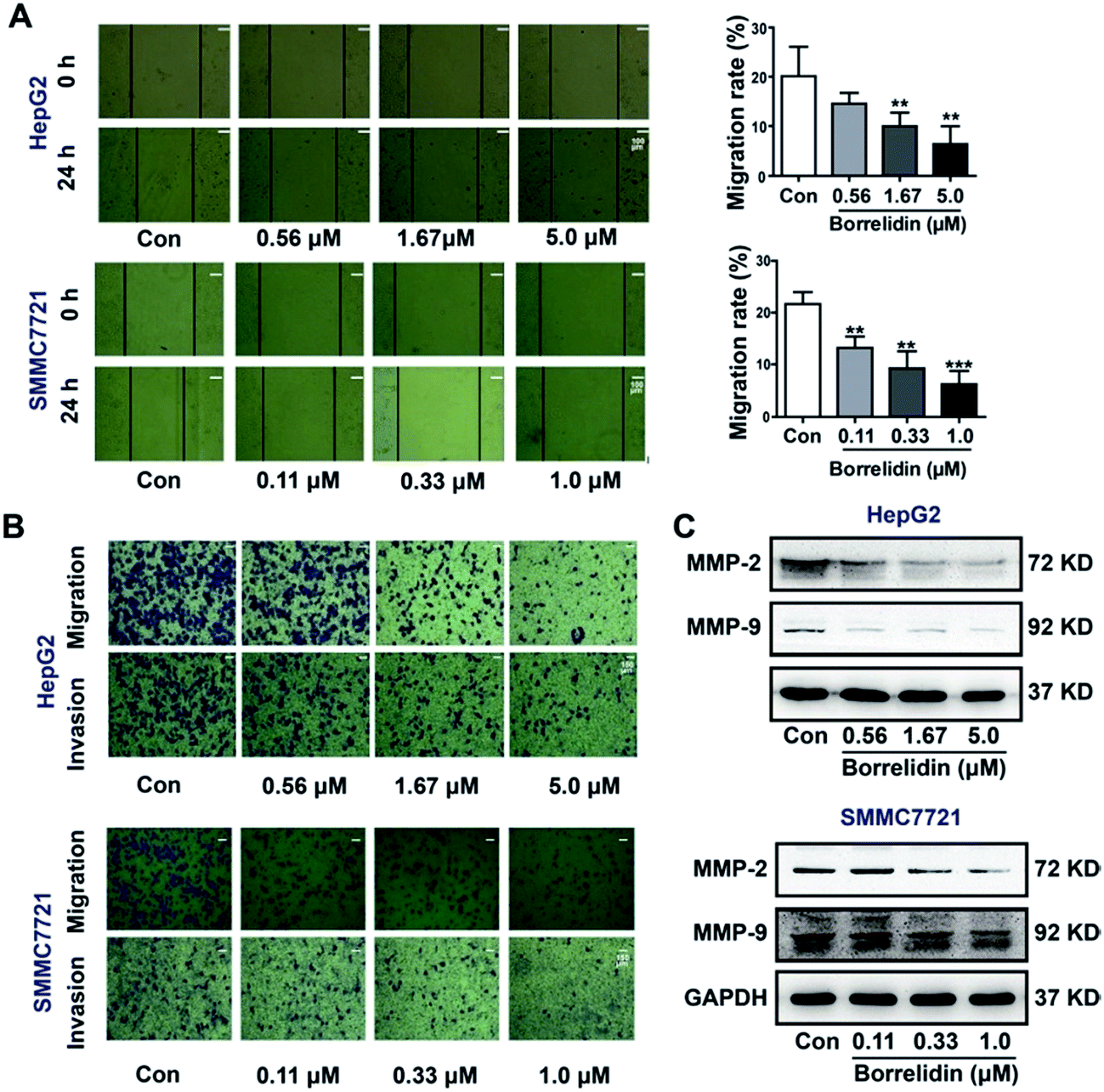
The progression of metastasis exacerbated the probability of morbidity and mortality in cancer populations.19 Cell migration and invasion were critical steps in the process of metastasis of cancer cells. In the current study, the effect of borrelidin on the migration and invasion of HCC cells were investigated. Firstly, a wound healing assay was conducted to detect cell mobility after borrelidin treatment. The results showed that borrelidin significantly decreased the movement ability of HepG2 and SMMC7721 cells in a dose-dependent manner (Fig. 5A). Then, the invasive and migratory behavior of borrelidin treated HepG2 and SMMC7721 cells were analyzed using Boyden chamber assays with or without Matrigel. The results demonstrated that inhibition occurred in a dose-dependent manner compared to control group (Fig. 5B). Matrix metalloproteinases (MMPs) exhibited the ability to degrade the extracellular matrix (ECM) and contributed to the invasion of cancer cells.20 Among them, MMP-2 and MMP-9 were crucial for degrading the main component of ECM.21–23 In order to investigate the effect of borrelidin on MMP-2 and MMP-9 expressions in HCC cells, we detected the intracellular protein levels of MMP-2 and MMP-9 of HepG2 and SMMC7721 cells by western blot analysis. As shown, the protein expressions of MMP-2 and MMP-9 were down-regulated in a dose-dependent manner (Fig. 5C).
