
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of unusual microphase-separated ultrathin films of poly(vinyl catechol-block-styrene) (PVCa-b-PSt) at the air–water interface by solution casting onto water†
Hiroshi Yabu *a and
Shusaku Naganob
*a and
Shusaku Naganob
aWPI-Advanced Institute for Materials Research (AIMR), Tohoku University, 2-1-1, Katahira, Aoba-Ku, Sendai 980-8577, Japan. E-mail: hiroshi.yabu.d5@tohoku.ac.jp
bNagoya University Venture Business Laboratory, Nagoya University, Furocho, Chikusa-Ku, Nagoya 464-8603, Japan
First published on 29th June 2017
Abstract
This report describes the preparation of ultrathin films possessing unusual fingerprint-like morphologies for poly(vinyl catechol-block-styrene) (PVCa-b-PSt) at the air–water interface by simply mixing a solution into water followed by solvent evaporation. Structural analysis and comparison with typical surface micelles of amphiphilic diblock copolymers indicated that precipitation and micelle aggregation played a significant role in the formation and unusual microphase separation of PVCa-b-PSt ultrathin films. In addition, the interior structures of the ultrathin films could be adjusted by adding homo-polystyrene (hPSt). Silver nanoparticle arrays were formed by immersing ultrathin films in aqueous AgNO3 and a microphase-separated structure in the ultrathin films could act as a template for the arrays.
Introduction
Block copolymers form nanoscale phase-separated structures in their bulk films. These phase-separated structures were applied to not only control bulk properties but also to act as templates for nanoscale patterning. Amphiphilic block copolymers, in particular, can modify the surfaces of materials, and act as templates for metal and inorganic nanostructures, drug carriers, and solid-state ion conductors due to the differing properties of the hydrophobic and hydrophilic polymer segments. To achieve these applications, the self-assembly of amphiphilic block copolymers has been investigated in detail.1The self-assembly of amphiphilic block copolymers at the air–water interface is of interest because it allows the formation of unique nanostructures based on surface micelles.2–5 Many types of amphiphilic copolymers have been examined to control nanostructures at the air–water interfaces by changing their composition and forming different types of morphology in thin films.6–8 In addition, surface patterns have been applied to templates for nanoparticle assemblies or block copolymer lithography.9 To extend the feasibility of these nanostructured thin films composed of amphiphilic block copolymers, ultrathin films formed from amphiphilic block copolymers with functional moieties are needed.
Catechol-containing polymers have been investigated because of their unique adhesive10–13 and reductive properties.14,15 Due to multiple interactions between catechol moieties and substrates, catechol-containing polymers adhere onto a wide variety of substrates, regardless of the composition. Furthermore, phenolic hydroxyl groups can reduce metal ions in the presence of catechol moieties to form solid metal. Many reports exist concerning synthesis of catechol-terminated polymers16,17 and random co-polymers18 containing catechol groups. However, few reports exist describing the synthesis and application of block copolymers containing catechol moieties.19
A recent report described the synthesis of poly(vinyl catechol-block-styrene) (PVCa-b-PSt) using the reversible addition-fragmentation transfer (RAFT) polymerization process with 3,4-dimethoxy styrene (DMSt) and styrene (St), followed by deprotection of methoxy groups with BBr3.20 This new type of catechol-containing diblock copolymer formed an inverse micelle structure in THF and formed silver nanoparticles in the core part of the micelles in the presence of silver ions. Furthermore, PVCa-b-PSt formed a variety of absolute microphase-separated structures due to its amphiphilic nature, and possessed conductivity and reductive properties toward metal ions in the water phase.21 Most recently, lamellae-structured PVCa-b-PSt films with silver nanoparticles in the PVCa domains exhibited relatively high proton conductivity under high humidity conditions.22
This report describes the preparation of ultrathin films possessing unusual morphologies for PVCa-b-PSt at the air–water interface by simply mixing a solution into water followed by solvent evaporation. A detailed analysis of the nanoscale phase-separated structures and the formation mechanism of the self-assembly process was performed. In addition, the control of nanoscale morphology by homopolymer blending is also described.
