
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Internal asymmetric induction by the C-6 substituent on the oxidation reaction of interglycosidic sulfur atom of thiodisaccharides†
J. P. Colomer*a,
A. B. Peñéñory a and
O. Varela*bc
a and
O. Varela*bc
aInstituto de Investigaciones en Fisico-Química de Córdoba – Universidad Nacional de Córdoba (INFIQC-UNC-CONICET), Facultad de Ciencias Químicas, Departamento de Química Orgánica, Edificio de Ciencias 2, Ciudad Universitaria, CP5016 – Córdoba, Argentina. E-mail: juanpablo@fcq.unc.edu.ar
bUniversidad de Buenos Aires, Facultad de Ciencias Exactas y Naturales, Departamento de Química Orgánica, Pabellón 2, Ciudad Universitaria, C1428EHA – Buenos Aires, Argentina. E-mail: varela@qo.fcen.uba.ar
cConsejo Nacional de Investigaciones Científicas y Técnicas (CONICET)-UBA, Centro de Investigación en Hidratos de Carbono (CIHIDECAR), Buenos Aires, Argentina
First published on 14th September 2017
Abstract
Since the enantio or diastereoselective preparation of sulfoxides is a current challenge, we explore the possibility of inducing diastereoselectivity in the oxidation of the sulfur atom of thiodisaccharides, according to their substitution patterns. Thus, a series of 3-deoxy-4-S-(β-D-glucopyranosyl)-4-thio-β-D-xylo-hexopyranoside derivatives, with different substituents at C-6 (OH, OAc or OTBS) of the reducing end, have been synthesized and treated with m-CPBA for the oxidation of the sulfur atom at C-4, which is vicinal to C-6. The absolute configuration at the sulfur stereocenter of the resulting sulfoxides was established taking into account shielding/deshielding anisotropic effects of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond on the chemical shift of the NMR signals of selected protons, in the most populated syn ϕ/syn ψ conformation of the thiodisaccharide S-oxides. The OAc and OTBS derivatives afforded diastereomeric mixtures of R and S sulfoxides in a similar ratio (1.4
O bond on the chemical shift of the NMR signals of selected protons, in the most populated syn ϕ/syn ψ conformation of the thiodisaccharide S-oxides. The OAc and OTBS derivatives afforded diastereomeric mixtures of R and S sulfoxides in a similar ratio (1.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1.6:1, respectively). In contrast, the oxidation of thiodisaccharide with a free hydroxyl group at C-6 was completely diastereoselective in favor of the R sulfoxide. The influence of the thiodisaccharide C-6 substituent on the stereochemical course of the oxidation is discussed.
:1 and 1.6:1, respectively). In contrast, the oxidation of thiodisaccharide with a free hydroxyl group at C-6 was completely diastereoselective in favor of the R sulfoxide. The influence of the thiodisaccharide C-6 substituent on the stereochemical course of the oxidation is discussed.
Introduction
Numerous organic molecules containing sulfoxide functionality show a wide range of interesting biological activities. Many compounds of this type are employed as pharmaceuticals (e.g., sulforaphane,1 esomeprazole,2 and armodafinil3) and present a specific configuration at the sulfur stereocenter, which is crucial to generate the desired biological effect. In addition, enantiomerically pure sulfoxides are employed as powerful chiral auxiliaries, ligands or catalysts in asymmetric synthesis.4–7 Therefore, the enantio or diastereoselective preparation of sulfoxides is a current challenge for synthetic organic chemists. In this regard, a number of procedures for the diastereoselective or enantioselective (including enzymes or microorganisms) sulfoxidations have been reported.8–11The oxidation of the sulfur atom of thioglycosides usually affords diastereomeric mixtures of glycoside sulfoxides, and the diastereoselectivity relies on the solvent, the temperature and the structure and substitution of the starting S-glycoside.12 In some particular cases, high diastereoselectivities in the oxidation of thioglycosides have been achieved.13 Witczak and coworkers14 reported the oxidation of thiodisaccharides to sulfoxides or sulfones, derivatives that inhibit the proliferation of selected murine and human tumor cell lines. However, the sulfoxides were obtained as diastereomeric mixtures and the absolute configuration of the SO group has not been established.
In connection with our work on the synthesis of thiodisaccharides and their inhibitory activity of specific glycosidases,15–19 we have studied the oxidation of these substrates to their respective diastereomeric sulfoxides. These compounds could be successfully separated in many cases and the absolute configuration of the sulfur stereocenter was determined by a procedure developed by us.20,21 This procedure is based on NMR techniques and takes into account anisotropic effects of the SO group. Our results were in agreement with those obtained using other methodologies employed to assign the sulfur absolute configuration of glycosyl sulfoxides.13,22–24
The diastereomers RS and SS of sugar sulfoxides are hydrolyzed with different kinetics by acids or glycosidases.12 Thus, we have prepared and assigned the configuration of sulfur for both isomers of benzyl 3-deoxy-4-S-(β-D-galactopyranosyl)-4-thio-β-D-threo-pentopyranoside S-oxides, which showed to be competitive inhibitors of the β-galactosidase from Escherichia coli.20,21 The key structural features of the molecular recognition process have been determined using NMR techniques and molecular modelling.25 The different configuration at sulfur resulted in dissimilar conformational behavior and in a distinctive geometrical disposition of the molecule in the active site of the enzyme. These structural modifications justify the different rates of hydrolysis observed for each diastereoisomer.
Due to the importance of a specific configuration at the sulfur stereocenter of sulfoxides and as continuation of our previous work on this subject, we report herein the study of the oxidation of 3-deoxy-S-(1→4)-disaccharides, with the aim of achieving high diastereoselectivity in the reaction.
Results and discussion
In order to determine the influence of the C-6 substituent of the reducing end, vicinal to the sulfur at C-4, on the stereochemical course of the oxidation of this atom, a series of 3-deoxy-S-(1→4)-disaccharides was synthesized. Thus, the synthetic route employed involved the conjugate addition of 1-thioaldose derivatives to sugar enones followed by reduction of the carbonyl group, which proved to be highly diastereoselective and high yielding.17,19 As Michael acceptors, the sugar enones 1–3, with the C-6 hydroxyl group unprotected (2) or substituted as acetyl ester (1) or tert-butyldimethylsilyl ether (TBS, 3), were prepared (Scheme 1). The enone 1 was obtained by the methodology previously described.26 The deacetylation of 1 to afford 2 was conducted under mild conditions using bis(tributyltin) oxide27 (TBTO) in order to avoid the elimination of the AcO group at C-6. The silylation of the free hydroxyl group of 2 was performed with tert-butyldimethylsilyl chloride (TBSCl) in MeCN to give the enone 3. | ||
| Scheme 1 Synthetic sequence to obtain 2H-pyran-3(6H)-ones 1–3. | ||
The conjugate addition of 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranose28 (4) to 2H-pyran-3(6H)-ones 1 or 3 was conducted at −18 °C for 1.5 h, in the presence of a catalytic amount of triethylamine (Et3N). As already described in previous work, and to avoid the retro-Michael reaction,19 the crude mixture of 2-keto thiodisaccharides 5 (Scheme 2) was treated with sodium borohydride in MeOH (0 °C, 30 min) for the reduction of the carbonyl group, to afford the 3-deoxy-4-S-glycosyl-4-thiohexopyranosides 6–8 (from 1) and 9–11 (from 3). The control of the temperature was important in order to increase the diastereoselectivity during the sequence. The NaBH4 reduction requires also a short time, as longer reaction times led to partial de-O-acetylation of the products.
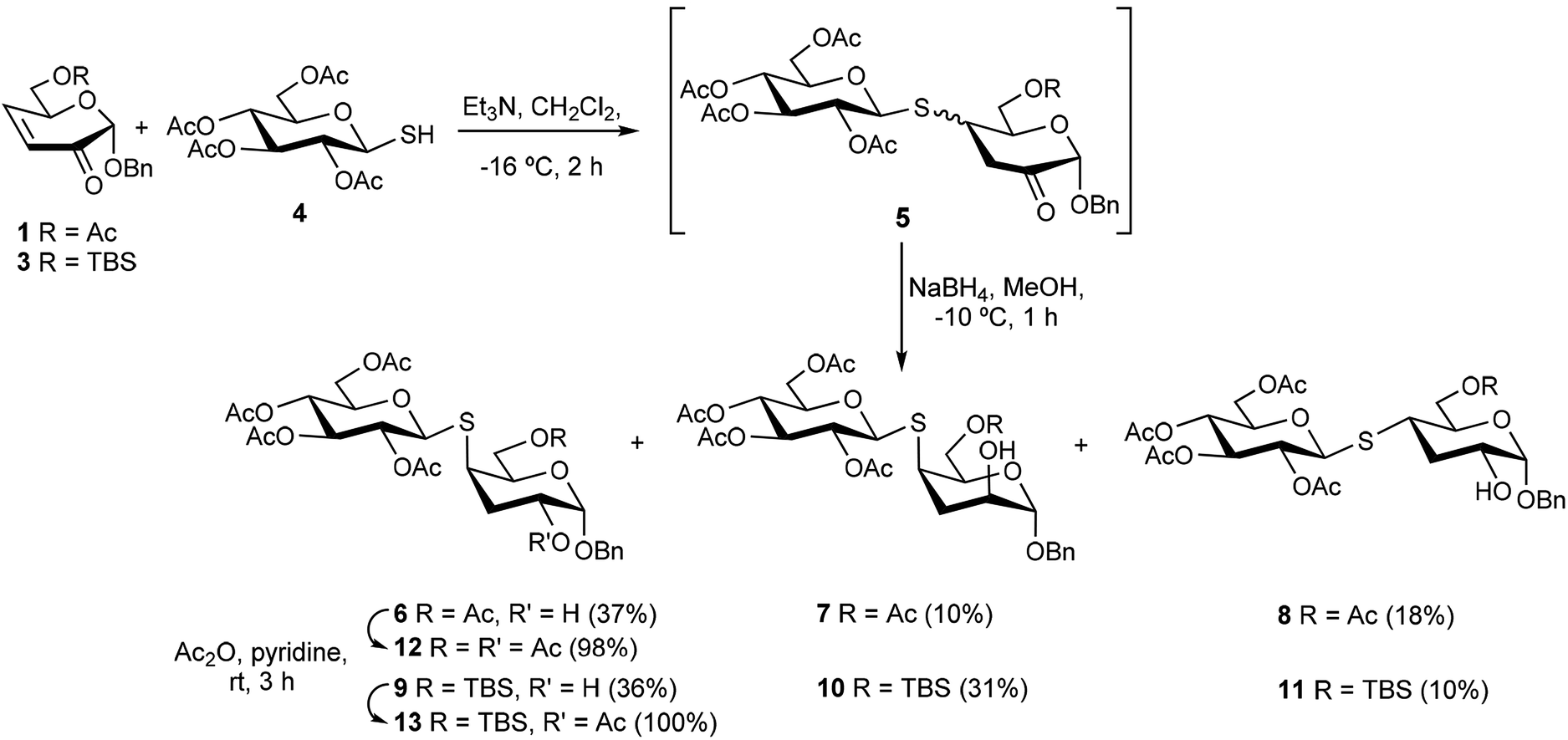 | ||
| Scheme 2 Michael addition of thioaldose 4 to 2H-pyran-3(6H)-ones 1 and 3, followed by reduction, to afford thiodisaccharides 6–11. | ||
The mixtures obtained were subjected to column chromatography. The major product isolated starting from 1 was the thiodisaccharide 6. The 1H NMR spectrum of 6 revealed small coupling constant values for 4-H, which appeared as a broad singlet, and for 2-H (J1,2 = 3.5 Hz, J2,3eq.= J3eq,4 = 3.7 Hz) indicating the R configuration for the new stereocenters at C-4 and C-2 (3-deoxy-D-xylo-hexopyranoside configuration for the reducing end). Similarly, the coupling constant values determined for 4-H (J3ax,4 = J4,5 = 12.8, J3eq.4 = 3.9 Hz) and for 2-H (J1,2 = 1.8, J2,3eq = 4.3, J2,3ax = 12.3 Hz), are indicative that the thiodisaccharide 8, which was isolated as a minor product, possessed respectively the R and S configuration at C-2 and C-4 (3-deoxy-D-ribo-hexopyranoside). The large J values observed for J2,3ax, J3ax,4 and J4,5 are result of the axial disposition for both 2-H and 4-H, in the preferred chair conformation of the 3-deoxy-pyranoside unit. A third minor component of the mixture was the thiodisaccharide 7, which was contaminated with a non-reduced 2-keto thiodisaccharide 5. However, some signals such as 1-H (broad singlet), 2-H (J2,3a = J2,3b = 4.6 Hz) and 4-H (broad singlet) allowed us to assign the configuration of the reducing end of 7 as 3-deoxy-D-lyxo-hexopyranoside.