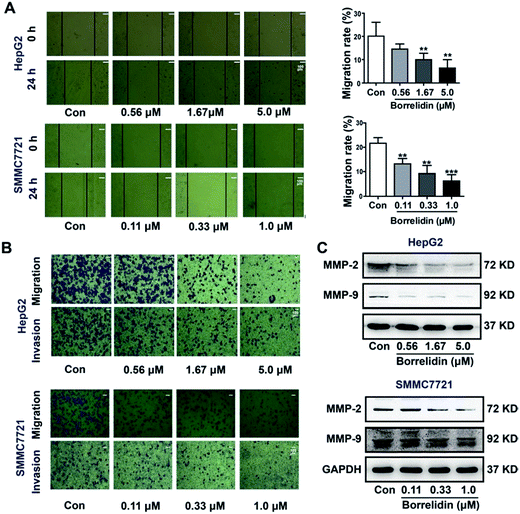 |
| | Fig. 5 Borrelidin inhibited the migration and invasion potential of HCC cells. (A) The wound healing assay for evaluating the effect of borrelidin on mobility of HepG2 cells and SMMC7721 cells. The average scraped width per well at 0 h and 24 h was measured and calculated as migration rate. (B) The transwell migration assay with or without Matrigel for assessing the effect of borrelidin on invasive or migratory potential of HepG2 cells and SMMC7721 cells. (C) The intracellular MMP-2 and MMP-9 protein expression levels of HepG2 and SMMC7721 cells treated with borrelidin were assessed by western blot. The vehicle-treated group was the control (Con). Data were represented as means ± SD (n = 3). *p < 0.05 vs. control; **p < 0.01 vs. control; ***p < 0.001 vs. control. | |
2.5. MAPKs signaling pathway was regulated by borrelidin in HCC cells
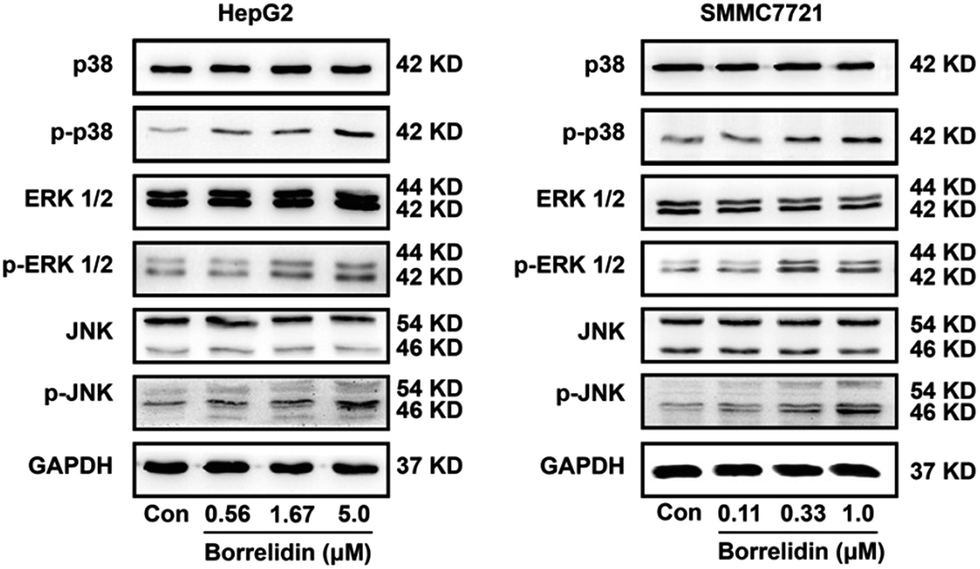
MAPKs family proteins, including the p38, extracellular signal-regulated kinases 1/2 (ERK1/2), and c-Jun amino (N)-terminal kinases 1/2 (JNK), played essential roles in anti-tumor agent-induced apoptosis and cell cycle arrest.24–27 JNK and p38 kinase could distort the balance of Bcl-2 family proteins, leading to cell apoptosis.28 The activation of ERK signaling pathway was responsible for the cleavage of caspases.29,30 Besides, activation of ERK could increase levels of p21 and p27 as well as reduce cyclin D and cyclin E expressions to cause cell cycle arrest.31,32 To investigate the effect of borrelidin on MAPKs signaling pathway, we performed western blot analysis to investigate the expressions of p38, ERK1/2 and JNK in the borrelidin treated cells. As shown in Fig. 6, phosphorylation levels of p38, ERK1/2 and JNK were obviously increased in borrelidin-treated HCC cells in a dose-dependent manner. The results indicated that the anti-tumor effect of borrelidin on HCC cells was related to up-regulating MAPKs signaling pathway.