Experimental
Materials
The PVCa-b-PSt [Mn PDMSt = 11.6 kg mol−1, Mn PSt = 23.8 kg mol−1, Mw/Mn = 1.16 (GPC, PSt standard)] was synthesized using reversible-addition fragmentation transfer (RAFT) polymerization of 3,4-dimethoxy styrene (DMSt) and styrene (St), followed by deprotection of the methoxy group with BBr3, according to a previously reported method.21,23 The PVCa-ran-PSt was synthesized by free radical copolymerization of DMSt and St (Mn = 25.5 kg mol−1, Mw = 42.6 kg mol−1, Mw/Mn = 1.7, see ESI, S1†). The PSt homopolymer (hPSt, Mn = 12.6 kg mol−1) and tetrahydrofuran (THF, GR) were purchased from Polymer Source Inc. and Wako Chemical Industries, Inc., respectively.Preparation of ultrathin films
Fig. 1 shows the procedure used to prepare ultrathin films of PVCa-b-PSt. A 10 mg mL−1 solution of PVCa-b-PSt was prepared in THF. Millipore-membrane filtered water was added to a glass dish (f = 80 mm), and then a small amount of the PVCa-b-PSt THF solution (ca. 200 μL) was dropped into the water. After THF evaporation, ultrathin films at the air–water interface were collected using a Cu grid covered with a collodion membrane or an epoxy resin plate prepared from EPOC (Ohken Shoji, Tokyo). The samples were dried at room temperature. The ultrathin films and colloidal assemblies of PVCa-b-PSt were stained with OsO4 vapor for 2 hours. Nanoscale features of these samples were observed by transmission electron microscopy (TEM, H-7650, Hitachi), with an acceleration voltage of 100 kV. To observe the cross-sectional image, the ultrathin film on an epoxy resin plate was coated with epoxy resin and then cured at 70 °C overnight. After curing, ultrathin cross-sections with a thickness of 100 nm were obtained using an ultramicrotome (UC-7, Lica, Germany). The fast Fourier transformation, which is the image analysis method to obtain a reciprocal lattice space image by Fourier transformation of the original images, were obtained by using imaging software, Image J®, and FFT image was obtained from original image shown in Fig. 3(a).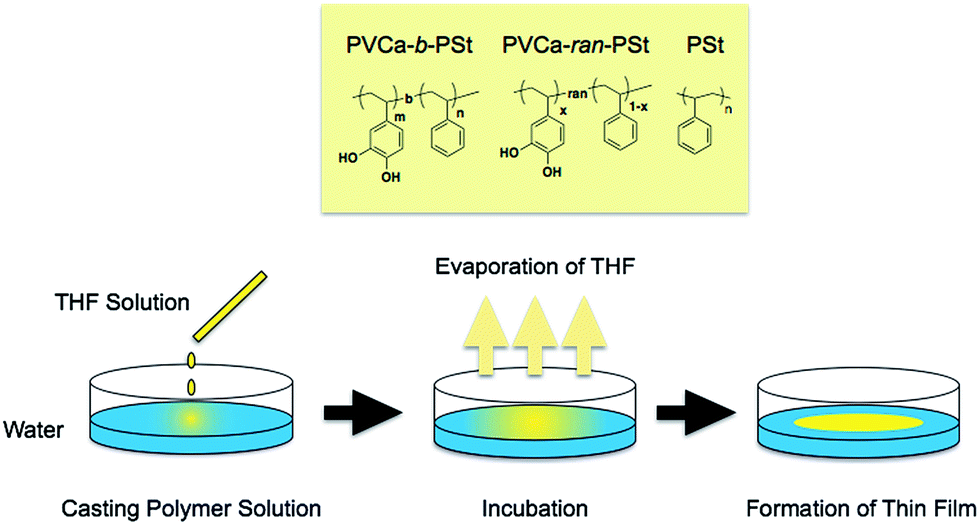 | ||
| Fig. 1 Chemical structures of the polymers used in this report and a schematic illustration of ultrathin film formation. | ||
Preparation of nanoscale colloidal assemblies of PVCa-b-PSt dispersed in the water phase
Nanoscale colloidal assemblies of PVCa-b-PSt dispersed in the water phase was prepared at the same time of ultrathin films and also sampled and cast onto a Cu grid. The top view of the sample was observed as same manner as the ultrathin films.Preparation of ultrathin films of PVCa-b-PSt and PSt blends
A THF solution of PSt (5–10 mg mL−1) was prepared and then mixed with a THF solution of PVCa-b-PSt (5–10 mg mL−1) at different mixing ratios. The PVCa-b-PSt and PSt blended ultrathin films were prepared using the same method.Preparation of Langmuir films