Thiodisaccharide 9 was the main product obtained from the enone 3. This compound showed the same configuration at C-4 and C-2 as that of 6, with equatorial disposition for 4-H (broad doublet) and axial for 2-H (J1,2 = 3.7, J2,3eq = 3.3 Hz).
The thiodisaccharides 10 and 11 were also isolated as an inseparable mixture, in a 3.3:1 ratio, respectively. The configuration of the new stereocenters at C-4 and C-2 of 10 and 11 were also assigned on the basis of 1H NMR signals that appeared in a clean region of the spectrum (no overlapped with other resonances). In the case of 10, the small coupling constant values for 4-H (broad doublet) and 1-H (broad singlet) suggested an equatorial disposition for both protons (3-deoxy-D-lyxo-hexopyranoside configuration). The NMR spectrum of thiodisaccharide 11 showed coupling constant values for 4-H (J3ax,4 = 13.5, J3eq,4 = 4.2, J4,5 = 11.1 Hz) and 2-H (J1,2 = 3.5, J2,3ax = 12.4 Hz) in agreement with the axial disposition of 4-H and 2-H (3-deoxy-D-ribo-hexopyranoside configuration).
The diastereofacial selectivity observed for the conjugate addition reaction was attributed to the stereocontrol exerted by the axial disposition of the benzyloxy group of dihydropyranones 1 and 3, in the preferred 0H5 conformation.29 This conformation is stabilized by the anomeric effect, increased by the carbonyl group vicinal to the anomeric position,30 and also because of the equatorial orientation of the bulky substituted hydroxymethyl group. Similarly, the diastereofacial selectivity in the borohydride reduction, in favor of the thiodisaccharides 6 and 8, was also attributed to the stereocontrol produced by the axial benzyloxy group, which induced the approach of the hydride from the Si face of the carbonyl. On the other hand, the reduction of the 6-O-TBS substituted intermediate 5, led to an approximately 1:1 ratio of 9:10. Probably the steric hindrance generated by the silyloxymethyl group (even in an equatorial disposition) in combination with the axially disposed thioglycoside unit, both opposite to the benzyloxy group, could encumber the stereocontrol exerted by this anomeric substituent, leading to a higher proportion of 10.
The free hydroxyl group of thiodisaccharides 6 and 9 was subjected to acetylation leading to compounds 12 and 13, which differ in the substitution at C-6. Furthermore, in order to obtain an analogue with the 6-OH unprotected, the TBS group of compound 13 was removed using a solution of tetrabutylammonium fluoride (TBAF) in THF to afford the desired thiodisaccharide 14 (Scheme 3).
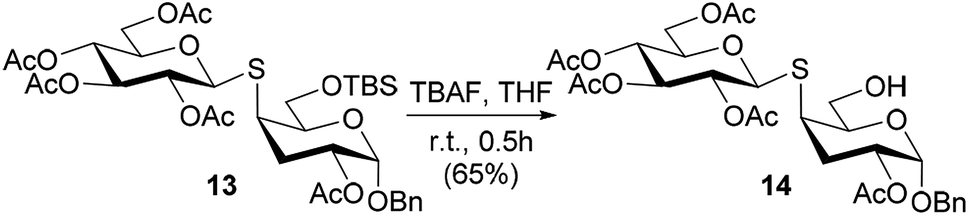 | ||
| Scheme 3 Desilylation reaction of compound 13 to obtain the thiodisaccharide 14. | ||
Having in hand the thiodisaccharides 12–14, with the same configuration of all the stereocenters but different substitution at 6-C (OAc, OTBS and OH), they were subjected to oxidation of the sulfur atom employing m-chloroperoxybenzoic acid (m-CPBA) (Scheme 4). The reaction was conducted in 1:1 CH2Cl2/Et2O at 0 °C for 30 min, in order to prevent the oxidation to the sulfone. Thus, the oxidation of 12 or 13 gave the diastereomeric mixture of sulfoxides 15R,S or 16R,S, respectively. These poor selectivity is in agreement with the results of Crich and coworkers,31 who reported that oxidation of equatorial thioglicosides (β configuration) afforded mixtures of sulfoxides, while the oxidation of axial thioglycosides (α configuration) are usually highly diastereoselective.
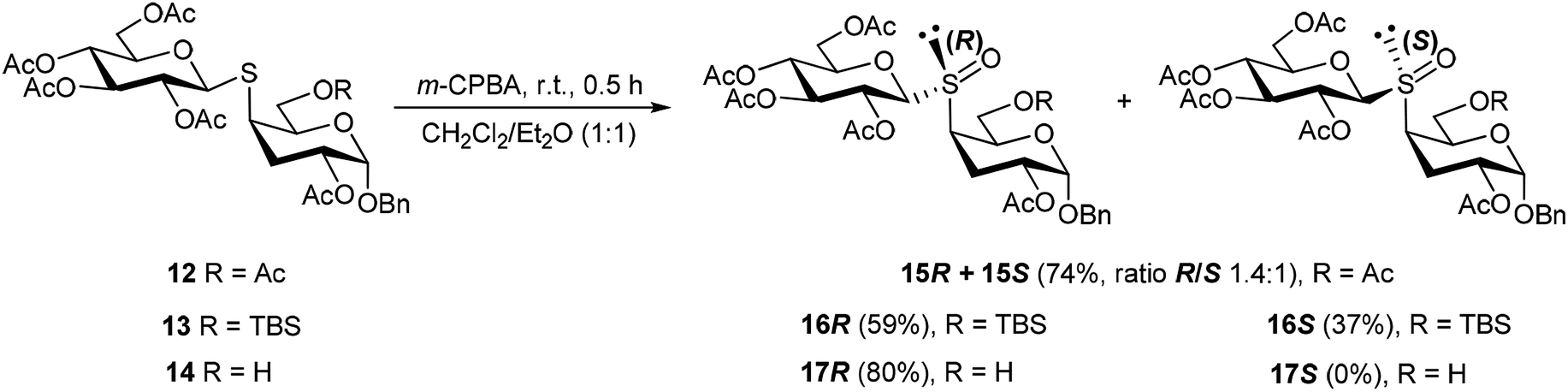 | ||
| Scheme 4 Oxidation reaction of thiodisaccharides 12–14. | ||
All the efforts to separate the diastereomeric mixture 15R,S were unsuccessful. However, isomer 15R could be isolated in pure form by means of the procedure described below. Fortunately, the mixture of sulfoxides 16R and 16S, obtained from 13 in 96% overall yield, could be separated by column chromatography. Interestingly, the oxidation of 14 was highly diastereoselective to afford 17 as a single product in 80% yield. The stereoisomer 15R was obtained through a short synthetic route starting from 16R, which was treated with TBAF for O-desilylation to afford 17R. The NMR spectra of 17R were identical to the product of oxidation of 14. Acetylation of 17R led to 15R.
The absolute configuration at the sulfur stereocenter of each individual sulfoxide was determined following the procedure reported from our laboratory.20,21 For this purpose, it was necessary to establish the predominant conformations adopted for each diastereoisomer, as result of the rotation of the thioglycosidic linkage. These conformations (rotamers) can be described according to the torsion angles ϕ (H-1′–C-1′–S–C-4) and ψ (C-1′–S–C-4–4-H) defined for such a linkage. The presence of a given rotamer may be determined by the experimental detection of characteristic interresidue NOE interactions.16,32–34 Furthermore, the shielding/deshielding effects on the signals of specific protons in the 1H NMR spectrum were analyzed. Such effects are produced by the anisotropy of SO bond (considered to be of acetylenic character and with axial symmetry) and with the shielding cones oriented along the SO linkage.35 Additionally, the shielding effect on hydrogen atoms disposed α anti-axial to the lone pair of electrons of the sulfoxide group must be considered, as well as the deshielding of protons having a syn-axial orientation with respect to the SO bond.35–38
The oxidation of 12 afforded an inseparable mixture of sulfoxides 15R,S (which was later determined to be in a ratio 15R:15S 1.4:1). The 1H and 13C NMR spectra of the mixture were clear enough to assign all the signals of each individual sulfoxide. As explained above, the assignment of the sulfur atom configuration required the evaluation of the conformation of these molecules. The coupling constants values (J) observed for the vicinal protons of 15 indicated, as expected, a strong preference for the 4C1 conformation of both hexopyranose rings. These conformations were confirmed, for each diastereoisomer, by the presence of characteristic intraresidue NOE cross peaks (3ax-H–5-H; 1′-H–3′-H, 1′-H–5′-H, 3′-H–5′-H, and 2′-H–4′-H) in the NOESY spectrum of 15. Key interresidue NOE contacts were also detected, that allowed the assignment of the most populated rotamers around of the thioglycosidic linkage. Thus, the major isomeric sulfoxide (later assigned as 15R) showed an intense interresidue NOE contact between 3eq-H–1′-H, and a barely perceptible cross-peak between 4-H–1′H suggesting the presence of the syn ϕ/syn ψ rotamer. The lack of NOE interactions between 2-H–1′-H and 4-H–2′-H revealed that the respective syn ϕ/anti ψ and anti ϕ/syn ψ rotamers were practically absent. This result is in agreement with the reported preferred conformation adopted for (1→4)-thiodisaccharides.32,33 This rotamer is stabilized by the exo-anomeric effect generated by the sulfur lone-pair disposed anti to the polar C-1′–O-1′ bond. Similarly to 15R, the NOESY spectrum of 15S showed as major rotamer the syn ϕ/syn ψ (cross peak between 4-H–1′-H) and the anti ϕ/syn ψ (weak cross peak 4-H–2′-H) as minor conformer.