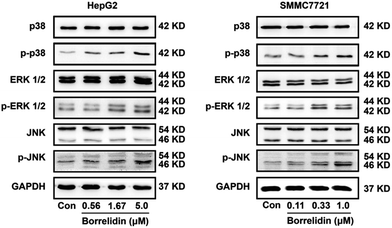 |
| | Fig. 6 Up-regulations of MAPKs pathway modulated cell cycle and apoptosis of HCC cells treated with borrelidin. The protein expression of p38, p-p38, ERK1/2, p-ERK1/2, JNK and p-JNK of HepG2 cells (A) and SMMC7721 cells (B) treated with borrelidin were assessed by western blot. The vehicle-treated group was the control (Con). | |
2.6. Borrelidin inhibited SMMC7721 cells xenograft growth in vivo
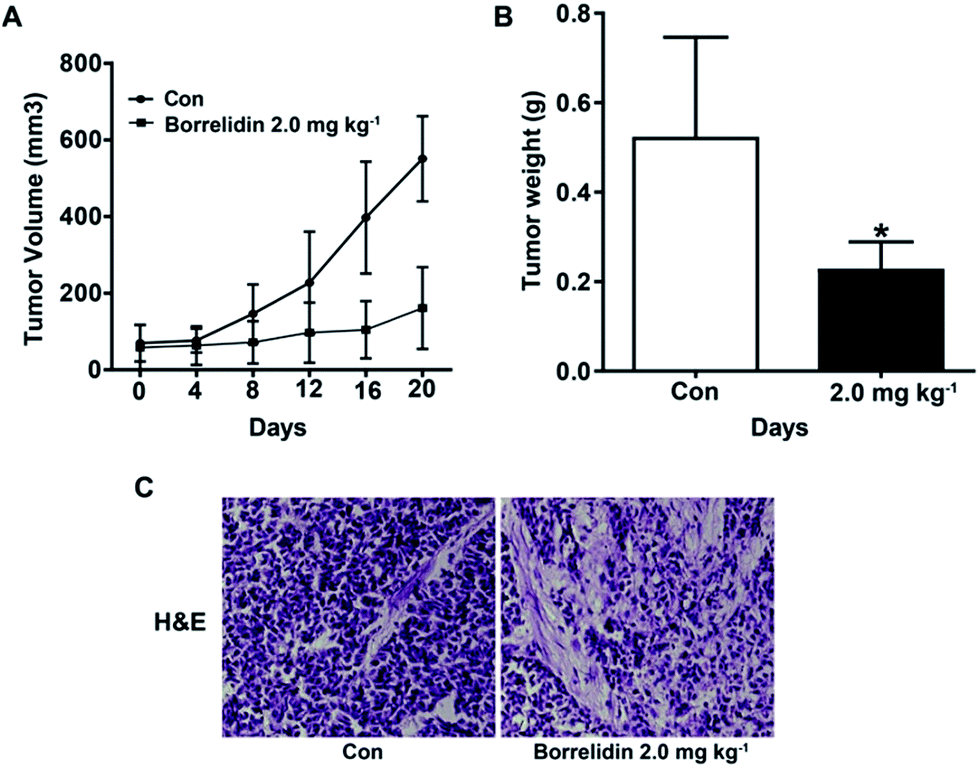
To investigate effect of borrelidin against HCC cells in vivo, a human hepatoma SMMC7721 xenograft model was established. As shown, borrelidin significantly inhibited the tumor growth compared to the control group (Fig. 7A and B). Besides, histological studies on tumor tissue sections with H&E staining showed that borrelidin changed the histology of human tumor xenografts in nude mice (Fig. 7C). Tumor tissues in the control group grew actively with deep-dyed big nucleolus. While tumor in the borrelidin treatment group showed distinct pathological features including fewer karyokinetic events, loss of cell–cell contacts and cell necrosis.
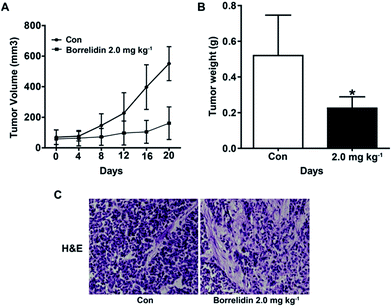 |
| | Fig. 7 The inhibitory effect of borrelidin on tumor growth in SMMC7721 cell xenograft model. (A) Tumor volumes were measured at the indicated days. (B) Tumor weight were recorded after mice were sacrificed. (C) Representative micrographs of tumor section of H&E staining. The vehicle-treated group was the control (Con). Data were represented as means ± SD (n = 5). Statistical differences were considered significantly at the level of *p < 0.05, vs. control. | |
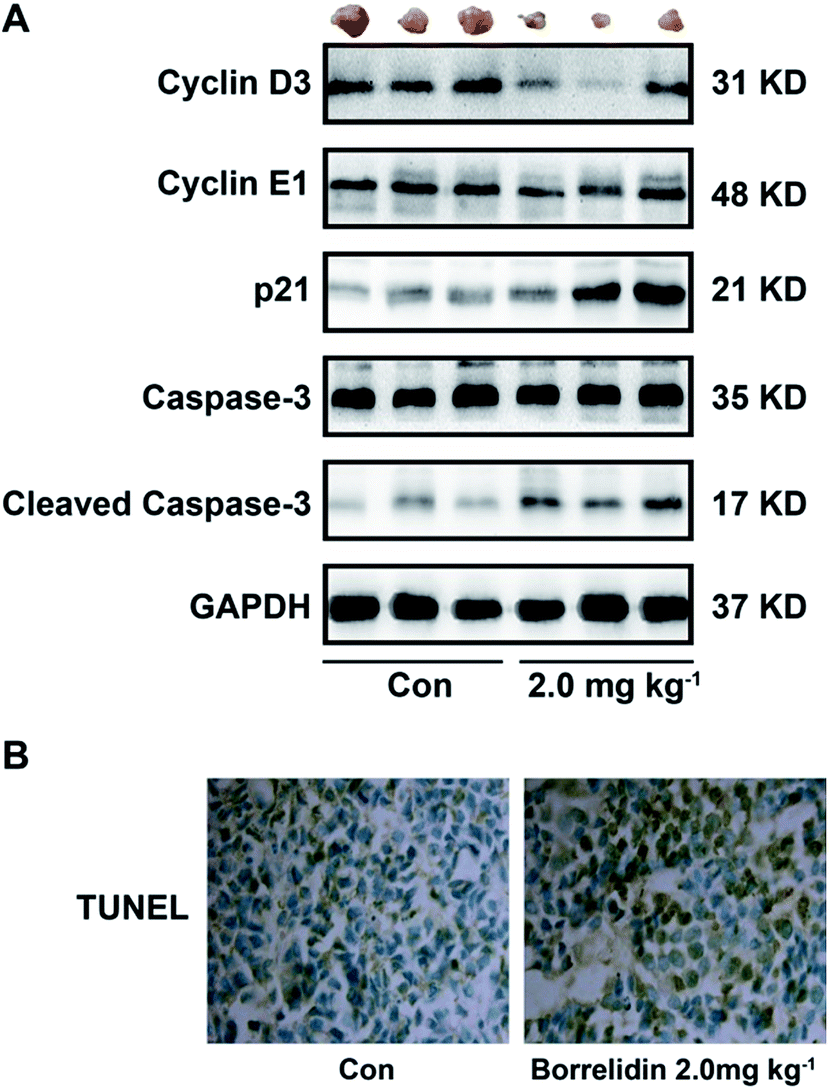
Based on the study in vitro, borrelidin inhibited the growth of SMMC7721 cells through causing cell cycle arrest and inducing apoptosis. Therefore, we performed western blot to verify the mechanism in vivo. As expected, the protein expressions of cyclin D3, cyclin E1 were reduced and p21 was increased, and cleavage of caspase-3 was activated in the tissues treated with borrelidin (Fig. 8A). Furthermore, TUNEL assay was performed in the xenografted tissues. Compared to vehicle treatment, the borrelidin treated tissues exhibited more TUNEL-positive cells, which demonstrated that borrelidin caused cell apoptosis accompanied by DNA breaks (Fig. 8B). Besides, there were no significant changes in body weight, liver index, spleen index, serum activities of AST and ALT between vehicle and borrelidin-treated group, which indicated that borrelidin did not cause severe toxicity in mice (Table 1).
