
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Moderate oxidation levels of Ru nanoparticles enhance molecular oxygen activation for cross-dehydrogenative-coupling reactions via single electron transfer†
Mu Lin‡
a,
Lin-Xiu Dai‡a,
Jun Gu‡a,
Li-Qun Kanga,
Yu-Hao Wanga,
Rui Si b,
Ze-Qiong Zhaoa,
Wen-Chi Liua,
Xuefeng Fua,
Ling-Dong Sun*a,
Ya-Wen Zhang*a and
Chun-Hua Yan*a
b,
Ze-Qiong Zhaoa,
Wen-Chi Liua,
Xuefeng Fua,
Ling-Dong Sun*a,
Ya-Wen Zhang*a and
Chun-Hua Yan*a
aBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: yan@pku.edu.cn; ywzhang@pku.edu.cn; sun@pku.edu.cn
bShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
First published on 29th June 2017
Abstract
For converting a homogeneous synthetic chemistry reaction to a heterogeneous one, using nanocatalysts, it is essential to investigate the underlying principles and the associated dominant factors of methodologies between inorganic nanoparticles and organic substrates. Here it is reported that novel, hydrothermally synthesized ruthenium (Ru) nanoparticles performed differently in molecular oxygen activation via single electron transfer for cross-dehydrogenative-coupling reactions, following a volcano shaped relationship between their oxidation levels and catalytic activity. Characterizations and a systematic mechanism study indicated that Ru3+ sites contributed to more superoxo- or peroxo-like species being formed from molecular oxygen on Ru nanoparticles with a moderate oxidation level, and the Ru3+ were the highly active species which accelerate this cross-dehydrogenative-coupling reaction via single electron transfer.
Introduction
With the development of controlled synthesis of nanocatalysts with different sizes, shapes, composition, structure, oxidation state and so on, remarkable progress on the investigation of the relationship between the structure of catalytic active sites and the catalytic performance has been made, which provides more opportunities to design and synthesize nanocatalysts with high activity, selectivity and stability.1–8 In recent years, nanocatalysts in organic chemistry have attracted much attention.9–14 Homogeneous catalysts possess higher catalytic activity than that of heterogeneous catalysts because of the easy accessibility of reactants to active sites and higher selectivity by tuning the steric and electronic properties of ligands. There is no doubt that because of the separation of the catalysts from the reaction solutions it is too difficult to reuse these catalysts, resulting in the contamination of the products and the waste of noble metal catalysts. For green and sustainable chemistry, it is of great significance to develop highly efficient nanocatalysts for important organic reactions.Carbon–carbon bond formation is the essential link in all organic molecules, and is a central part of many chemical syntheses, such as in natural products, medicines, and organic materials.15–20 Building a C–C linkage directly via the cleavage of dual C–H bonds using molecular oxygen as the terminal oxidant, called aerobic cross-dehydrogenative coupling (CDC), has been successfully developed into the most atom-economic, clean, and efficient strategy in synthetic organic chemistry.15,21,22 Homogeneous transition metal catalyzed activation of C–H bonds for C–C bond formation, has proved to be versatile for the CDC reaction, using metals such as cobalt (Co), copper (Cu), iron (Fe), nickel (Ni), palladium (Pd), rhodium (Rh), and ruthenium (Ru).23–25 However, for widely investigated Pd or Cu catalyzed CDC reactions, most of the reports only provided possible reaction mechanisms without enough experimental evidence. In past decades, the development of heterocatalysis means that nanoparticles (NPs) play more and more important roles in organic syntheses, and many NPs (e.g., Fe3O4, Cu6Se4.5, CuFe2O4, ruthenium(IV) oxide (RuO2), RuO2/graphene, and so) exhibit higher catalytic performance than their salts or complexes in CDC reactions.26–29 However, the complicated structure of the active sites makes the elucidation of the reaction mechanism in a heterogeneous system challenging.30–32 Thus, understanding the relationship between the structure effects (electronic and geometric) of NPs and the catalytic performance towards important organic reactions is necessary to illustrate the reaction mechanism and to enable the screening of highly efficient heterocatalysts.
In this paper it is reported that novel Ru NPs performed differently in triggering CDC reactions. The catalytic mechanism was studied and it was found that the content of Ru3+ and its oxidation levels were crucial for the catalytic activity, and a volcano shaped relationship was established. On the basis of systematic characterizations, adsorbed superoxo- or peroxo-like species depending on the content of Ru3+ drove the CDC reactions, which was the essence of the as-observed structure–activity relationship.