On the other hand, the oxidation reaction of thiodisaccharide 13 afforded the sulfoxides 16R (59% yield) and 16S (37%), which were separated by column chromatography. The coupling constants values (J) observed for the vicinal protons of 16R and 16S also indicated, the almost exclusive 4C1 conformation of both hexopyranose rings. The presence of some characteristic intraresidue NOE cross peaks in the NOESY spectra of these sulfoxides also confirmed this observation (1′-H–3′-H, 1′-H–5′-H, 3′-H–5′-H, and 2′-H–4′-H). The predominance of the syn ϕ/syn ψ form for 16R was established on the basis of the NOE interactions between 3eq-H–1′-H. The detection of a very weak interaction 4-H–2′-H suggested low population of the anti ϕ/syn ψ conformer. Similarly, the isomer 16S showed the same preference for the syn ϕ/syn ψ rotamer (strong interaction between 4-H–1′-H) and the barely perceptible cross-peak 4H–2′-H was indicative of a very low population of the anti ϕ/syn ψ form.
Interestingly, oxidation of the thiodisaccharide 14 was highly diastereoselective to give the sulfoxide 17R as the unique isomer, in 80% yield. As for the other sulfoxides already described, 17R showed a preference for the 4C1 conformation for both hexopyranose rings, and the intense cross peak 3eq-H–1′-H in the NOESY spectrum suggested the syn ϕ/syn ψ as the main rotamer.
Finally, the absolute configuration of the sulfur stereocenter of each sulfoxide was established according to the chemical shifts observed in their respective 1H NMR spectra. The chemical shift values for the signals of the more relevant protons are listed in Table 1. As can be seen, the protons 2-H and 3eq-H are more deshielded in 15S than in 15R, while 5-H, 6a-H and 6b-H are more shielded in 15S compared with 15R. These chemical shift differences could be explained considering the anisotropic effects of the SO bond on the given protons, according to their relative special orientation. Thus, as shown in Fig. 1, in the syn ϕ/syn ψ conformation of 15S (S configuration at the sulfur stereocenter) the protons 2-H and 3eq-H are located within the deshielding cone of SO bond; while 5-H, 6a-H and 6b-H are placed near to the sulfur lone pair, lying in a shielding zone. Similar effects operating in the syn ϕ/syn ψ conformation of isomer 15R justify the chemical shifts observed. Thus, the signals of 2-H and 3eq-H appeared upfield in 15R, with respect to those of 15S, as such protons are sited near to sulfur lone pair, in a protection zone. In contrast 5-H, 6a-H and 6b-H are located in the deshielding region of the SO bond.
| Sulfoxide | Chemical shift δ [ppm] | ||||
|---|---|---|---|---|---|
| 2-H | 3eq-H | 5-H | 6a-H | 6b-H | |
| 15R | 4.70 | 2.01 | 4.66 | 4.46 | 4.46 |
| 15S | 5.22 | 2.50 | 4.54 | 4.22 | 4.22 |
| 16R | 4.67 | 2.03 | 4.55 | 4.02 | 3.86 |
| 16S | 5.18 | 2.40 | 4.32 | 3.72 | 3.72 |
| 17R | 4.60 | 2.05 | 4.50 | 3.81 | 3.74 |
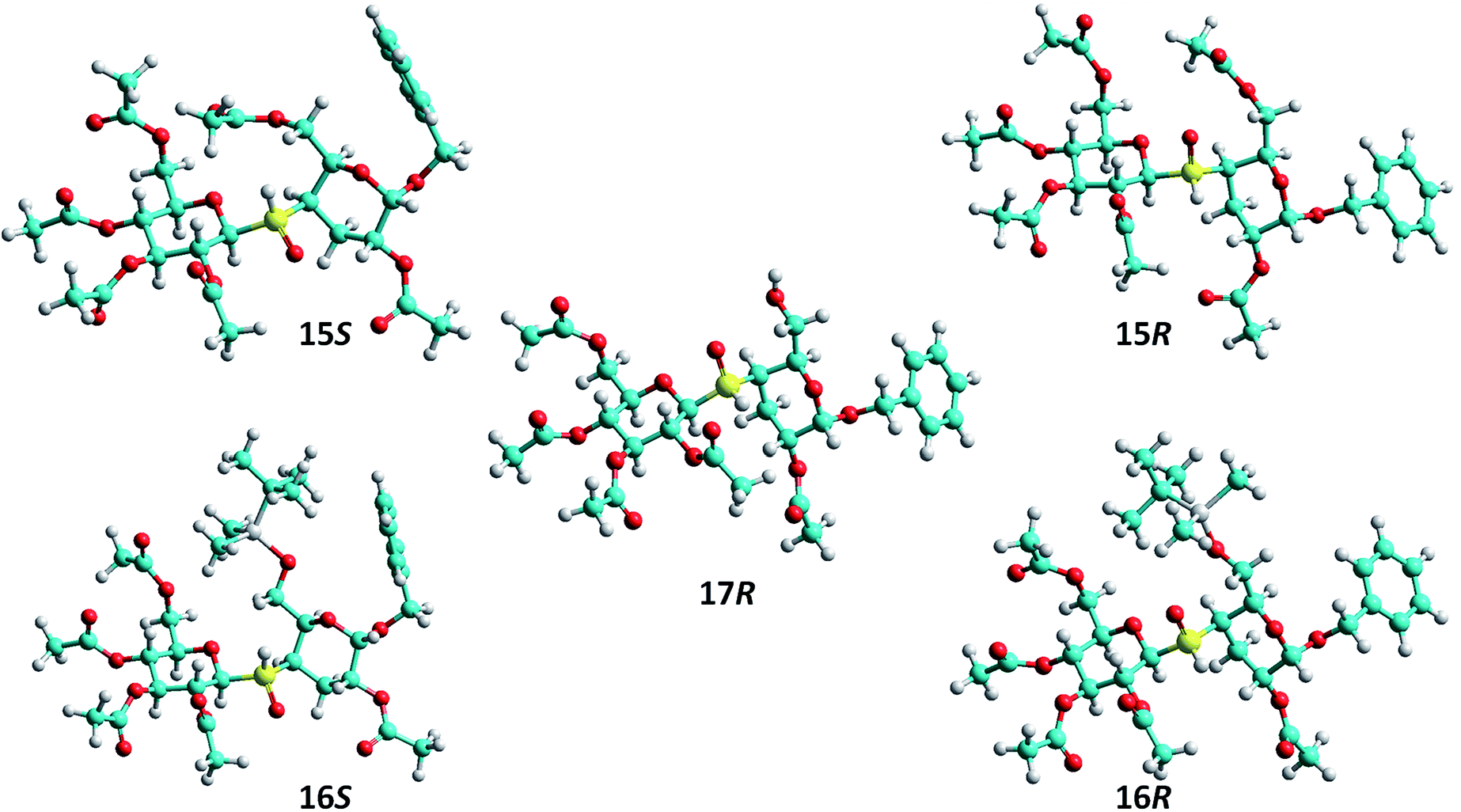 | ||
| Fig. 1 Conformations of thiodisaccharide sulfoxides 15S, 15R, 16S, 16R and 17R in the predominant conformation syn ϕ/syn ψ, based on the NOE interactions detected. | ||
The same analysis allowed us to establish the absolute configuration at the sulfur atom of 16S and 16R. As these isomers are populating the same rotamer conformations as found for 16S and 16R, respectively, the invoked anisotropic effects justify the chemical shifts of the protons located near to the SO group. Thus, the signal of 2-H and 3eq-H are shifted downfield in 16S respect to 16R, while the opposite displacements are observed for the signals of 5-H, 6a-H and 6b-H, which are shifted upfield in 16S respect to 16R. It is worth to mention that our assignments are in agreement with those predicted by the empirical rule which states that the signal of the anomeric carbon (C-1′) of the RS diastereoisomer is shielded upfield with respect to the same signal in the SS counterpart.23,39
The absolute configuration of 17R was assigned, as a first approximation, by comparison of the chemical shift values of the signals of 2-H and 3eq-H of 17R with those of sulfoxides 15S, 15R, 16S and 16R. These two protons, in contrast with 5-H, 6a-H and 6b-H, are located at a major distance of C-6, and therefore are less affected by the electronic properties of different substituents at C-6. Thus, the chemical shift values observed for the signals of 2-H and 3eq-H of 17R (Table 1), and also the δ1-C′, were very similar to those measured for 15R and 16R, suggesting that the absolute configuration of the sulfoxide was also R. It is important to mention that this analysis could be performed, because all the thiodisaccharide sulfoxides present the same preferred syn ϕ/syn ψ conformation. Therefore, the relative orientations and anisotropic effects of the SO bond are expected to be very similar for compounds with the same SO configuration. This assignment was confirmed by conducting the silylation of the OH-6 of 17R. The NMR spectra of the product obtained were identical to those of 15R, indicating that 17R possessed the same absolute R configuration at the sulfur stereocenter.
Next, in order to determine the diastereoselectivity observed in the oxidation reaction of 12–14, the conformations of these thiodisaccharides were assessed. As for the corresponding sulfoxides, the NOESY spectra of thiodisaccharides 12–14 exhibited clear 3eq-H–1′-H and 4-H–1′H interresidue cross peaks, suggesting again the syn ϕ/syn ψ as major rotamer in the conformational equilibrium. The diastereoselectivity in the oxidation of thiodisaccharides 12 and 13 were similar, even when the size of the substituents of C-6, vicinal to the sulfur atom, is different (tert-butyldimethylsilyloxy versus acetoxy). In order to explain this behavior, the spatial arrangement of the TBSO group of 13 was investigated by NOESY spectroscopy. The NOESY spectrum of 13 showed NOE interactions between the methyl and t-butyl protons of TBS with 5-H as well as between the TBS-t-butyl and PhCH2O protons. These cross peaks suggested that the TBS group was located remote to the sulfur reactive center (probably in a gt conformation for the substituted hydromethyl group) and hence the steric hindrance of TBS on sulfur should be negligible. Therefore, the chemical environment of sulfur should be similar in the preferred syn ϕ/syn ψ conformation of thiodisaccharides 12 and 13, giving rise to a similar diastereoselectivity during the oxidation. On the other hand, the NOESY spectrum of 14 showed a NOE interaction between OH and H-4, indicating that the hydroxyl group was disposed in the proximity of the sulfur reactive center. Therefore, as described for the epoxidation reaction of allylic alcohols (Henbest rule),40,41 the hydroxyl group at C-6 can interact by hydrogen bonding with m-CPBA, and then deliver the oxidant to the pro-(R) lone pair of electrons of S, which is the closer electron pair in space in the syn ϕ/syn ψ conformation, with complete diastereoselectivity (Scheme 5).
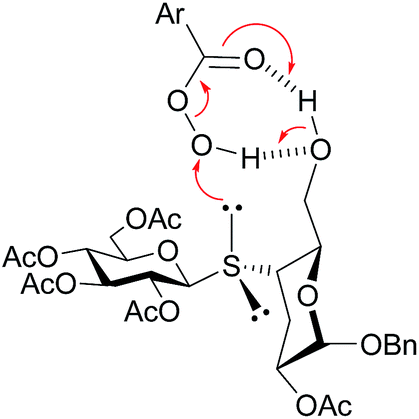 | ||
| Scheme 5 Diastereoselective delivery of oxygen from m-CPBA to the pro-(R) lone pair of electrons of sulfur of 14. | ||
Once stablished the configuration at the sulfur stereocenter of all thiodisaccharides S-oxides, we proceed with the O-deacetylation of the sulfoxide obtained with complete diastereoselectivity 17R. The hydrolysis of the acetyl protecting groups was performed with a mixture of MeOH/Et3N/H2O (4:1:5) to lead the free 3-deoxy-S-(1→4)-disaccharide S-oxide 18R in very good yield (Scheme 6). The evaluation of the free glycosyl sulfoxide as enzyme inhibitor, as well as antitumor agents, is underway.