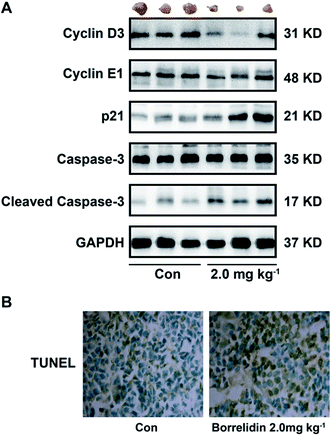 |
| | Fig. 8 Borrelidin caused cell cycle arrest and induced apoptosis in SMMC7721 cell xenograft model. (A) Protein expressions of cyclin D3, cyclin E1, p21, caspase-3 and cleaved caspase-3 of tumor tissues treated with borrelidin (0 and 2 mg kg−1) were assessed by western blot. (B) TUNEL staining of representative micrographs of tumor section treated with borrelidin (0 and 2 mg kg−1). The vehicle-treated group was the control (Con). | |
Table 1 Physiological and biochemical index of xenograft nude mice
| |
Body weight (g) |
Liver index (mg g−1) |
Spleen index (mg g−1) |
AST (karU) |
ALT (karU) |
| Con |
25.1 ± 1.0 |
53.7 ± 3.0 |
7.5 ± 1.7 |
97.2 ± 4.7 |
76.2 ± 3.6 |
| Borrelidin (2 mg kg−1) |
22.7 ± 2.5 |
54.9 ± 1.8 |
7.2 ± 1.8 |
97.9 ± 4.2 |
75.9 ± 2.9 |
3. Conclusions
Taken together, borrelidin caused G0/G1 cell cycle arrest, induced caspase-dependent apoptosis and inhibited the growth of HCC cells through up-regulating MAPKs signaling pathway. Moreover, it could attenuate migration and metastasis behavior of HCC cells. Borrelidin may serve as a potential lead for development of anti-tumor agent for HCC treatment.
4. Experimental
4.1. General experimental procedures
NMR spectra were collected on a Bruker AV-600 spectrometer, δ in ppm rel. to TMS, J in Hz. ESIMS were recorded on an Agilent 1290-6420 Triple Quadrupole LC-MS spectrometer. Silica gel (100–200 mesh, 200–300 mesh, Qingdao Marine Chemical Ltd., Qingdao, China), Sephadex LH-20 (GE Healthcare Bio-sciences AB, Uppsala, Sweden), YMC*GEL ODS-A (S-50 μm, 12 nm) (YMC Co., Ltd, Kyoto, Japan) were used for column chromatography. Flow cytometric analysis was used a LSR-Fortessa flow cytometer (BD, Franklin Lakes, NJ, USA). MTT, protein quantification and serum analysis were analyzed using a microplate reader (BioTek Synergy H1, BioTek Instruments, Inc., Vermont, USA). Human hepatocellular carcinoma cell lines HepG2 and SMMC7721 were purchased from Chinese Academy of Sciences (Shanghai, China). Fetal bovine serum (FBS), L-glutamine and penicillin-streptomycin were purchased from Gibco BRL Co., Ltd. (Gaithersburg, MD, USA). Antibodies of cyclin D1, cyclin D3, cyclin E1, CDK2, CDK4, CDK6, p21, Bax, Bcl-2, caspase-3, caspase-9, MMP-2, MMP-9, p38, p-p38 (Thr180/Tyr182), ERK1/2, p-ERK1/2 (Thr202/Tyr204), JNK, p-JNK (Thr183/Tyr185) and GAPDH were purchased from Cell Signaling Technology (Dancers, MA, USA).
4.2. Microbial material
The organism for producing borrelidin was isolated from fresh fecal samples excreted by healthy adult Hylobates hoolock living in Yunnan Wild Animal Park, Kunming, Yunnan province, in October 2013. The strain was identified as Streptomyces vinaceusdrappus by Dr Xiu Chen based on morphological characteristics and 16S rRNA gene sequences. The BLAST result showed that the sequence was most similar (100.0%) to the sequence of S. vinaceusdrappus (strain: NBRC 13099T, GenBank accession no. AB184311). The strain (no. YIM 100880) was deposited at the Yunnan Institute of Microbiology, Yunnan University, China.
4.3. Fermentation for Streptomyces vinaceusdrappus
A slant culture of the strain was used to inoculate 500 mL Erlenmeyer flasks containing 100 mL of seed medium composed of 4 g L−1 yeast extract, 4 g L−1 glucose, 5 g L−1 malt extract, 1.0 mL multiple vitamin solution, and 1.0 mL trace element solution at a pH of 7.2 with no adjustment. The flasks were cultured for 2 days at 28 °C on a rotary shaker at 180 rpm. This seed culture was used to inoculate fermentation medium with a 10% volume. The fermentation was carried out in a 500 mL Erlenmeyer flask containing 100 mL of fermentation medium composed of 10 g L−1 soybean meal, 2 g L−1 peptone, 5 g L−1 glucose, and 5 g L−1 soluble starch at a pH of 7.0 with no adjustment, and it was incubated for 7 days at 28 °C on a rotary shaker at 180 rpm.