THF solutions of PVCa-ran-PS and PVCa-b-PS of ca. 1 mmol (monomer unit) dm−3 were prepared. Spreading behavior of the random copolymer and block copolymer was characterized using a Lauda FW-1 film balance filled with pure water (Milli-Q grade, 18 MΩ cm) at 20 °C. The solution was spread onto the water surface in the Langmuir trough. Surface pressure vs. area (π–A) isotherms were obtained upon constant compression at a speed of 20 cm2 min−1 with the sliding barrier. The ultrathin films floating on the water were transferred onto a freshly cleaved mica substrate using the conventional Langmuir–Blodgett method (vertical dipping method) for atomic force microscopy (AFM) studies. Topographical AFM observations were obtained using a SPA300/SPI3700 system (Seiko Instruments, Tokyo) in noncontact mode.Results and discussion
Fig. 2(a) and (b) show TEM images of PVCa-ran-PSt aggregates dispersed around the air–water interface and the water phase. Spherical aggregates were most commonly observed, although some network-like aggregates formed around the air–water interface. These results are identical to those previously reported for polymer particle formation by self-organized precipitation (SORP).28–30 Since PVCa-ran-PSt is basically a hydrophobic polymer, it precipitated in the water phase after complete evaporation of THF to formed dispersed particles. | ||
| Fig. 2 TEM images of PVCa-ran-PSt network-like (a) and spherical (b) aggregates formed at the air–water interface. The PVCa domains were stained with OsO4. | ||
In contrast, a high reflection region at the air–water interface was observed for PVCa-b-PSt after THF evaporation. After collecting the highly-reflected region on a Cu grid and staining the film with OsO4, the area was observed using TEM. Fig. 3 shows TEM images of the ultrathin films obtained. Fingerprint-like morphologies were clearly seen. Dark and bright regions were attributed to stained PVCa domains and non-stained PSt domains, respectively. The surface area indicated the copolymerization ratio between PVCa and PSt segments, showing a very uniform periodic structure. Fast Fourier transformation (FFT) indicated that the average periodicity of the phase-separated structure was 41 nm. Recently, Kim et al. reported that fingerprint-like morphologies formed at the air–water interface when the spreading area of poly(styrene-block-2-vinyl pyridine) (PSt-b-P2VP) was restricted. However, the surface density of the optically reflective area obtained in the present study was very low, indicating that the morphology of PVCa-b-PSt ultrathin film was not identical with the previously reported morphology.24 Furthermore, a cross-sectional TEM image of this ultrathin film was also examined [inset image of Fig. 3(b), also see ESI, S2†]. Surprisingly, a monolayer of circular domains composed of PSt cores and PVCa shells was observed. This result is very different from that obtained for usual surface micelles because diblock copolymers often phase-separate at the air–water interface, with hydrophobic segments exposed to air and hydrophilic segments in the water phase.25 Note that the PVCa-b-PSt used in this experiment forms a cylindrical structure in its bulk state, with cores and shells composed of PVCa and PSt domains, respectively.21 These results indicate that microphase-separated structures were inverted by the process of mixing into water followed by solvent evaporation, and suggests that a different formation mechanism must exist for this phenomenon.