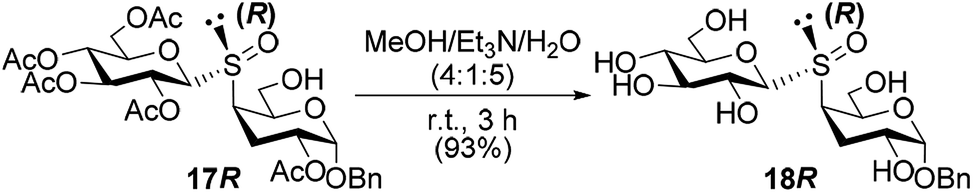 | ||
| Scheme 6 Deprotection of thiodisaccharide S-oxide 17R. | ||
Conclusions
The oxidation reaction of the sulfur atom of benzyl 2-O-acetyl-3-deoxy-4-S-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-4-thio-β-D-xylo-hexopyranosides, with different substituents on C-6 (12–14), was performed employing m-CPBA as oxidant. The absolute configuration at the sulfur stereocenter of each sulfoxide was established taking into account shielding/deshielding anisotropic effects of the SO bond on the chemical shift of the NMR signals of selected protons, in the most populated syn ϕ/syn ψ conformation of the thiodisaccharide S-oxides.
Oxidation of the sulfur atom of thiodisaccharides with the hydroxymethyl group (C-6) substituted with acetyl (12) or TBS (13) afforded the diastereomeric mixtures of sulfoxides 15R,S and 16R,S, in a similar ratio (1.4:1 and 1.6:1, respectively). In contrast, the oxidation of thiodisaccharide 14, with a free hydroxyl group at C-6 led to a single sulfoxide with R configuration (17R). Under the reaction conditions employed, the oxidation of all the thiodisaccharides studied was in favor of the R-isomer. The selectivity observed may be justified taking into account that, in the most populated syn ϕ/syn ψ conformer of the precursors, the pro-(R) lone pair of electrons of sulfur is less hindered that the pro-(S). The comparable selectivity in the oxidation reaction of 12 and 13 suggested a similar environment around the sulfur atom. In fact, the TBS substituent on 13 was located remote to this reactive center (as shown by NOE experiments) and the steric hindrance produced by the bulky group should be negligible. The asymmetric induction in the oxidation of 14 may be attributed to a hydrogen bonding interaction of the OH group with m-CPBA, delivering the oxidant to the pro-(R) lone pair of electrons, which is closer in space in the syn ϕ/syn ψ conformation of the thiodisaccharide. In agreement with this result, an enhanced diastereoselectivity was observed for the oxidation of thioglycosides when the vicinal OH group at C-2 was unprotected.12,39
Experimental section
General methods
Column chromatography was carried out with silica gel 60 (230–400 mesh). Analytical thin-layer chromatography (TLC) was carried out on silica gel 60 F254 aluminium backed plates (layer thickness 0.2 mm). Compounds were visualized by UV absorption at 254 nm, and by charring with a solution of (NH4)6Mo7O24·4H2O 25 g L−1, (NH4)4Ce(SO4)4·2H2O 10 g L−1 and 10% H2SO4 in H2O. Optical rotations were measured at 25 °C in a 1 dm cell in the solvent indicated. Nuclear magnetic resonance (NMR) spectra were recorded at 400 or 500 MHz (1H) and 100.0 or 125.7 MHz (13C). Chemical shifts were referred to tetramethylsilane or to the residual solvent peak (CHCl3: 1H: δ = 7.26 ppm, 13C: δ = 77.2 ppm). Assignments of 1H and 13C NMR spectra were supported by 2D 1H–COSY, NOESY and 2D 1H–13C HSQC and HMBC experiments. High-resolution mass spectra (HRMS) were obtained using the electrospray ionization (ESI) technique and Q-TOF detection.:1), until complete conversion of the starting material (Rf = 0.80) into a more polar product (Rf = 0.52). The mixture was concentrated and the residue dissolved in MeCN (60 mL) and extracted with hexane (4 × 30 mL). The acetonitrile phase was concentrated and the residue was purified by column chromatography using hexane/EtOAc (2:1 → 1:1) as eluent, to give enone 2 (213 mg, 90%) as a light yellow syrup. [α]28D = +65.5 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 7.37–7.30 (m, 5H, PhCH2O), 6.98 (dd, J3,4 = 10.6, J4,5 = 1.6 Hz, 1H, 4-H), 6.20 (dd, J3,4 = 10.6, J3,5 = 2.3 Hz, 1H, 3-H), 5.00 (s, 1H, 1-H), 4.83 (d, J = 11.8 Hz, 1H, PhCH2O), 4.74 (d, J = 11.8 Hz, 1H, PhCH2O), 4.61 (m, 1H, 5-H), 3.85 (br. d, J6a,6b = 11.3 Hz, 1H, 6a-H), 3.77 (dd, J6a,6b = 10.9, J5,6b = 4.4 Hz, 1H, 6b-H), 1.90 (br. s, 1H, OH) ppm. 13C NMR (100 MHz, CDCl3): δ = 188.6 (C-2), 148.2 (C-4), 136.8, 128.7, 128.3 (×2) (C-aromatic), 126.6 (C-3), 97.3 (C-1), 71.3 (PhCH2O), 69.7 (C-5), 64.4 (C-6) ppm. HRMS (ESI): calcd for C13H14NaO4 257.0784 [M + Na]+; found 257.0793.:1) showed until complete conversion of the starting material (Rf = 0.28) into a faster moving compound (Rf = 0.82). The reaction mixture was diluted with CH2Cl2 (50 mL) and washed with water (3 × 40 mL). The organic layer was dried (MgSO4) and filtered. The evaporation of the solvent gave an oily residue, which was purified by column chromatography employing hexane/EtOAc (97:3 → 90:10). The enone 3 was isolated as a syrup (592 mg, 65%). [α]28D = +11.5 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ = 7.37–7.30 (m, 5H, PhCH2O), 7.11 (dd, J3,4 = 10.6, J4,5 = 1.6 Hz, 1H, 4-H), 6.16 (dd, J3,4 = 10.6, J3,5 = 2.4 Hz, 1H, 3-H), 4.95 (s, 1H, 1-H), 4.83 (d, J = 11.7, 1H, PhCH2O), 4.72 (d, J = 11.7, 1H, PhCH2O), 4.53 (m, 1H, 5-H), 3.86 (dd, J6a,6b = 10.2, J5,6a = 5.6 Hz, 1H, 6a-H), 3.67 (dd, J6a,6b = 10.0, J5,6b = 7.0 Hz, 1H, 6b-H), 0.91 (s, 9H, Si(CH3)2C(CH3)3), 0.10, 0.09 (2 s, 6H, Si(CH3)2C(CH3)3) ppm. 13C NMR (125.7 MHz, CDCl3): δ = 189.0 (C-2), 149.5 (C-4), 136.9, 128.6, 128.2 (×2) (C-aromatic), 125.4 (C-3), 97.1 (C-1), 71.0 (OCH2Ph), 69.6 (C-5), 64.6 (C-6), 25.9 (Si(CH3)2C(CH3)3), 18.4 (Si(CH3)2C(CH3)3), −5.2 (×2) (Si(CH3)2C(CH3)3)) ppm. HRMS (ESI): calcd for C19H28NaO4Si 371.1649 [M + Na]+; found 371.1663.General procedure for the conjugate addition and NaBH4 reduction reactions
The corresponding 2H-pyran-3(6H)-one (0.50 mmol) and thioaldose 4 (182 mg, 0.50 mmol) were dissolved in anhydrous CH2Cl2 (1.0 mL) and cooled to −18 °C. A solution of NEt3 (3.8 μL) in CH2Cl2 (0.2 mL) was added, and the mixture was stirred at −18 °C for 1.5 h, when TLC showed complete consumption of the starting materials. The mixture was concentrated and the residue was dissolved in dry MeOH (2.7 mL) and treated with sodium borohydride (21 mg, 0.56 mmol) at 0 °C for 30 min. Then, the mixture was stirred in batch with Dowex 50W (H+) resin, filtered, and concentrated. Column chromatography of the residue with pentane/EtOAc (4:1 → 1:1) afforded the desired thiodisaccharides.