4.4. Extraction, isolation and identification of borrelidin
The completed fermentation broth (80 L) was separated into filtrate and mycelium by centrifugation. The filtrate was extracted with EtOAc three times. The combined EtOAc extract was subjected to open silica gel (100–200 mesh) column chromatography eluting with a CH2Cl2–MeOH solvent system (from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1, and MeOH at last) to yield eight fractions, Fr.1–Fr.8. Fraction 4 (4.5 g) was subjected to Sephadex LH-20 chromatography (MeOH) to produce 9 subfractions (Fr.4.1–Fr.4.9). Fr.4.4 (0.3 g) and Fr.4.5 (2.0 g) containing borrelidin were collected and combined on the basis of their TLC profile (CH2Cl2–MeOH = 15:1, Rf = 0.48) and further separated by ODS column chromatography, eluting with water–methanol (25:75) to give a purified borrelidin (about 1.25 g).
:1 to 1:1, and MeOH at last) to yield eight fractions, Fr.1–Fr.8. Fraction 4 (4.5 g) was subjected to Sephadex LH-20 chromatography (MeOH) to produce 9 subfractions (Fr.4.1–Fr.4.9). Fr.4.4 (0.3 g) and Fr.4.5 (2.0 g) containing borrelidin were collected and combined on the basis of their TLC profile (CH2Cl2–MeOH = 15:1, Rf = 0.48) and further separated by ODS column chromatography, eluting with water–methanol (25:75) to give a purified borrelidin (about 1.25 g).
Borrelidin: white amorphous power, [α]25D = −12.53° (c 0.76, MeOH); ESI-MS m/z: 490 [M + H]+, 512 [M + Na]+. 1H-NMR (600 MHz, DMSO-d6) δ (ppm): 6.95 (1H, d, J = 11.4 Hz), 6.46 (1H, brt, J = 13.0 Hz), 6.31 (1H, ddd, J = 14.7, 10.5, 4.3 Hz), 4.88 (1H, dt, J = 10.7, 3.6 Hz), 4.06 (1H, d, J = 9.6 Hz), 3.75 (1H, dt, J = 9.8, 3.0 Hz), 2.47–2.54 (2H, m), 2.37 (1H, ddd, J = 14.9, 10.7, 4.1 Hz), 2.29 (1H, q, J = 7.9 Hz), 2.25 (1H, dd, J = 15.4, 3.2 Hz), 2.07 (1H, dd, J = 15.4, 9.8 Hz), 1.84–1.90 (2H, m), 1.80 (1H, m), 1.60–1.73 (5H, m), 1.55 (1H, m), 1.35 (1H, m), 1.10 (2H, m), 1.00 (1H, brt, J = 13.0 Hz), 0.93 (1H, m), 0.92 (3H, d, J = 6.4 Hz, CH3), 0.86 (1H, brt, J = 11.1 Hz), 0.78 (3H, d, J = 6.4 Hz, CH3), 0.77 (3H, d, J = 6.4 Hz, CH3), 0.74 (3H, d, J = 7.2 Hz, CH3), 0.60 (1H, brt, J = 11.5 Hz); 13C-NMR (150 MHz, DMSO-d6) δ (ppm): 177.6, 171.3, 143.6, 139.2, 127.8, 119.5, 116.8, 75.3, 71.0, 70.6, 48.5, 48.1, 45.9, 43.4, 37.6, 37.4, 35.9, 35.7, 34.8, 31.2, 29.3, 27.3, 26.4, 25.2, 20.8, 18.8, 18.6, 15.4. The spectral properties were identical with those reported in the literature.33,34
4.5. Cell culture
Human hepatocellular carcinoma cell lines HepG2 and SMMC7721 were cultured in DMEM and RPIM-1640 culture medium, respectively, with 10% fetal bovine serum (FBS), 1% L-glutamine, 100 units per mL penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified 5% CO2 atmosphere incubator (Thermo Fisher Scientific, Waltham, MA, USA).
4.6. MTT assay
The cytotoxicity and proliferation inhibition were quantified by the MTT assay. HepG2 and SMMC7721 cells (7 × 103 cells per well) were seeded in 96-well plates respectively and exposed to borrelidin at six concentrations (0.33, 1.0, 3.3, 10.0, 33.0 and 100.0 μM). After 48 h incubation at 37 °C, 20 μL MTT was added into each well and incubated another 4 h. And then the liquid in the wells was removed. After that, 150 μL DMSO was added into each well. Use a microplate reader at 570 nm to measure the absorbance of each well. The results were represented as the mean ± SD from three independent experiments. The inhibition rate (%) = [(A570 (control)−A570 (borrelidin))]/A570 (control) × 100% and the IC50 values were calculated by a non-linear regression analysis using SPSS 13.0 (SPSS Inc., Chicago, IL, USA).
4.7. Measurement of cell cycle arrest
HepG2 and SMMC7721 cells (5 × 105 cells per well) were seeded in 6-well plates with 5.0 μM and 1.0 μM of borrelidin treatment for 24 h, respectively, then trypsinized, centrifuged and washed with cold PBS, and fixed with 75% ethanol left overnight at −20 °C. Following adequate washing with PBS, the fixed cells were incubated with PI working fluid (Dingguo Changsheng Biotechnology Co. Ltd., Beijing, China) for 30 min in the dark at room temperature. Samples were immediately run on a LSR-Fortessa flow cytometer with a total of 20000 cells collected and analyzed with the PI-based cell cycle phase. The results were analyzed using ModFit LT 4.1 (Verity Software House, Topsham, ME, USA) and represented as the mean ± SD from three independent experiments.