 | ||
| Fig. 3 TEM images of PVCa-b-PSt ultrathin films formed at the air–water interface at different magnifications (a) 8k, (b) 20k and (c) 40k, respectively. The PVCa domains were stained with OsO4. Inset images show an (a) FFT image and (b) cross-sectional TEM image of the ultrathin film. | ||
To investigate the morphological difference between the PVCa-ran-PSt and PVCa-b-PSt ultrathin films obtained by solution casting onto water, π–A isotherms for the floating polymer films were measured using a Langmuir film balance. A THF solution of PVCa-b-PSt or PVCa-ran-PSt was cast onto the surface of the water subphase, and then was compressed with the sliding barrier. Fig. 4(a) and inset show the π–A isotherms and close-up π–A isotherms of PVCa-b-PSt and PVCa-ran-PSt, respectively. The PVCa-ran-PSt possessed no significant surface pressure, indicating that the random copolymer could not form stable floating films or surface micelles on the water surface. Thus, no clear aggregation formed from the random copolymers and they precipitated as superfine spherical aggregates in the water phase. In contrast, the PVCa-b-PSt floating film experienced pressure beginning at ca. 0.03 nm2 with a quasi-plateau region. Further compression produced a sharp pressure increase above 60 mN m−1 within an area less than 0.02 nm2. The clear π–A isotherm suggests that the block copolymer could form a stable ultrathin film on a water surface. Fig. 4(b) and (c) show AFM images of ultrathin films transferred at 1 mN m−1 and 10 mN m−1 on a mica substrate using the Langmuir–Blodgett technique. The AFM images showed tubular and spherical surface micelles; spacing among the aggregates became closer with increasing surface pressure. According to data from the π–A isotherms and AFM observations, hydrophilic PVCa blocks monomolecularly extended onto the water with an aggregated PS core to form surface micelles in a large molecular area (greater than 0.03 nm2). With compression, the extended hydrophobic chains exhibited a phase transition from the two-dimensional to the three-dimensional state on water in the quasi-plateau region.5,26,27 Further compression produced a sharp pressure increase above 60 mN m−1 around an area of 0.02 nm2, originating from overlapping of PS cores at the water surface.2–4 These are structures similar to those of ultrathin films obtained by solution mixing, but the thickness of the surface micelles was almost half of that of the ultrathin films (ca. 20 nm). This result also supports double layers of PVCa-b-PSt molecules in the ultrathin films prepared by mixing a solution into water.
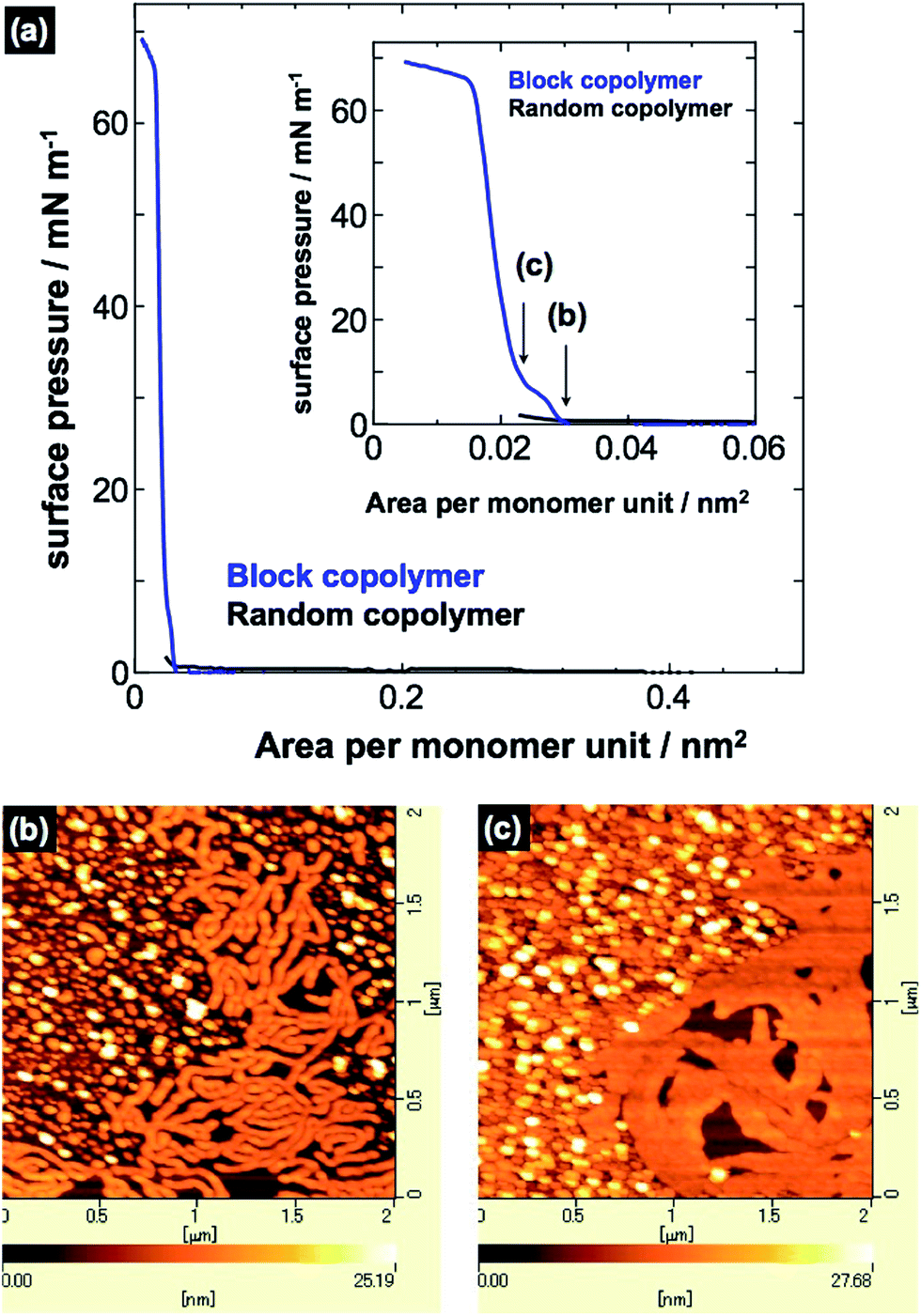 | ||
| Fig. 4 (a) π–A isotherms of PVCa-ran-PSt and PVCa-b-PSt. AFM images of surface micelles of PVCa-b-PSt transferred onto mica substrates at (b) 1 mN m−1 and (c) 10 mN m−1. | ||