Michael addition of thioaldose 4 to enone 1
:1) which were identified as a mixture of non-reduced 2-keto thiodisaccharide 5 and thiodisaccharide 7 (56 mg, 17%, ratio 5:7, 0.7:1), 1H NMR (400 MHz, CDCl3) data for 5: δ = 7.37–7.30 (m, 5H, PhCH2O overlapping with PhCH2O of 7), 5.19 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.03 (t, J3′,4′ = J4′,5′ = 9.7 Hz, 1H, 4′-H), 4.95 (dd, J1′,2′ = 10.0, J2′,3′ = 9.4 Hz, 2′-H), 4.78 (s, 1H, 1-H), 4.77 (d, J = 11.7 Hz, 1H, PhCH2O), 4.62 (d, J = 12.5 Hz, 1H, PhCH2O), 4.61 (d, J1′,2′ = 9.9 Hz, 1H, 1′-H), 4.52 (dd, J6a,6b = 11.7, J5,6a = 1.4 Hz, 1H, 6a-H), 4.34 (dd, J6a,6b = 12.0, J5,6b = 5.3 Hz, 1H, 6b-H), 4.29–4.19 (m, 1H, 5-H overlapping with 6a-H, 6b-H, 6′a-H of 7), 4.14 (m, 2H, 6′a-H, 6′b-H overlapping with 6′b-H of 7), 3.63 (ddd, J4′,5′ = 9.9, J = 5.2, J = 2.3 Hz, 1H, 5′-H), 3.32 (m, 1H, 4-H overlapping with 4-H of 7), 2.91 (dd, J3a,3b = 15.0, J2,3a = 12.0 Hz, 1H, 3a-H), 2.85 (dd, J3a,3b = 15.0, J2,3b = 6.4 Hz, 1H, 3b-H), 2.11–1.99 (5s, 15H, COCH3 overlapping with COCH3 of 7) ppm. 13C NMR (CDCl3, 100.0 MHz) data for 5: δ = 198.8 (C-2), 170.7–169.4 (COCH3 × 5 overlapping with COCH3 of 7), 136.3, 128.8, 128.3, 128.1 (C-aromatic), 98.2 (C-1), 83.0 (C-1′), 76.0 (C-5′), 73.7 (C-3′), 70.3 (C-5), 70.2 (C-2′), 70.1 (PhCH2O), 68.2 (C-4′), 63.7 (C-6), 62.1 (C-6′), 43.4 (C-3), 42.6 (C-4), 20.9–20.7 (COCH3 × 5 overlapping with COCH3 of 7) ppm. HRMS (ESI) data for 5: calcd for C29H36NaO14S 663.1718 [M + Na]+; found 663.1720.1H NMR (400 MHz, CDCl3) data for disaccharide 7: δ = 7.37–7.30 (m, 5H, PhCH2O overlapping with PhCH2O of 5), 5.23 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.09 (t, J3′,4′ = J4′,5′ = 9.1 Hz, 1H, 4′-H), 5.04 (t, J1′,2′ = J2′,3′ = 9.1 Hz, 1H, 2′-H), 4.85 (br. s, 1H, 1-H), 4.73 (d, J = 11.6 Hz, 1H, PhCH2O), 4.67 (d, J1′,2′ = 10.1 Hz, 1H, 1′-H), 4.50 (d, J = 11.6 Hz, 1H, PhCH2O), 4.42 (m, 1H, 5-H), 4.29–4.19 (m, 3H, 6a-H, 6b-H, 6′a-H, overlapping with 5-H of 5), 4.10 (m, 1H, 6′b-H overlapping with 6′a-H and 6′b-H of 5), 3.70 (m, 2H, 2-H, 5′-H), 3.50 (d, JOH,2 = 9.9 Hz, 1H, OH) 3.32 (br. s, 1H, 4-H overlapping with 4-H of 5), 2.29 (m, 2H, 3a-H, 3b-H), 2.11–1.99 (5s, 15H, COCH3 overlapping with COCH3 of 5) ppm. 13C NMR (CDCl3, 100.0 MHz) data for disaccharide 7: δ = 170.7–169.4 (COCH3 × 5 overlapping with COCH3 of 5), 137.1, 128.7, 128.5, 128.3 (C-aromatic), 99.9 (C-1), 82.2 (C-1′), 76.3 (C-5′), 73.9 (C-3′), 69.9 (C-2′), 69.2 (PhCH2O), 68.5 (C-5), 68.2 (C-4′), 67.1 (C-2), 65.3 (C-6), 62.0 (C-6′), 39.1 (C-4), 30.9 (C-3), 20.9–20.7 (COCH3 × 5 overlapping with COCH3 of 7) ppm. HRMS (ESI) data for 7: calcd for C29H38NaO14S 665.1874 [M + Na]+; found 665.1867.:1), which was identified as thiodisaccharide 6 (135 mg, 37%). 1H NMR (400 MHz, CDCl3): δ = 7.38–7.30 (m, 5H, PhCH2O), 5.20 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.07 (t, J3′,4′ = J4′,5′ = 9.7 Hz, 1H, 4′-H), 5.00 (t, J1′,2′ = J2′,3′ = 9.6 Hz, 1H, 2′-H), 4.91 (d, J1,2 = 3.5 Hz, 1H, 1-H), 4.76 (d, J = 11.5 Hz, 1H, PhCH2O), 4.58 (d, J1′,2′ = 10.1 Hz, 1H, 1′-H), 4.51 (d, J = 11.5 Hz, 1H, PhCH2O), 4.27 (m, 1H, 5 H), 4.23–4.11 (m, 4H, 6a-H, 6b-H, 6′a-H, 6′b-H), 4.06 (m, 1H, 2-H), 3.66 (ddd, J4′,5′ = 9.9, J5′,6′a = 4.6, J5′,6′b = 2.4 Hz, 1H, 5′-H), 3.32 (br. s, 1H, 4-H), 2.19 (dt, J3a,3b = 12.6, J2,3a = J3a,4 = 3.7 Hz, 1H, 3a-H), 2.09 (m, 1H, 3b-H), 2.07, 2.05 (×2), 2.01, 1.99 (5s, 15H, CH3CO) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.8, 170.7, 170.2, 169.5 (×2) (COCH3), 137.0, 128.7, 128.3 (×2) (C-aromatic), 97.2 (C-1), 83.1 (C-1′), 76.1 (C-5′), 74.0 (C-3′), 70.1 (C-2′), 69.5 (PhCH2O), 68.4 (×2, C-5, C-4′), 65.5 (C-6), 64.4 (C-2), 62.1 (C-6′), 43.2 (C-4), 35.0 (C-3), 20.9, 20.8, 20.7 (×3) (COCH3) ppm. HRMS (ESI): calcd for C29H38NaO14S 665.1874 [M + Na]+; found 665.1875.:1) and identified as thiodisaccharide 8 (66 mg, 18%) and was obtained as a white solid, mp at 113.6 °C (dec.).[α]25D = +30.4 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 7.38–7.30 (m, 5H, PhCH2O), 5.18 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.03 (t, J3′,4′ = J4′,5′ = 9.8 Hz, 1H, 4′-H), 4.95 (t, J1′,2′ = J2′,3′ = 9.5 Hz, 1H, 2′-H), 4.93 (d, J1,2 = 1.8 Hz, 1H, 1-H), 4.75 (d, J = 11.7 Hz, 1H, PhCH2O), 4.58 (d, J1′,2′ = 10.3 Hz, 1H, 1′-H), 4.55 (d, J = 11.8 Hz, 1H, PhCH2O), 4.45 (dd, J6a,6b = 11.9, J5,6a = 1.3 Hz, 1H, 6a-H), 4.28 (dd, J6a,6b = 12.0, J5,6b = 5.4 Hz, 1H, 6b-H), 4.16 (dd, J6′a,6′b = 12.1, J5′,6′a = 5.1 Hz, 1H, 6′a-H), 4.05 (dd, J6′a,6′b = 12.4, J5′,6′b = 1.9 Hz, 1H, 6′b-H), 3.80 (ddd, J4,5 = 11.0, J5,6b = 5.4, J5,6a = 1.4 Hz, 1H, 5-H), 3.75 (m, 1H, 2-H), 3.59 (ddd, J4′,5′ = 9.5, J5′,6′a = 4.7, J5′,6′b 2.0 Hz, 1H, 5′-H), 2.90 (td, J3b,4 = J4,5 = 12.8, J3a,4 = 3.9 Hz, 1H, 4-H), 2.26 (dt, J3a,3b = 11.8, J2,3a = J3a,4 = 4.3 Hz, 1H, 3a-H), 2.10, 2.07, 2.03, 2.01, 1.99 (5s, 15H, CH3CO), 1.88 (q, J2,3b = J3a,3b = J3b,4 = 12.3 Hz, 1H, 3b-H) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.7, 170.6, 170.2, 169.4 (×2) (COCH3), 137.1, 128.7, 128.3, 128.2 (C-aromatic), 97.1 (C-1), 82.8 (C-1′), 75.9 (C-5′), 73.8 (C-3′), 70.2 (C-5), 70.1 (C-2′), 69.7 (PhCH2O), 68.2 (C-4′), 67.8 (C-2), 64.2 (C-6), 62.1 (C-6′), 39.8 (C-4), 35.9 (C-3), 21.0, 20.8, 20.7 (×3) (COCH3) ppm. HRMS (ESI): calcd for C29H38NaO14S 665.1874 [M + Na]+; found 665.1872.Michael addition of thioaldose 4 to enone 3
:1). They were identified as 10 and 11 (235 mg, 44%, ratio 10/11 3.3:1.0). 1H NMR (400 MHz, CDCl3) data for disaccharide 10: δ = 7.37–7.28 (m, 5H, PhCH2O overlapping with PhCH2O of 11), 5.23 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.09 (t, J3′,4′ = J4′,5′ = 9.8 Hz, 1H, 4′-H), 5.03 (t, J1′,2′ = J2′,3′ = 9.8 Hz, 1H, 2′-H overlapping with 4′-H of 11), 4.85 (br. s, 1H, 1-H), 4.76 (d, J = 11.7 Hz, 1H, PhCH2O), 4.71 (d, J1′,2′ = 10.0 Hz, 1H, 1′-H), 4.51 (d, J = 11.7 Hz, 1H, PhCH2O), 4.26 (m, 1H, 5-H), 4.23 (dd, J6′a,6′b = 12.5, J5′,6′a = 4.8 Hz, 1H, 6′a-H), 4.09 (dd, J6′a,6′b = 12.4, J5′,6′b = 2.2 Hz, 1H, 6′b-H), 3.76–3.64 (m, 4H, 6a-H, 6b-H, 5′-H, 2-H overlapping with 2-H of 11), 3.36 (br. d, J = 2.5 Hz, 1H, 4-H), 2.26 (m, 2H, 3a-H, 3b-H overlapping with 3a-H of 11), 2.05–1.98 (4 s, 12H, CH3CO overlapping with CH3CO of 11), 0.88 (s, 9H, Si(CH3)2C(CH3)3), 0.08, 0.07 (2 s, 6H, Si(CH3)2C(CH3)3 overlapping with Si(CH3)2C(CH3)3 of 11) ppm. 13C NMR (CDCl3, 100.0 MHz) data for isomer 10: δ = 170.6, 170.2, 169.5, 169.4 (CH3CO), 137.4, 128.6, 128.3, 128.0 (C-aromatic), 100.0 (C-1), 81.6 (C-1′), 76.1 (C-5′), 74.0 (C-3′ overlapping with C-3′ of 11), 71.1 (C-5), 70.2 (C-2′), 69.0 (PhCH2O), 68.3 (C-4′), 67.5 (C-2), 64.3 (C-6), 62.1 (C6′), 38.8 (C-4), 30.2 (C-3), 26.0 (Si(CH3)2C(CH3)3), 20.8–20.6 (CH3CO), 18.3 (Si(CH3)2C(CH3)3), −5.2, −5.3 (Si(CH3)2C(CH3)3) ppm. 1H NMR (400 MHz, CDCl3) data for disaccharide 11: δ = 7.37–7.28 (m, 5H, PhCH2O overlapping with PhCH2O of 10), 5.15 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.03 (t, J3′,4′ = J4′,5′ = 9.6 Hz, 1H, 4′-H overlapping with 2′-H of 10), 4.93 (dd, J1′,2′ = 10.1, J2′,3′ = 9.4 Hz, 2′-H), 4.89 (d, J1,2 = 3.5 Hz, 1H, 1-H), 4.76 (d, J1′,2′ = 10.2 Hz, 1H, 1′-H), 4.73 (d, J = 9.9 Hz, 1H, PhCH2O), 4.54 (d, J = 9.9 Hz, 1H, PhCH2O), 4.18 (dd, J6′a,6′b = 12.4, J5′,6′a = 5.1 Hz, 1H, 6′a-H), 4.05 (dd, J6′a,6′b = 12.5, J5′,6′b = 2.1 Hz, 1H, 6′b-H), 3.93 (dd, J6a,6b = 11.5, J5,6a = 3.7 Hz, 1H, 6a-H), 3.79 (dd, J6a,6b = 11.3, J5,6a = 0.8 Hz, 1H, 6b-H), 3.76–3.64 (m, 1H, 2-H, overlapping with 6a-H, 6b-H, 5′-H, 2 H of 10), 3.58 (ddd, J4′,5′ = 10.0, J5′,6′a = 5.1, J5′,6′b = 2.2 Hz, 1H, 5′-H), 3.52 (br. dd, J4,5 = 