Results and discussion
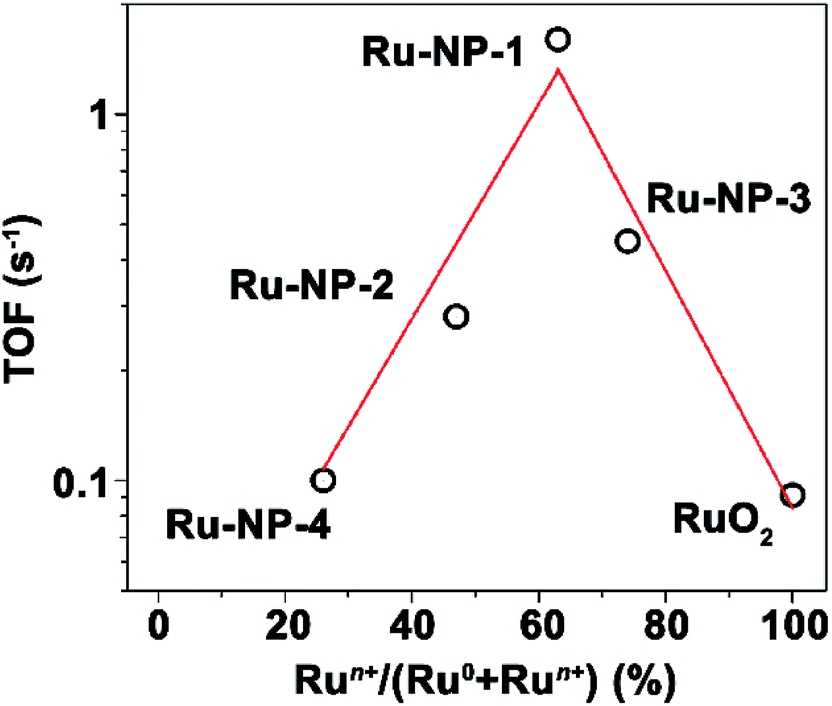
Ru NPs used in this work were prepared using a hydrothermal strategy, in which ruthenium chloride (RuCl3·xH2O), formaldehyde and poly(vinylpyrrolidone) (PVP) were used as precursor, reducing agent and particle stabilizer, respectively. Sodium oxalate (Na2C2O4), which can form stable complexes with Ru2+ and Ru3+ ions,33 was also introduced to avoid the rapid and complete reduction of the Ru precursor. Therefore, it was expected that Ru NPs with a considerable amount of Ru atoms with a positive valency would be obtained. The Ru NPs obtained after 8 h (denoted as Ru-NP-1) mainly contains 70 wt% of hexagonal-closest-packed (hcp) ultra-thin (thickness of 1.4 nm) plates with two {0001} facets as a basal surface, and the other 30 wt% of the NPs was comprised of poorly crystallized nanospheres. Transmission electron spectroscopy (TEM) images, and X-ray diffraction (XRD) patterns were presented in the ESI; Fig. S1 and S2,† and the size distributions of all the samples are listed in Table S1.†The Ru NPs were introduced into the CDC reaction of tetrahydroisoquinolines via oxidative C(sp3)–H functionalization.34–36 With the optimization of reaction conditions (Table S5; ESI†), the Ru-nanocatalyzed CDC reaction of tetrahydroisoquinolines and nucleophiles such as indole and nitromethane performed with excellent selectivity and reactivity (Fig. 1). These Ru NPs converted the traditionally homogeneous CDC reaction with tough oxidants, energy supplement or/and high temperature into a heterogeneous process with a simple procedure, namely, just stirring under ambient conditions (room temperature, cheap solvents methanol (CH3OH)/water (H2O), open flask, and free of light or thermal energy supplement). Turnover frequency (TOF) was as high as 33.6 s−1 under 60 °C. The recycling capability of Ru NPs was also examined, and the yields of recovered Ru NPs changed slightly. The listed yields of product 4a were 97%, 93%, 87%, 96%, 86%, 77% after six catalytic cycles.