Structures of PVCa-b-PSt formed in the water phase were also observed by TEM. Fig. 5(a) shows TEM images of PVCa-b-PSt aggregations formed inside the water phase. The aggregates were composed of spherical micelles, whose surfaces were covered with hydrophilic PVCa domains and were of various sizes. The PVCa-b-PSt precipitated as spherical micelles and form aggregates in water phase. At this stage, the aggregates still had some THF molecules and floated to the air–water interface. Fig. 5(b) shows the TEM image of aggregates obtained from the region that did not exhibit high reflectance at the air–water interface. The small aggregates gradually increase in size with rearrangement of phase-separated structures. These results indicate that the morphology of self-assembled ultrathin films gradually change from a single micellar structure to assembled ultrathin films. The aggregates self-assembled at the air–water interface to form larger assemblies during the THF evaporation. After complete evaporation of THF, unusual microphase separated ultrathin films were finally obtained.
 | ||
| Fig. 5 TEM images of PVCa-b-PSt aggregates obtained from the (a) water phase and (b) low reflective area at the air–water interface. The PVCa domains were stained with OsO4. | ||
The mechanism for formation of the phase-separated ultrathin films at the air–water interface is thought to occur as follows. After mixing the solution into the water phase, block copolymer micelles with a surface covered with hydrophilic PVCa domains formed spontaneously. During the solvent evaporation process, the micelles aggregated into larger structure and moved to the air–water interface due to their amphiphilic nature. At the air–water interface, the micelles grew in two dimensions and annealed during the evaporation of THF, followed by self-assembly into ultrathin films having well-developed phase-separated structures. This process is very different from surface micelle formation and provides a new technique to fabricate phase-separated ultrathin films.
Phase-separated structures of diblock copolymers can be controlled by mixing homopolymers. Altering the mixing ratio and molecular weight of the homopolymers can provide unique nanoscale phase-separated structures. When the molecular weight of a mixed homopolymer is comparable with that of the same segment in the diblock copolymer, PSt homopolymer lies in the PSt domain of block copolymer, lengthening the periodicity of microphase separation. Homopolymer polystyrene (hPSt), with a molecular weight comparable to that of the PSt segment of PVCa-b-PSt, was mixed. Fig. 6 shows TEM images obtained from a mixed solution of PVCa-b-PSt and hPSt at mixing ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (a)–(c), 1:1 (d)–(f), and 1:2 (g)–(i). At a low mixing ratio, the phase-separated structure had the fingerprint morphology; however, the PVCa layer was thinner than that of the original phase and the periodicity of phase separation elongated from 41 nm to 56 nm. The spacing of phase-separated structures increased with the amount of hPSt, finally forming a thin platelet-like PVCa phase at a ratio of 1:2. These results indicate that interior morphologies and periodicities of ultrathin films can be controlled by adjusting the mixing ratio of the homopolymers, even though they form at the air–water interface.