10.8, J5,6b = 1.9 Hz, 1H, 5-H), 3.01 (ddd, J3b,4 = 13.5, J4,5 = 11.1, J3a,4 = 4.2 Hz, 1H, 4-H), 2.26 (m, 1H, 3a-H overlapping with 3a-H and 3b-H of 10), 2.05–1.98 (4 s, 12H, CH3CO overlapping with CH3CO of 10), 1.83 (q, J2,3b = J3a,3b = J3b,4 = 12.4 Hz, 1H, 3b-H), 0.91 (s, 9H, Si(CH3)2C(CH3)3), 0.09, 0.07 (2s, 6H, Si(CH3)2C(CH3)3 overlapping with Si(CH3)2C(CH3)3 of 10) ppm. 13C NMR (CDCl3, 100.0 MHz) data for isomer 11: δ = 170.7, 170.2, 169.5, 169.3 (CH3CO), 137.5, 128.6, 128.1, 128.0 (C-aromatic), 97.2 (C-1), 83.3 (C-1′), 75.8 (C-5′), 74.0 (C-3′ overlapping with C-3′ of 10), 73.2 (C-5), 70.4 (C-2′), 69.4 (PhCH2O), 68.3 (C-4′), 68.1 (C-2), 62.8 (C-6), 62.2 (C6′), 39.1 (C-4), 35.7 (C-3), 26.1 (Si(CH3)2C(CH3)3), 20.8–20.6 (CH3CO), 18.5 (Si(CH3)2C(CH3)3), −4.9, −5.1 (Si(CH3)2C(CH3)3) ppm. HRMS (ESI): calcd for C33H50NaO13SSi 737.2634 [M + Na]+; found 737.2661.:1), which was obtained as a colourless syrupy and identified as thiodisaccharide 9 (168 mg, 31%). [α]20D = +16.8 (c = 1.1, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 7.36–7.28 (m, 5H, PhCH2O), 5.19 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.07 (t, J3′,4′ = J4′,5′ = 9.7 Hz, 1H, 4′-H), 5.00 (t, J1′,2′ = J2′,3′ = 9.6 Hz, 1H, 2′-H), 4.89 (d, J1,2 = 3.7 Hz, 1H, 1-H), 4.79 (d, J = 11.6 Hz, 1H, PhCH2O), 4.58 (d, J1′,2′ = 10.1 Hz, 1H, 1′-H), 4.51 (d, J = 11.5 Hz, 1H, PhCH2O), 4.21 (dd, J6′a,6′b = 12.4, J5′,6′a = 4.7 Hz, 1H, 6′a-H), 4.11–4.06 (m, 3H, 6′b-H, 5-H, 2-H), 3.67 (m, 2H, 6a-H, 6b-H), 3.64 (ddd, J4′,5′ = 9.7, J5′,6′a = 4.6, J5′,6′b = 2.4 Hz, 1H, 5′-H), 3.34 (br. d, J = 2.0 Hz, 1H, 4-H), 2.15 (dt, J3a,3b = 11.7, J2,3a = J3a,4 = 3.3 Hz, 1H, 3a-H), 2.05, 2.03, 2.00, 1.98 (4s, 13H, CH3CO overlapping with 3b-H), 0.88 (s, 9H, Si(CH3)2C(CH3)3), 0.07, 0.06 (2s, 6H, Si(CH3)2C(CH3)3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.7, 170.2, 169.5, 169.4 (COCH3), 137.3, 128.6, 128.3, 128.1 (C-aromatic), 97.1 (C-1), 82.7 (C-1′), 76.0 (C-5′), 74.0 (C-3′), 71.3 (C-5), 70.4 (C-2′), 69.1 (PhCH2O), 68.3 (C-4′), 64.6 (C-2), 64.4 (C-6), 62.1 (C-6′), 42.8 (C-4), 34.5 (C-3), 26.0 (Si(CH3)2C(CH3)3), 20.8, 20.7, 20.6 (×2) (COCH3), 18.3 (Si(CH3)2C(CH3)3), −5.2 (×2) (Si(CH3)2C(CH3)3) ppm. HRMS (ESI): calcd for C33H50NaO13SSi 737.2634 [M + Na]+; found 737.2623.General procedure for the O-acetylation of thiodisaccharides
To a solution of thiodisaccaride (0.16 mmol) in pyridine (1 mL) was added dropwise Ac2O (1 mL). The reaction was stirred at room temperature for 3 h. Then, the mixture was cooled in an ice/water bath and MeOH was added dropwise (1 mL) and stirred for 30 min. The evaporation of the solvent followed by co-evaporation with toluene (3 × 1 mL) afforded the desired pure product.:1). [α]25D = +28.5 (c = 1.2, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 7.36–7.29 (m, 5H, PhCH2O), 5.21 (m, 2H, 2-H, 3′-H), 5.08 (t, J3′,4′ = J4′,5′ 9.7 Hz, 1H, 4′-H), 5.01 (t, J1′,2′ = J2′,3′ = 9.6 Hz, 1H, 2′-H), 5.01 (d, J1,2 = 3.2 Hz, 1H, 1-H), 4.74 (d, J = 12.0 Hz, 1H, PhCH2O), 4.61 (d, J1′,2′ = 10.0 Hz, 1H, 1′-H), 4.52 (d, J = 12.0 Hz, 1H, PhCH2O), 4.36 (m, 1H, 5-H), 4.21 (dd, J6′a,6′b = 12.5, J5′,6′a = 4.6 Hz, 1H, 6′a-H), 4.16 (m, 2H, 6a-H, 6b-H), 4.13 (dd, J6′a,6′b = 12.5, J5′,6′b = 2.3 Hz, 1H, 6′b-H), 3.67 (ddd, J4′,5′ = 9.9, J5′,6′a = 4.4, J5′,6′b = 2.4 Hz, 1H, 5′-H), 3.39 (br. d, J = 2.1 Hz, 1H, 4-H), 2.34 (td, J2,3a = J3a,3b = 12.6, J3a,4 = 3.6 Hz, 1H, 3a-H), 2.14 (m, 1H, 3b-H), 2.07, 2.06 (×2), 2.03, 2.01, 1.99 (COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.7, 170.6, 170.2 (×2), 169.7, 169.4 (COCH3), 137.3, 128.6, 128.1, 128.0 (C-aromatic), 94.9 (C-1), 82.8 (C-1′), 76.1 (C-5′), 73.9 (C-3′), 70.0 (C-2′), 69.2 (PhCH2O), 68.3 (×2, C-4′, C-5), 66.9 (C-2), 65.4 (C-6), 62.0 (C-6′), 42.9 (C-4), 30.9 (C-3), 21.1, 20.9, 20.8, 20.7 (×3) (COCH3). HRMS (ESI): calcd for C31H40NaO15S 707.1980 [M + Na]+; found 707.1959.:1). [α]20D = +21.5 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 7.34–7.28 (m, 5H, PhCH2O), 5.24 (m, 1H, 2-H), 5.21 (t, J2′,3′ = J3′,4′ = 9.4 Hz, 1H, 3′-H), 5.08 (t, J3′,4′ = J4′,5′ = 9.7 Hz, 1H, 4′-H), 5.01 (t, J1′,2′ = J2′,3′ = 9.9 Hz, 1H, 2′-H), 4.98 (d, J1,2 = 3.3 Hz, 1H, 1-H), 4.77 (d, J = 12.1 Hz, 1H, PhCH2O), 4.60 (d, J1′,2′ = 10.1 Hz, 1H, 1′-H), 4.53 (d, J = 12.1 Hz, 1H, PhCH2O), 4.22 (dd, J6′a,6′b = 12.3, J5′,6′a = 4.7 Hz, 1H, 6′a-H), 4.20 (m, 1H, 5-H), 4.09 (dd, J6′a,6′b = 12.3, J5′,6′b = 2.2 Hz, 1H, 6′b-H), 3.66 (m, 3H, 5′-H, 6a-H, 6b-H), 3.41 (br. d, J = 2.1 Hz, 1H, 4-H), 2.31 (td, J2,3a = J3a,3b = 12.3, J3,4 = 3.3 Hz, 1H, 3a-H), 2.12 (m, 1H, 3b-H), 2.06, 2.05, 2.04, 2.01, 1.99 (COCH3), 0.89 (s, 9H, Si(CH3)2C(CH3)3), 0.08, 0.07 (2s, 6H, Si(CH3)2C(CH3)3). 13C NMR (100 MHz, CDCl3): δ = 170.6, 170.3, 170.2, 169.9, 169.5 (COCH3), 137.5, 128.5, 128.0, 127.9 (C-aromatic), 94.7 (C-1), 82.4 (C-1′), 76.0 (C-5′), 74.0 (C-3′), 71.2 (C-5), 70.2 (C-2′), 68.9 (OCH2Ph), 68.4 (C-4′), 67.2 (C-2), 64.4 (C-6), 62.1 (C-6′), 42.5 (C-4), 30.3 (C-3), 26.0 (Si(CH3)2C(CH3)3), 21.1, 20.8, 20.7 (×3) (COCH3), 18.4 (Si(CH3)2C(CH3)3), −5.2 (×2, Si(CH3)2C(CH3)3) ppm. HRMS (ESI): calcd for C35H52NaO14SSi 779.2739 [M + Na]+; found 779.2721.Desilylation reaction of thiodisaccharide 13
:1 → 1:1) to give the thiodisaccharide 14 (153 mg, 65%) as a more polar product (Rf = 0.29, hexane/EtOAc, 1:1), and a white solid, mp at 146 °C (dec.) [α]28D = +32.9 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ = 7.35–7.27 (m, 5H, PhCH2O), 5.21 (t, J2′,3′ = J3′,4′ = 9.4 Hz, 1H, 3′-H), 5.10 (ddd, J2,3a = 12.0, J2,3b = 4.6, J1,2 = 3.8 Hz, 1H, 2-H), 5.04 (dd, J4′,5′ = 10.0, J3′,4′ = 9.6 Hz, 1H, 4′-H), 5.00 (dd, J1′,2′ = 10.0, J2′,3′ = 9.4 Hz, 1H, 2′-H), 4.96 (br. d, J1,2 = 3.6 Hz, 1H, 1-H), 4.72 (d, J = 12.2 Hz, 1H, PhCH2O), 4.58 (d, J1′,2′ = 10.1 Hz, 1H, 1′-H), 4.54 (d, J = 12.2 Hz, 1H, PhCH2O), 4.18 (m, 1H, 5-H), 4.14 (m, 2H, 6′a-H, 6′b-H), 3.71 (dd, J6a,6b = 11.6, J5,6a = 7.7 Hz, 1H, 6a-H), 3.70 (m, 1H, 5′-H), 3.56 (dd, J6a,6b = 11.6, J5,6b = 6.2 Hz, 1H, 6b-H), 3.39 (br d, J = 2.5 Hz, 1H, 4-H), 2.36 (td, J2,3a = J3a,3b = 12.4, J3a,4 = 3.6 Hz, 1H, 3a-H), 2.17 (br. td, J3a,3b = 12.8, J2,3b = J2,3b = 3.8 Hz, 1H, 3b-H), 2.08, 2.07, 2.03, 2.01, 1.99 (COCH3) ppm. 13C NMR (125.7 MHz, CDCl3): δ = 170.7, 170.2 (×2), 169.9, 169.4 (COCH3), 137.4, 128.5, 128.0 (×2) (C-aromatic), 95.2 (C-1), 83.8 (C-1′), 76.1 (C-5′), 73.7 (C-3′), 70.2 (C-2′), 69.7 (C-5), 69.6 (PhCH2O), 68.3 (C-4′), 67.5 (C-2), 62.3 (C-6), 62.1 (C-6′), 43.6 (C-4), 31.3 (C-3), 21.1, 20.8 (×2), 20.7 (×2) (COCH3). HRMS (ESI): calcd for C29H38NaO14S 665.1875 [M + Na]+; found 665.1885.General procedure for the oxidation reaction of S-(1→4)-thiodisaccharides (12–14) to sulfoxides (15–17)
A solution of mCPBA (77%, 32 mg, 0.141 mmol) in 2 mL of dry CH2Cl2 was added dropwise to another solution of the thiodisaccharide (0.072 mmol) in 2 mL of ethyl ether at 0 °C. The reaction was allowed to reach room temperature and after 30 min of stirring was diluted with EtOAc (30 mL). To the mixture was added a solution of NaHSO3 (satd. aq.; 10 mL) and stirred for 30 min. The organic phase was separated and stirred with NaHCO3 (satd. aq.; 10 mL) for 30 min. The organic extract was washed with NaHCO3 (satd. aq.; 10 mL) and brine (10 mL), dried (MgSO4), and filtered. The evaporation of the solvent, followed by co-evaporation with 1:1 toluene/EtOH (5 × 10 mL) afforded an oily residue that was purified by column chromatography with pentane/EtOAc (3:2 → 1:1) or CH2Cl2/EtOAc (8:1 → 4:1), to give the diasteromeric mixture of sulfoxides, which in some cases could be separated.