4.8. Hoechst 33258 staining
Cells stained Hoechst 33258 (Beyotime Biotechnology Co. Ltd, Shanghai, China) was used to evaluate the morphological changes. HepG2 and SMMC7721 cells (105 cells per well) were seeded in 6-well plates and treated with 5.0 μM and 1.0 μM of borrelidin for 24 h, respectively. Then cells were fixed with paraformaldehyde for 5 min, washed three times with PBS and stained with 500 μL Hoechst 33258 for 5 min in the dark at room temperature. Morphological changes were evaluated and photographed by fluorescence microscope (DMi8, Leica, Heidelberg, Germany).
4.9. Measurement of apoptosis
HepG2 and SMMC7721 cells (5 × 105 cells per well) were seeded in 6-well plates and treated with 5.0 μM and 1.0 μM of borrelidin for 24 h, respectively. Then trypsinized, centrifuged and washed with cold PBS, and re-suspended in 490 μL of the provided binding buffer. 5 μL FITC-labeled Annexin V and 5 μL PI (BD, Franklin Lakes, NJ, USA) were added to the samples and mixed gently. After 15 min incubation in the dark at room temperature, samples were immediately run on a LSR-Fortessa flow cytometer with a total of 20000 cells collected and analyzed with the Annexin V-FITC/PI apoptosis method. Cytographs were performed using BD FACSDiva Software (BD, Franklin Lakes, NJ, USA). The results were represented as the mean ± SD from three independent experiments.
4.10. Wound healing assay
HepG2 and SMMC7721 cells (106 cells per well) were seeded in 6-well plates. After cells were confluent, two vertical linear “wounds” were carefully scratched by a 200 μL plastic pipette tip. Then washed with PBS and added into 2 mL serum-free medium with 0, 0.56, 1.67 and 5.0 μM of borrelidin for HepG2 cell, and 0, 0.11, 0.33 and 1.0 μM of SMMC7721 cell, respectively, then incubated at 37 °C and photographed every 12 h by inverted microscope.
4.11. The transwell assay and tumor invasion assay
A Boyden chamber (Thermo Fisher Scientific, Roskilde, Denmark) was used to examine the cell migration and invasion. For the invasion assay, BD Matrigel™ Basement Membrane Matrix (BD, Franklin Lakes, NJ, USA) was thawed overnight at 4 °C and diluted with cold serum-free medium (1:3). 200 μL of DMEM (for HepG2 cells) or RPMI-1640 (for SMMC7721 cells) medium with 20% FBS was added to the lower chamber of the transwell plate. The upper chamber of transwell was coated with 100 μL Matrigel. After Matrigel solidified, 200 μL of DMEM or RPMI-1640 medium with 1% FBS and 5 × 105 cells and different concentrations of borrelidin (0, 0.56, 1.67 and 5.0 μM for HepG2 cells; 0, 0.11, 0.33 and 1.0 μM for SMMC7721 cells) were added to the upper chamber of the transwell plates, respectively. After 24 h incubation at 37 °C, removed non-invaded cells with a cotton swab and fixed invaded cells in methanol and stained with crystal violet. For migration, the procedure was the same as the invasion assay except absence of Matrigel. The pictures were photographed by inverted microscope. The results were represented as the mean ± SD from three independent experiments.
4.12. RNA extraction and real-time qPCR
The effects of borrelidin on the gene expression were examined in HepG2 and SMMC7721 cells by SYBR Green real-time quantitative PCR (Applied Biosystems, Waltham, MA, USA). HepG2 and SMMC7721 cells (8 × 105 cells per well) were seeded in 6-well plates and treated with 5.0 μM and 1.0 μM of borrelidin for 24 h, respectively. The total RNA from cells was extracted using TRIzol Reagent (Ambion, Foster City, CA, USA) according to the manufacturer's instructions. The cDNA was synthesized from the extracted total RNA using a reverse transcriptase kit (Promega, Madison, WI, USA). The forward and reverse primers for Cyclin D1 were 5-CTGTGCTGCGAAGTGGAAACCAT-3 and 5-TTCATGGCCAGCGGGAAGACCTC-3, respectively; Cyclin D3 were 5-AGCGCCTTTCCCAACTCTA-3 and 5-CCAGGGTTACCACCACTTGT-3; Cyclin E1 were 5-GCTCGGGCTTTGTCCAG-3 and 5-CTTCCTGGACACGCTGGT-3; CDK2 were 5-CCTTGTTTGTCCCTTCTAC-3 and 5-CAAATCCACCCACTATGA-3; CDK4 were 5-CCTCTTTGGCAGCTGGTCAC-3 and 5-ACCGACACCAATTTCAGCCA-3; CDK6 were 5-CTGAATGCTCTTGCTCCTTT-3 and 5-AAAGTTTTGGTGGTCCTTGA-3; p21 were 5-AGGCACCGAGGCACTCAGAG-3 and 5-TGACAGGTCCACATGGTCTTCC-3; Bax were 5-CTGAGCAGATCATGAAGACA-3 and 5-AGTTTGCTGGCAAAGTAGAA-3; Bcl-2 were 5-CTTTGAGTTCGGTGGGGTCA-3 and 5-GGGCCGTACAGTTCCACAAA-3; GAPDH were 5-GGCAAGTTCAATGGCACAGT-3 and 5-TGGTGAAGACGCCAGTAGACTC-3. Real-time qPCR was performed using at 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 20 s for total 35 cycles. GAPDH was used as an internal control. The 2−ΔΔCT method was used to analyze relative changes in gene expressions for statistical analysis.35
4.13. Western blot analysis
In vitro experiment, HepG2 and SMMC7721 cells (8 × 105 cells per well) were seeded in 6-well plates and treated with different concentrations (0, 0.56, 1.67 and 5.0 μM for HepG2 cell, and 0, 0.11, 0.33 and 1.0 μM for SMMC7721 cell) of borrelidin for 24 h, then washed with cold PBS and lysed with RIPA lysis buffer (Beyotime Biotechnology Co. Ltd, Shanghai, China) containing 2% protease inhibitor cocktail (Promega, Madison, WI, USA). In vivo experiment, tumor tissue homogenate was lysed for western blot analysis. The protein concentration of the lysates was determined by a BCA Protein Assay Kit (Beyotime Biotechnology Co. Ltd, Shanghai, China). Equal quantities of protein (40–60 μg) were separated by SDS-PAGE, transferred to PVDF membranes (Millipore, Billerica, MA, USA), and blocked with 5% non-fat milk for 90 min at room temperature. The membranes were incubated with specific primary antibodies overnight at 4 °C and subsequently probed with corresponding secondary antibodies for 1 h at room temperature. Then, protein bands were detected with ECL select western Blot detection reagent (Millipore, Billerica, MA, USA) by chemiluminescence (Tanon 5500, Shang Hai, China). The relative amounts of protein loaded was normalized to the respective GAPDH bands.