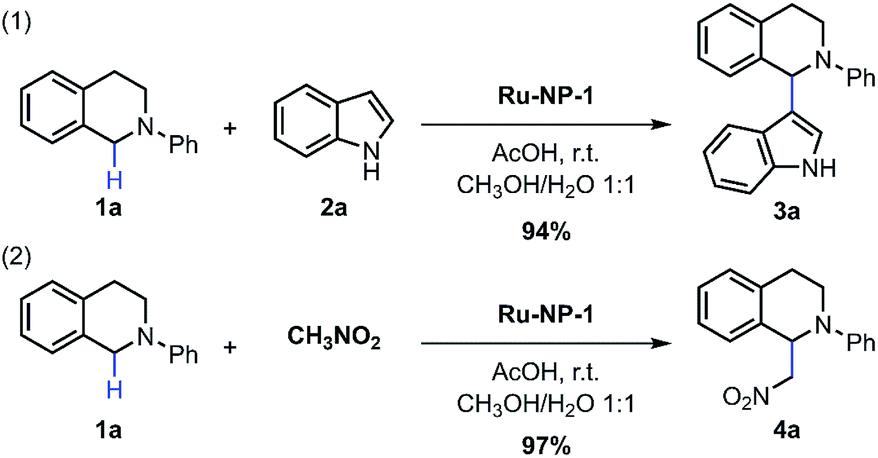 | ||
| Fig. 1 Ru-nanocatalyzed CDC reactions. Reaction conditions: 8 mol% of Ru NPs, 1 equiv. of 1, 4 equiv. of 2, 48 μL acetic acid (AcOH), 0.025 M in concentration. Isolated yield of 3a after column chromatography and the yield of product 4a was determined using 1H-NMR with 4-chloroacetophenone as an internal standard. | ||
It is difficult to distinguish homogeneous and heterogeneous catalysis, because of the possibility that metal ions may leach from a heterogeneous catalyst into solution and act as the homogeneous active species. Therefore, it was necessary to test the Ru ions leached from NPs in the reaction system. Firstly, as revealed by using in situ X-ray adsorption spectroscopy (see Fig. 2), the spectra of the sample at different reaction times did not show obvious differences, indicating that the oxidation level of the Ru element in the NPs remained unchanged during the catalytic reaction. Secondly, no Ru element was detected using inductively coupled plasma – atomic emission spectroscopy (ICP-AES) in the reaction solvent mixture of CH3OH–H2O after removal of Ru NPs. Thirdly, no significant changes in the morphology and oxidation level were observed, as revealed by the TEM image and X-ray photoelectron spectroscopy (XPS) spectra shown in Fig. S3 and S4 (ESI†), indicating that Ru NPs were stable during this reaction. Finally, the recovered solution was also used after removing Ru NPs to catalyse the model substrates and no reaction occurred. Thus, it was verified that the Ru NPs did not leach to form a homogeneously active catalyst.
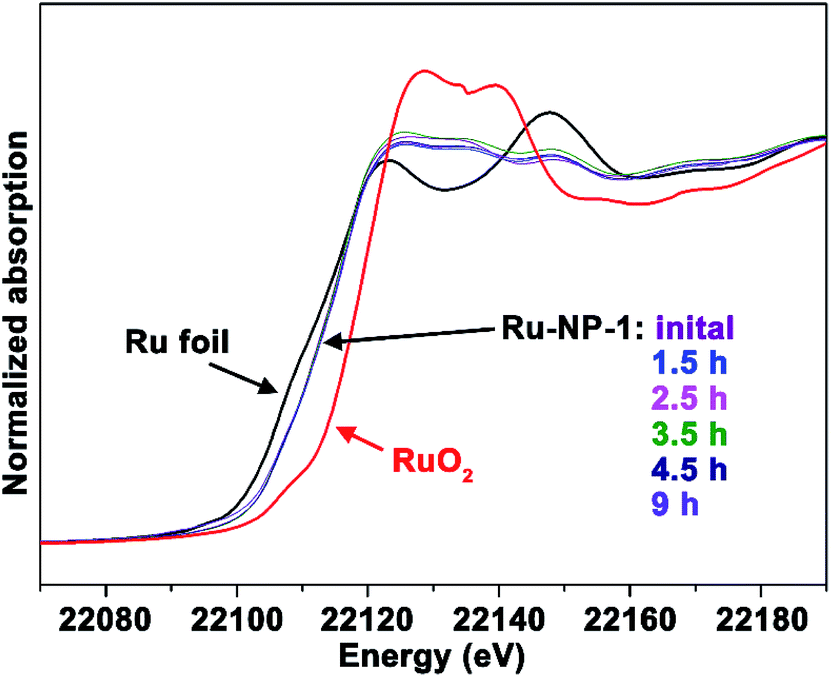 | ||
| Fig. 2 Ru K-edge in situ X-ray adsorption spectroscopy monitoring of Ru-NP-1 during the catalytic reaction. The spectra were taken before the reaction and during the reaction at of 1.5 h, 2.5 h, 3.5 h, 4.5 h and 9 h. The spectra of the Ru foil and RuO2 powders were used as references. | ||
A study of the mechanism of the heterogeneous CDC reaction helped to understand the catalytic behaviour and function of NPs towards organic substrates. The results of control experiments used to test the importance of oxygen are presented in Table S7.† The results confirmed that oxygen is necessary in this reaction and more oxygen yields a faster conversion rate of substrates. Furthermore, electron paramagnetic resonance (EPR) spectroscopy showed the existence of superoxide radicals (O2˙−) that were generated when oxygen molecules captured one electron in this reactive system, but no radicals were detected when the substrates or the catalyst was absent (Fig. 3).37 Thus, O2 + e− = O2˙−, and this process not only suggests that this CDC reaction proceeds as a single-electron-transfer (SET) catalysis, but also confirmed that oxygen was the electron acceptor.