Oxidation reaction of thiodisaccharide 12 to sulfoxides 15R and 15S
:1) showed a single spot by TLC (Rf = 0.29, pentane/EtOAc, 1:1). The isomer 15R was obtained pure by the procedure described below; but the spectral data described for 15S were obtained from the mixture 15R,S. 1H NMR (400 MHz, CDCl3) data for S isomer: δ = 7.39–7.30 (m, 5H, PhCH2O overlapping with PhCH2O of 15R), 5.43 (d, J1′,2′ = J2′,3′ = 9.4 Hz, 1H, 2′-H), 5.28 (t, J2′,3′ = J3′,4′ = 9.2 Hz, 1H, 3′-H), 5.22 (m, 1H, 2-H), 5.20 (d, J1,2 = 1.6 Hz, 1H, 1-H), 5.09 (t, J3′,4′ = J4′,5′ = 9.7 Hz, 1H, 4′-H), 4.74 (d, J = 11.9 Hz, 1H, PhCH2O), 4.55–4.53 (m, 2H, PhCH2O, 5-H overlapping with 1′-H of 15R), 4.32 (d, J1′,2′ = 9.7 Hz, 1H, 1′-H), 4.26–4.16 (m, 4H, 6a-H, 6b-H, 6′a-H, 6′b-H overlapping with 6′a-H, 6′b-H of 15R), 3.79 (m, 1H, 5′-H overlapping with 5′-H of 15R), 3.45 (m, 1H, 4-H), 2.50 (ddd, J3a,3b = 13.3, J2,3a = 5.7, J3a,4 = 2.6 Hz, 1H, 3a-H), 2.28 (m, 1H, 3b-H overlapping with 3a-H of 15R), 2.09–2.01 (6 s, 18H, COCH3 overlapping with COCH3 and 3b-H of 15R) ppm. 13C NMR (CDCl3, 100.0 MHz) data for S isomer: δ = 170.7–168.8 (CH3CO), 137.2, 128.6, 128.1, 128.0 (C-aromatic), 95.1 (C-1), 90.4 (C-1′), 77.0 (C-5′), 73.4 (C-3′), 69.9 (OCH2Ph), 68.6 (C-2′), 67.7 (C-4′), 66.5 (C-2), 66.2 (C-5), 63.7 (C-6), 61.6 (C-6′), 55.0 (C-4), 22.9 (C-3), 21.1–20.6 (COCH3) ppm. HRMS (ESI): calcd for C31H41O16S 701.2110 [M + H]+; found 701.2085.:1, 0.4 mL) at room temperature for 2 h led, after column chromatography, to syrupy 15R (18 mg, 61% from 16R). 1H NMR (400 MHz, CDCl3): δ = 7.39–7.30 (m, 5H, PhCH2O), 5.47 (t, J1′,2′ = J2′,3′ = 9.5 Hz, 1H, 2′-H), 5.40 (t, J2′,3′ = J3′,4′ = 9.2 Hz, 1H, 3′-H), 5.12 (t, J4′,5′ = J3′,4′ = 9.7 Hz, 1H, 4′-H), 5.08 (d, J1,2 = 3.3 Hz, 1H, 1-H), 4.82 (d, J = 11.8 Hz, 1H, PhCH2O), 4.70 (dt, J2,3a = 8.8, J1,2 = J2,3b = 3.9 Hz, 1H, 2-H), 4.66 (m, 1H, 5-H), 4.58 (d, J = 11.7 Hz, 1H, PhCH2O), 4.56 (d, J1′,2′ = 9.6 Hz, 1H, 1′-H), 4.46 (m, 2H, 6a-H, 6b-H), 4.23 (dd, J6′a,6′b = 12.4, J6′a,5′ = 2.3 Hz, 1H, 6′a-H), 4.17 (dd, J6′a,6′b = 12.5, J6′b,5′ = 5.6 Hz, 1H, 6′b-H) 3.80 (ddd, J4′,5′ = 9.9, J6′b,5′ = 5.5, J6′a,5′ = 2.3 Hz, 1H, 5′-H), 3.62 (br. s, 1H, 4-H), 2.34 (td, J2,3a = J3a,3b = 13.6, J3a,4 = 3.9 Hz, 1H, 3a-H), 2.08–2.02 (6 s, 19H, COCH3, 3b-H) ppm. 13C NMR (CDCl3, 100.0 MHz): δ = 170.7–168.8 (CH3CO), 136.8, 128.7, 128.3, 128.1 (C-aromatic), 94.9 (C-1), 86.8 (C-1′), 77.4 (C-5′), 74.0 (C-3′), 69.4 (OCH2Ph), 69.2 (C-5), 68.0 (C-4′), 67.1 (C-2), 66.4 (C-2′), 64.7 (C-6), 62.1 (C-6′), 58.8 (C-4), 25.9 (C-3), 21.0–20.6 (COCH3) ppm.Oxidation reaction of thiodisaccaride 13 to sulfoxides 16R and 16S
:1). [α]25D = +25.4 (c = 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ = 7.39–7.29 (m, 5H, PhCH2O), 5.46 (t, J1′,2′ = J2′,3′ = 9.5 Hz, 1H, 2′-H), 5.40 (t, J2′,3′ = J3′,4′ = 9.2 Hz, 1H, 3′-H), 5.10 (dd, J4′,5′ = 10.1, J3′,4′ = 9.2 Hz, 1H, 4′-H), 5.07 (d, J1,2 = 3.5 Hz, 1H, 1-H), 4.91 (d, J = 11.9 Hz, 1H, OCH2Ph), 4.67 (dt, J2,3b = 12.7, J1,2 = J2,3a = 4.0 Hz, 1H, 2-H), 4.62 (d, J = 11.9 Hz, 1H, OCH2Ph), 4.56 (d, J1′,2′ = 9.7 Hz, 1H, 1′-H), 4.55 (dt, J5,6a = 8.7, J4,5 = J5,6b = 1.8 Hz, 1H, 5-H), 4.21 (dd, J6′a,6′b = 12.5, J5′,6′a = 3.0 Hz, 1H, 6′a-H), 4.18 (dd, J6′a,6′b = 12.5, J5′,6′b = 5.6 Hz, 1H, 6′b-H), 4.02 (dd, J6a,6b = 11.9, J5,6a = 8.9 Hz, 1H, 6a-H), 3.86 (dd, J6a,6b = 11.9, J5,6b = 2.0 Hz, 1H, 6b-H), 3.80 (ddd, J4′,5′ = 10.1, J5′,6′b = 5.5, J5′,6′a = 2.9 Hz, 1H, 5′-H), 3.53 (br. S, 1H, H-4), 2.36 (td, J2,3a = J3a,3b = 13.5, J3a,4 = 4.0 Hz, 1H, 3a-H), 2.08 (×2), 2.05, 2.04, 2.02 (5 s, 16H, CH3CO overlapping with 3b-H), 0.90 (s, 9H, Si(CH3)2C(CH3)3), 0.09, 0.07 (2 s, 6H, Si(CH3)2C(CH3)3) ppm. 13C NMR (125.7 MHz, CDCl3): δ = 170.8, 170.5 (×2), 169.3, 168.9 (COCH3), 137.1, 128.6, 128.3, 128.1 (C-aromatic), 94.5 (C-1), 86.6 (C-1′), 77.4 (C-5′), 74.1 (C-3′), 72.7 (C-5), 68.7 (OCH2Ph), 68.0 (C-4′), 67.4 (C-2), 66.4 (C-2′), 64.3 (C-6), 62.2 (C-6′), 59.4 (C-4), 26.2 (C-3), 26.1 (Si(CH3)2C(CH3)3), 21.1, 20.9, 20.8, 20.7 (×2) (COCH3), 18.4 (Si(CH3)2C(CH3)3), −5.0, −5.2 (Si(CH3)2C(CH3)3) ppm. HRMS (ESI): calcd for C35H52NaO15SSi 795.2688 [M + Na]+; found 795.2699.:1). [α]25D = +31.1 (c = 0.7, CHCl3). 1H NMR (500 MHz, CDCl3): δ = 7.38–7.28 (m, 5H, PhCH2O), 5.49 (t, J1′,2′ = J2′,3′ = 9.5 Hz, 1H, 2′-H), 5.28 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.20 (d, J1,2 = 2.8 Hz, 1H, 1-H), 5.18 (m, 1H, 2-H), 5.09 (dd, J4′,5′ = 10.0, J3′,4′ = 9.5 Hz, 1H, 4′-H), 4.72 (d, J = 12.1 Hz, 1H, OCH2Ph), 4.59 (d, J1′,2′ = 9.7 Hz, 1H, 1′-H), 4.56 (d, J = 12.1 Hz, 1H, OCH2Ph), 4.32 (td, J5,6a = J5,6b = 6.6, J4,5 = 2.6 Hz, 1H, 5-H), 4.22 (dd, J6′a,6′b = 12.6, J5′,6′a = 4.9 Hz, 1H, 6′a-H), 4.13 (dd, J6′a,6′b = 12.6, J5′,6′b = 2.2 Hz, 1H, 6′b-H), 3.74 (ddd, J4′,5′ = 10.0, J5′,6′a = 4.9, J5′,6′b = 2.3 Hz, 1H, 5′-H), 3.72 (m, 2H, 6a-H, 6b-H), 3.57 (dt, J3b,4 = 5.0, J3a,4 = J4,5 = 2.5 Hz, 1H, 4-H), 2.40 (ddd, J3a,3b = 13.2, J2,3a = 5.1, J3a,4 = 2.5 Hz, 1H, 3a-H), 2.24 (ddd, J3a,3b = 13.5, J2,3b = 11.6, J3b,4 = 5.4 Hz, 1H, 3b-H), 2.07, 2.03, 2.02 (×2), 2.01 (COCH3), 0.92 (s, 9H, Si(CH3)2C(CH3)3), 0.10 (×2) (2s, 6H, Si(CH3)2C(CH3)3) ppm. 13C NMR (125.7 MHz, CDCl3): δ = 170.6, 170.4, 170.0, 169.3, 168.9 (COCH3), 137.5, 128.6, 128.1, 128.0 (C-aromatic), 95.2 (C-1), 90.7 (C-1′), 77.0 (C-5′), 73.9 (C-3′), 70.0 (OCH2Ph), 68.2 (C-5), 67.7 (C-2′), 67.6 (C-4′), 66.8 (C-2′), 62.2 (C-6), 61.8 (C-6′), 53.7 (C-4), 26.0 (Si(CH3)2C(CH3)3), 22.7 (C-3), 21.0, 20.8 (×2), 20.7 (×2) (COCH3), 18.4 (Si(CH3)2C(CH3)3), −5.2 (×2) (Si(CH3)2C(CH3)3) ppm. HRMS (ESI): calcd for C35H52NaO15SSi 795.2688 [M + Na]+; found 795.2705.:1). [α]22D = +4.0 (c = 0.8, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.37–7.29 (m, 5H, PhCH2O), 5.50 (t, J1′,2′ = J2′,3′ = 9.5 Hz, 1H, 2′-H), 5.41 (t, J2′,3′ = J3′,4′ = 9.3 Hz, 1H, 3′-H), 5.12 (t, J3′,4′ = J4′,5′ = 9.7 Hz, 1H, 4′-H), 5.04 (d, J1,2 = 3.1 Hz, 1H, 1-H), 4.77 (d, J = 12.1 Hz, 1H, PhCH2O), 4.65 (d, J = 12.2 Hz, 1H, PhCH2O), 4.61 (d, J1′,2′ = 9.7 Hz, 1H, 1′-H), 4.60 (m, 1H, 2-H), 4.50 (m, 1H, 5-H), 4.24 (dd, J6′a,6′b = 12.3, J5′,6′a = 1.3 Hz, 1H, 6′a-H), 4.16 (dd, J6′a,6′b = 12.4, J5′,6′b = 5.6 Hz, 1H, 6′b-H), 3.81 (m, 2H, 6a-H, 5′-H), 3.74 (m, 2H, 4-H, 6b-H), 2.37 (td, J3a,3b = 13.9, J2,3a = 3.8 Hz, 1H, 3a-H), 2.08 (×2), 2.06, 2.04, 2.02 (5 s, 16H, CH3CO overlapping with 3b-H). 