:1 (a)–(c), 1:1 (d)–(f), and 1:2 (g)–(i). At a low mixing ratio, the phase-separated structure had the fingerprint morphology; however, the PVCa layer was thinner than that of the original phase and the periodicity of phase separation elongated from 41 nm to 56 nm. The spacing of phase-separated structures increased with the amount of hPSt, finally forming a thin platelet-like PVCa phase at a ratio of 1:2. These results indicate that interior morphologies and periodicities of ultrathin films can be controlled by adjusting the mixing ratio of the homopolymers, even though they form at the air–water interface.
 | ||
| Fig. 6 TEM images of ultrathin films of PVCa-b-PSt and hPSt blends at a mixing ratio of (a)–(c) 2:1, (d)–(f) 1:1, and (g)–(i) 1:2 at different magnifications. The PVCa domains were stained with OsO4. | ||
Since PVCa domains are rich in catechol groups, which have the ability to reduce metal ions, the PVCa-b-PSt ultrathin film can be used as a template for patterning silver nanoparticle arrays. Fig. 7(a)–(c) show TEM images of ultrathin PVCa-b-PSt films after immersion in aq. AgNO3 for 1 h, 2 h, and 4 days, respectively. Early after immersion, tiny black dots identified as silver nanoparticles, were seen. The black dots gradually enlarged with immersion time and became aligned along the PVCa domains stained with OsO4. It is important to note that silver nanoparticles were located only PVCa regions and formed arrays along to the PVCa domains (ESI, S3†). Furthermore, due to its unique morphology, PVCa domains were exposed to the surface of ultrathin films. From the top view, some silver nanoparticles located on the middle of PSt phases (ESI, Fig. S4(a)†), however, from the cross-sectional TEM image (ESI, Fig. S4(b)†), there is no silver nanoparticles located inside of the PSt phase but located on the surface and bottom of the film. These results indicate that the PVCa ultrathin films can act as reductive nanoscale templates for metal nanoparticle arrangement.
 | ||
| Fig. 7 TEM images of ultrathin films of PVCa-b-PSt immersed in aq. AgNO3 for (a) 1 h, (b) 2 h, and (c) 4 days. The PVCa domains were stained with OsO4. | ||
Conclusions
Formation of ultrathin films of PVCa-b-PSt at the air–water interface was accomplished by mixing a THF solution with the water phase followed by solvent evaporation. Structural analysis and comparison with typical surface micelles of amphiphilic diblock copolymers indicated that precipitation and micelle aggregation played a significant role in the formation and unusual microphase separation of PVCa-b-PSt ultrathin films. In addition, the interior structures of the ultrathin films could be adjusted by adding hPSt. Silver nanoparticle arrays were formed by immersing ultrathin films in aqueous AgNO3 and a microphase-separated structure in the ultrathin films could act as a template for the arrays.These findings provide a new technique for fabricating nanoscale features based on phase separation of amphiphilic diblock copolymers and will contribute to new microfabrication technologies. In addition, the formation process of the ultrathin films in this experiment provides insight into understanding molecular aggregation behaviors of amphiphilic block copolymers at the air–water interface.
Acknowledgements
This work was partially supported by JSPS KAKENHI (No. 17H01223 and 16K14071) and Nanotechnology Platform Program (Nagoya University, Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. H. Y. would like to thank Ms. Minori Suzuki, WPI-AIMR, Tohoku University, for the TEM observations. H. Y. also would like to thank Dr Y. Saito and Mr H. Ohshima, Graduate School of Engineering, Tohoku University for helping the synthesis of copolymers.References
- J. Li, K. Kamata, S. Watanabe and T. Iyoda, Adv. Mater., 2007, 19, 1267–1271 CrossRef CAS.
- J. Y. Zhu, A. Eisenberg and R. B. Lennox, J. Am. Chem. Soc., 1991, 113, 5583–5588 CrossRef CAS.
- X. Wang, X. Ma and D. Zang, Soft Matter, 2013, 9, 443–453 RSC.
- A. G. da Silva and A. S. Gamboa, Langmuir, 1998, 14(18), 5327–5330 CrossRef.