4.14. Animal and tumor xenograft studies
BALB/c nude mice (weighing 18–20 g) were used for building a xenograft model. Mice were administrated with 100 μL SMMC7721 cell suspension (5 × 106 cells) by subcutaneous injection, and when tumor volumes reaching 50 mm3, mice was randomly allocated to two groups (n = 5 per group). Animals of borrelidin treated group was administered borrelidin (2 mg kg−1, dissolved in 60% 1,2-propanediol) once every other day by intraperitoneal injection. Control group were treated with vehicle in the same way. Body weight and tumor size (tumor volume = length (mm) × width (mm) × depth (mm) × π/6) were measured every other day. After 3 weeks of treatment, whole blood of each mouse was collected from the orbit for serum analysis. Then, the mice were sacrificed, and the tumor was excised. The wet weight of internal organs and tumor of each mouse were recorded. A part of tumor was prepared for western blot and other part for paraffin block. The study was performed in strict accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006), and all experimental protocols were approved by the Laboratory Ethics Committees of College of Life and Health Sciences, Northeastern University (Shenyang, China).
4.15. Hematoxylin and eosin (H&E) staining
Tumor tissues were fixed in 4% paraformaldehyde, paraffin-embedded and section at 5 μm. Afterwards, the tissue section was deparaffinized in xylene and rehydrated in a series of graded alcohols. To visualize histology, the tissue section was stained with hematoxylin and eosin and photographed using a microscope (Olympus, Model BX40F4, Tokyo, Japan).
4.16. Detection of apoptotic cells in situ
The number of apoptosis cells was identified by Tdt-mediated dUTP nick end labelling (TUNEL) method with an apoptosis detection kit (Millipore, Billerica, MA, USA) according to the manufacturer's instructions.36
4.17. Serum analysis
Serum from each mouse was analyzed to observe changes of aspartate aminotransferase (AST) and L-alanine 2-oxoglutarate aminotransferase (ALT) levels. Serum AST and ALT analysis were conducted in 96-well plates using Aspartate aminotransferase Assay Kit and Alanine aminotransferase Assay Kit (Jiancheng Bioengineering Ins., Nanjing, China) respectively according to the manufacture's instruction. The enzyme activity was monitored by measuring the optical density (510 nm) of the plates.
5. Data analysis
All results are presented graphically as the group mean ± standard deviation (SD) from at least three independent experiments. Student's t-test was used to determine differences between two groups using SPSS 13.0. The threshold value for acceptance of differences was 5% (p ≤ 0.05).
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was funded by National Natural Science Foundation of China (No. 81573327, 81500934, and 31460005) and Basic Scientific Research Fund of Northeastern University, China (No. N142002001).
References
- J. L. Petrick, M. Braunlin, M. Laversanne, P. C. Valery, F. Bray and K. A. McGlynn, Int. J. Cancer, 2016, 139, 1534–1545 CrossRef CAS PubMed.
- J. U. Marquardt, P. R. Galle and A. Teufel, J. Hepatol., 2012, 56, 267–275 CrossRef PubMed.
- M. A. Avila, C. Berasain, B. Sangro and J. Prieto, Oncogene, 2006, 25, 3866–3884 CrossRef CAS PubMed.
- J. M. Llovet and J. Bruix, J. Hepatol., 2008, 48(suppl 1), 20–37 CrossRef PubMed.
- D. J. Trader and E. E. Carlson, Bioorg. Med. Chem. Lett., 2015, 25, 4767–4769 CrossRef CAS PubMed.
- T. Wakabayashi, R. Kageyama, N. Naruse, N. Tsukahara, Y. Funahashi, K. Kitoh and Y. Watanabe, J. Antibiot., 1997, 50, 671–676 CrossRef CAS PubMed.
- D. Habibi, N. Ogloff, R. B. Jalili, A. Yost, A. P. Weng, A. Ghahary and C. J. Ong, Invest. New Drugs, 2012, 30, 1361–1370 CrossRef CAS PubMed.
- A. Sidhu, J. R. Miller, A. Tripathi, D. M. Garshott, A. L. Brownell, D. J. Chiego, C. Arevang, Q. Zeng, L. C. Jackson, S. A. Bechler, M. U. Callaghan, G. H. Yoo, S. Sethi, H. S. Lin, J. H. Callaghan, G. Tamayo-Castillo, D. H. Sherman, R. J. Kaufman and A. M. Fribley, ACS Med. Chem. Lett., 2015, 6, 1122–1127 CrossRef CAS PubMed.