13C NMR (CDCl3, 100.0 MHz) δ = 170.8, 170.5, 170.4, 169.3, 168.9 (COCH3), 137.1, 128.7, 128.3, 128.2 (C-aromatic), 95.3 (C-1), 86.5 (C-1′), 77.4 (C-5′), 74.0 (C-3′), 70.3 (PhCH2O), 69.5 (C-5), 68.0 (C-4′), 67.7 (C-2), 66.2 (C-2′), 62.0 (C-6), 61.8 (C-6′), 58.9 (C-4), 25.4 (C-3), 21.1, 20.8, 20.7 (×2), 20.6 (COCH3) ppm. HRMS (ESI): calcd for C29H38NaO15S 681.1824 [M + Na]+; found 681.1830.:1:5; 0.46 mL) was stirred at room temperature for 3 h. The mixture was concentrated, and the resulting residue was dissolved in water (1 mL) and eluted through a column filled with a Dowex MR-3C mixed-bed ion-exchange resin. The deionized solution was concentrated, and the unprotected compound was purified by dissolution in water (1 mL) and filtration through an octadecyl C18 minicolumn (Amprep, Amersham Biosciences). Evaporation of the water gave the unprotected thiodisaccharide S-oxides as colourless syrupy that was identified as 18R (31 mg, 93%). Rf = 0.72, BuOH/EtOH/H2O (10:4:4). [α]23D = +32.3 (c = 1.0, MeOH). 1H NMR (400 MHz, D2O) δ = 7.51–7.40 (m, 5H, PhCH2O), 5.10 (d, J1,2 = 3.3 Hz, 1H, 1-H), 4.88 (d, J = 11.6 Hz, 1H, PhCH2O), 4.69 (d, J = 11.7 Hz, 1H, PhCH2O), 4.56 (m, 1H, 5-H), 4.50 (d, J1′,2′ = 9.2 Hz, 1H, 1′-H), 3.96–3.89 (m, 3H, 2-H, 6′a-H, 6′b-H), 3.84 (m, 2H, 4-H, 6a-H), 3.76 (m, 2H, 6b-H, 2′-H), 3.69 (t, J2′,3′ = J3′,4′ = 8.8 Hz, 1H, 3′-H), 3.66 (m, 1H, 5′-H), 3.50 (t, J3′,4′ = J4′,5′ = 9.3 Hz, 1H, 4′-H), 2.30 (td, J2,3a = J3a,3b = 14.3, J3a,4 = 3.9 Hz, 1H, 3a-H) 1.96 (br dt, J3a,3b = 14.2, J2,3b = J3b,4 = 3.5 Hz, 1H, 3b-H) ppm. 13C NMR (CDCl3, 100.0 MHz) δ = 136.9, 128.8, 128.6, 128.4 (C-aromatic), 97.1 (C-1), 87.2 (C-1′), 80.5 (C-5′), 76.8 (C-3′), 71.2 (C-5), 69.6 (PhCH2O), 68.6 (C-4′), 67.2 (C-2′), 63.6 (C-2), 62.0 (C-6), 60.5 (C-6′), 56.7 (C-4), 27.9 (C-3) ppm. HRMS (ESI): calcd for C19H28NaO10S 471.1295 [M + Na]+; found 471.1275.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are indebted to the Secretaría de Ciencia y Tecnología (SECyT), Universidad Nacional de Córdoba (UNC), Universidad de Buenos Aires (UBA), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Fondo para la investigación Científica y Tecnológica Argentina (FONCyT) for financial support. J. P. C., A. B. P. and O. V. are research members from CONICET.Notes and references
- A. Tarozzi, C. Angeloni, M. Malaguti, F. Morroni, S. Hrelia and P. Hrelia, Oxid. Med. Cell. Longevity, 2013, 415078 Search PubMed.
- J. Li, J. Zhao, J. E. Hamer-Maansson, T. Andersson, R. Fulmer, M. Illueca and P. Lundborg, Clin. Ther., 2006, 28, 419–427 CrossRef CAS PubMed.
- M. Darwish, M. Kirby, E. T. Hellriegel and P. Robertson Jr, Clin. Drug Invest., 2009, 29, 613–623 CrossRef CAS PubMed.
- S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed.
- N. Khiar, A. Salvador, V. Valdivia, A. Chelouan, A. Alcudia, E. Álvarez and I. Fernández, J. Org. Chem., 2013, 78, 6510–6521 CrossRef CAS PubMed.
- H. Pellissier, Tetrahedron, 2006, 62, 5559–5601 CrossRef CAS.
- M. C. Carreño, Chem. Rev., 1995, 95, 1717–1760 CrossRef.
- F. Zhong, A. Pçthig and T. Bach, Chem. – Eur. J., 2015, 21, 10310–10313 CrossRef CAS PubMed.
- Z. Z. Li, S. U. Yao and B. H. Ye, ChemPlusChem, 2015, 80, 141–150 CrossRef CAS.
- A. A. Elkina, T. I. Kylosova, V. V. Grishko and I. B. Ivshina, J. Mol. Catal. B: Enzym., 2013, 89, 82–85 CrossRef.
- I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3705 CrossRef PubMed.
- N. Khiar, I. Alonso, N. Rodriguez, A. Fernandez-Mayoralas, J. Jimenez-Barbero, O. Nieto, F. Cano, C. Foces-Foces and M. Martin-Lomas, Tetrahedron Lett., 1997, 38, 8267–8270 CrossRef CAS.
- N. Khiar, I. Fernández, C. S. Araújo, J. A. Rodríguez, B. Suárez and E. Álvarez, J. Org. Chem., 2003, 68, 1433–1442 CrossRef CAS PubMed.
- Z. J. Witczak, P. Kaplon and P. M. Dey, Carbohydr. Res., 2003, 338, 11–18 CrossRef CAS PubMed.
- E. Repetto, C. Marino and O. Varela, Bioorg. Med. Chem., 2013, 21, 3327–3333 CrossRef CAS PubMed.
- V. E. Manzano, M. L. Uhrig and O. Varela, Org. Biomol. Chem., 2012, 10, 8884–8894 CAS.
- A. J. Cagnoni, M. L. Uhrig and O. Varela, Bioorg. Med. Chem., 2009, 17, 6203–6212 CrossRef CAS PubMed.
- V. E. Manzano, M. L. Uhrig and O. Varela, J. Org. Chem., 2008, 7224–7235 CrossRef CAS PubMed.
- M. L. Uhrig, V. E. Manzano and O. Varela, Eur. J. Org. Chem., 2006, 162–168 CrossRef CAS.
- J. P. Colomer, M. Á. Canales Mayordomo, B. Fernández de Toro, J. Jiménez-Barbero and O. Varela, Eur. J. Org. Chem., 2015, 1448–1455 CrossRef CAS.
- J. P. Colomer, V. E. Manzano and O. Varela, Eur. J. Org. Chem., 2013, 7343–7353 CrossRef CAS.
- C. A. Sanhueza, A. C. Arias, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2010, 21, 1830–1832 CrossRef CAS.
- C. A. Sanhueza, R. L. Dorta and J. T. Vázquez, J. Org. Chem., 2011, 76, 7769–7780 CrossRef CAS PubMed.
- C. A. Sanhueza, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2013, 24, 582–593 CrossRef CAS.
- J. P. Colomer, B. Fernández de Toro, F. J. Cañada, F. Corzana, J. Jiménez-Barbero, A. Canales and O. Varela, Eur. J. Org. Chem., 2016, 5117–5122 CrossRef CAS.
- M. L. Uhrig and O. Varela, Aust. J. Chem., 2002, 55, 155–160 CrossRef CAS.
- C. J. Salomon, E. G. Mata and O. A. Mascaretti, Tetrahedron Lett., 1991, 32, 4239–4242 CrossRef CAS.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 CrossRef CAS PubMed.
- W. G. Dauben, B. A. Kowalczyk and F. W. Lichtenthaler, J. Org. Chem., 1990, 55, 2391–2398 CrossRef CAS.
- F. W. Lichtenthaler, S. Nishiyama, P. Kohler and H. J. Lindner, Carbohydr. Res., 1985, 136, 13–26 CrossRef CAS.
- D. Crich, J. Mataka, L. N. Zakharov, A. L. Rheingold and D. J. Wink, J. Am. Chem. Soc., 2002, 124, 6028–6036 CrossRef CAS PubMed , and references therein.
- L. Calle, V. Roldós, F. J. Cañada, M. L. Uhrig, A. J. Cagnoni, V. E. Manzano, O. Varela and J. Jiménez-Barbero, Chem. – Eur. J., 2013, 19, 4262–4270 CrossRef CAS PubMed.
- E. Montero, A. García-Herrero, J. L. Asensio, K. Hirai, S. Ogawa, F. Santoyo-González, F. J. Cañada and J. Jiménez-Barbero, Eur. J. Org. Chem., 2000, 1945–1952 CrossRef CAS.
- A. García-Herrero, E. Montero, J. L. Muñoz, J. F. Espinosa, A. Vián, J. L. García, J. L. Asensio, F. J. Cañada and J. Jiménez-Barbero, J. Am. Chem. Soc., 2002, 124, 4804–4810 CrossRef.
- D. G. H. C. H. Green, J. Chem. Soc., Perkin Trans. 2, 1972, 458–463 RSC.
- R. D. G. Cooper, P. V. DeMarco, J. C. Cheng and N. D. Jones, J. Am. Chem. Soc., 1969, 91, 1408–1415 CrossRef CAS PubMed.
- R. D. G. Cooper, P. V. DeMarco and D. O. Spry, J. Am. Chem. Soc., 1969, 91, 1528–1529 CrossRef CAS PubMed.
- D. H. R. Barton, F. Comer and P. G. Sammes, J. Am. Chem. Soc., 1969, 91, 1529–1530 CrossRef CAS.
- N. Khiar, Tetrahedron Lett., 2000, 41, 9059–9063 CrossRef CAS.
- H. B. Henbest and R. A. L. Wilson, J. Chem. Soc., 1957, 1958–1965 RSC.
- P. Chamberlain, M. L. Roberts and G. H. Whitham, J. Chem. Soc. B, 1970, 1374–1381 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07225b |
| This journal is © The Royal Society of Chemistry 2017 |