- B. Chung, M. Choi, M. Ree, J. C. Jung, W. C. Zin and T. Chang, Macromolecules, 2006, 39, 684–689 CrossRef CAS.
- M. Sakuma, Y. Kumashiro, M. Nakayama, N. Tanaka, K. Umemura, M. Yamato and T. Okano, Biomacromolecules, 2014, 15, 4160–4167 CrossRef CAS PubMed.
- S. Nagano, Y. Matsushita, Y. Ohnuma, S. Shinma and T. Seki, Langmuir, 2006, 22, 5233–5236 CrossRef CAS PubMed.
- I. I. Perepichka, A. Badia and C. G. Bazuin, ACS Nano, 2010, 4, 6825–6835 CrossRef CAS PubMed.
- M. V. Meli, A. Badia, P. Grütter and R. B. Lennox, Nano Lett., 2002, 2, 131–135 CrossRef CAS.
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed.
- H. Zhang, L. P. Bré, T. Zhao, Y. Zheng, B. Newland and W. Wang, Biomaterials, 2014, 35, 711–719 CrossRef CAS PubMed.
- S.-B. Lee, C. González-Cabezas, K.-M. Kim, K.-N. Kim and K. Kuroda, Biomacromolecules, 2015, 16, 2265–2269 CrossRef CAS PubMed.
- X. Fan, L. Lin, J. L. Dalsin and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 15843–15847 CrossRef CAS PubMed.
- G. Fichman, L. Adler-Abramovich, S. Manohar, I. Mironi-Harpaz, T. Guterman, D. Seliktar, P. B. Messersmith and E. Gazit, ACS Nano, 2014, 8, 7220–7228 CrossRef CAS PubMed.
- L. Guo, Q. Liu, G. Li, J. Shi, J. Liu, T. Wang and G. Jiang, Nanoscale, 2012, 4, 5864 RSC.
- Y. Zhu, H. S. Sundaram, S. Liu, L. Zhang, X. Xu, Q. Yu, J. Xu and S. Jiang, Biomacromolecules, 2014, 15, 1845–1851 CrossRef CAS PubMed.
- J. L. Dalsin and P. B. Messersmith, Mater. Today, 2005, 8, 38–46 CrossRef CAS.
- J. D. White and J. J. Wilker, Macromolecules, 2011, 44, 5085–5088 CrossRef CAS.
- D. Leibig, A. H. E. Müller and H. Frey, Macromolecules, 2016, 49, 4792–4801 CrossRef CAS.
- Y. Saito and H. Yabu, Chem. Commun., 2015, 51, 3743–3746 RSC.
- Y. Saito, T. Higuchi, H. Jinnai, M. Hara, S. Nagano, Y. Matsuo and H. Yabu, Macromol. Chem. Phys., 2016, 217, 726–734 CrossRef CAS.
- H. Yabu, J. Matsui, M. Hara, S. Nagano, Y. Matsuo and Y. Nagao, Langmuir, 2016, 32, 9484–9491 CrossRef CAS PubMed.
- Y. Saito and H. Yabu, Chem. Commun., 2015, 51, 3743–3746 RSC.
- D. H. Kim and S. Y. Kim, J. Phys. Chem. Lett., 2017, 8, 1865–1871 CrossRef CAS PubMed.
- B. Yoon, J. Huh, H. Ito, J. Frommer, B. H. Sohn, J. H. Kim, E. L. Thomas, C. Park and H. C. Kim, Adv. Mater., 2007, 19, 3342–3348 CrossRef CAS.
- S. M. Baker, K. A. Leach, C. E. Devereaux and D. E. Gragson, Macromolecules, 2000, 33, 5432–5436 CrossRef CAS.
- Y. Seo, K. Paeng and S. Park, Macromolecules, 2001, 34, 8735–8744 CrossRef CAS.
- H. Yabu, T. Higuchi and M. Shimomura, Adv. Mater., 2005, 17, 2062–2065 CrossRef CAS.
- H. Yabu, Polym. J., 2013, 45, 261–268 CrossRef CAS.
- T. Higuchi, M. Shimomura and H. Yabu, Macromolecules, 2013, 46, 4064–4068 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of PVCa-ran-PSt. See DOI: 10.1039/c7ra06574d |
| This journal is © The Royal Society of Chemistry 2017 |