- T. Kawamura, D. Liu, M. J. Towle, R. Kageyama, N. Tsukahara, T. Wakabayashi and B. A. Littlefield, J. Antibiot., 2003, 56, 709–715 CrossRef CAS PubMed.
- M. Malumbres and M. Barbacid, Nat. Rev. Cancer, 2009, 9, 153–166 CrossRef CAS PubMed.
- H. Gali-Muhtasib and N. Bakkar, Curr. Cancer Drug Targets, 2002, 2, 309–336 CrossRef CAS PubMed.
- S. K. Mantena, S. D. Sharma and S. K. Katiyar, Carcinogenesis, 2006, 27, 2018–2027 CrossRef CAS PubMed.
- A. Deshpande, P. Sicinski and P. W. Hinds, Oncogene, 2005, 24, 2909–2915 CrossRef CAS PubMed.
- D. Gafiuc, M. Weiß, I. Mylonas and A. Brüning, J. Appl. Toxicol., 2014, 34, 1109–1113 CrossRef CAS PubMed.
- A. Gross, J. M. Mcdonnell and S. J. Korsmeyer, Genes Dev., 1999, 13, 1899–1911 CrossRef CAS PubMed.
- I. Budihardjo, H. Oliver, M. Lutter, X. Luo and X. Wang, Annu. Rev. Cell Dev. Biol., 1999, 15, 269–290 CrossRef CAS PubMed.
- S. K. Mantena, S. D. Sharma and S. K. Katlyar, Mol. Cancer Ther., 2006, 5, 296–308 CrossRef CAS PubMed.
- E. M. Creagh, H. Conroy and S. J. Martin, Immunol. Rev., 2003, 193, 10–21 CrossRef CAS PubMed.
- P. Friedl and K. Wolf, Nat. Rev. Cancer, 2003, 3, 362–374 CrossRef CAS PubMed.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS PubMed.
- N. Ramos-DeSimone, E. Hahn-Dantona, J. Sipley, H. Nagase, D. L. French and J. P. Quigley, J. Biol. Chem., 1999, 274, 13066–13076 CrossRef CAS PubMed.
- H. Zheng, H. Takahashi, Y. Murai, Z. Cui, K. Nomoto, H. Niwa, K. Tsuneyama and Y. Takano, Anticancer Res., 2006, 26, 3579–3583 CAS.
- A. Moroz, F. K. Delella, R. Almeida, L. M. Lacorte, W. J. Fávaro, E. Deffune and S. L. Felisbino, PLoS One, 2013, 8, e84757 Search PubMed.
- J. Wen, Y. Zhang, X. Chen, L. Shen, G. C. Li and M. Xu, Biochem. Pharmacol., 2004, 68, 323–331 CrossRef CAS PubMed.
- H. J. Kim, N. Chakravarti, N. Oridate, C. Choe, F. X. Claret and R. Lotan, Oncogene, 2006, 25, 2785–2794 CrossRef CAS PubMed.
- H. S. Park, K. I. Park, D. H. Lee, S. R. Kang, A. Nagappan, J. A. Kim, E. H. Kim, W. S. Lee, S. C. Shin, Y. S. Hah and G. S. Kim, Food Chem. Toxicol., 2012, 50, 2407–2416 CrossRef CAS PubMed.
- C. W. Phang, S. A. Karsani, G. Sethi, S. N. A. Malek and S. N. Abd Malek, PLoS One, 2016, 11, e0148775 Search PubMed.
- X. Sui, N. Kong, L. Ye, W. Han, J. Zhou, Q. Zhang, C. He and H. Pan, Cancer Lett., 2014, 344, 174–179 CrossRef CAS PubMed.
- S. J. Kim, N. W. Ju, C. D. Oh, Y. M. Yoon, W. K. Song, J. H. Kim, Y. J. Yoo, O. S. Bang, S. S. Kang and J. S. Chun, J. Biol. Chem., 2002, 277, 1332–1339 CrossRef CAS PubMed.
- C. Stefanelli, B. Tantini, M. Fattori, I. Stanic', C. Pignatti, C. Clo, C. Guarnieri, C. M. Caldarera, C. A. Mackintosh, A. E. Pegg and F. Flamigni, FEBS Lett., 2002, 527, 223–228 CrossRef CAS PubMed.
- L. Brown and S. Benchimol, J. Biol. Chem., 2006, 281, 3832–3840 CrossRef CAS PubMed.
- H. Zhang and I. Rosdahl, Int. J. Oncol., 2004, 25, 1241–1248 CAS.
- M. S. Kuo, D. A. Yurek and D. A. Kloosterman, J. Antibiot., 1989, 42, 1006–1007 CrossRef CAS PubMed.
- Z. Cao, G. Khodakaramian, K. Arakawa and H. Kinashi, Biosci., Biotechnol., Biochem., 2012, 76, 353–357 CrossRef CAS PubMed.
- M. W. Pfaffl, Nucleic Acids Res., 2001, 29, e45 CrossRef CAS PubMed.
- X. Jiang, X. Zhu, N. Liu, H. Xu, Z. Zhao, S. Li, J. Cai and J. Cao, Int. J. Biol. Sci., 2017, 13, 167–178 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, 13C-NMR, ESI-MS spectra and HPLC chromatogram of borrelidin, and the relative quantitative analysis of related proteins assessed by western blot in Fig. 3(C and D), 4(E and F), 5(C), and 6. See DOI: 10.1039/c7ra08290h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yi Jiangb,
Li Han*a,
Xiu Chenb,
Caijuan Hua,
Hao Sua,
Yu Mua,
Peipei Guana and
Xueshi Huang
a,
Yi Jiangb,
Li Han*a,
Xiu Chenb,
Caijuan Hua,
Hao Sua,
Yu Mua,
Peipei Guana and
Xueshi Huang