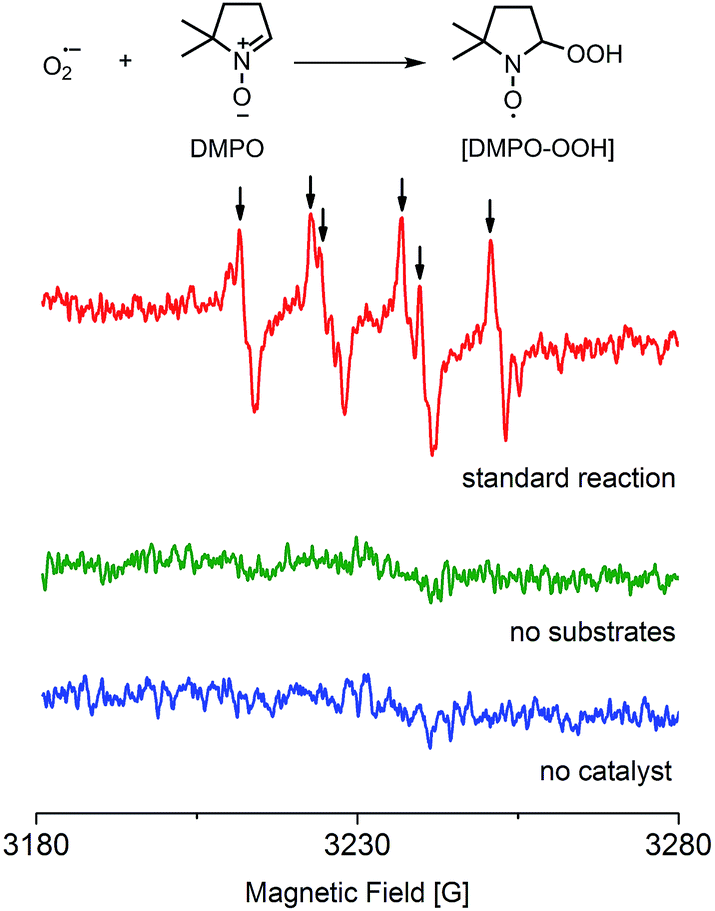 | ||
| Fig. 3 EPR spectrum of [DMPO–OOH] formed in the standard reactive system of Ru-nanocatalyzed CDC reactions (microwave frequency: 9056.103 MHz, microwave power: 4.0 mW). There are classical six peaks of the signals corresponding to [DMPO–OOH]. The calculated hyperfine splittings are g0 (2.002), αH (12.5 G), αN (14.0 G). | ||
Some previous work has shown that some adsorbed radicals, especially reactive oxygen species, can form on Ru-based hetero- or homogeneous catalysts and are active sites for the catalytic reaction.38,39 In order to study the relationship between the structure and the activity of the Ru NPs in the solution, EPR was used to detect radicals on the surface of Ru NPs which possibly mediated the SET process (Fig. 4). First, very broad and intensive signals with g tensor components g1 = 2.19, g2 = 2.04 and g3 = 1.88 ascribed to the Ru3+ species were observed on Ru-NP-1. If the as-synthesized Ru-NP-1 was post-reduced using hydrogen (H2) under hydrothermal conditions (denoted as Ru-NP-2), the signal of Ru3+ became weaker than that of Ru-NP-1 with the same amount of Ru element. The RuO2 powders considered as the over-oxidized sample showed little sign of Ru3+ species. Meanwhile, the SET reactions were used to test the catalytic activity of the three Ru catalysts. Among the Ru nanocatalysts, Ru-NP-1 showed the highest TOF value (up to 33.6 s−1), and the reaction achieved the final product with full conversion and an excellent yield as high as 94% (Table 1). Thus, it was supposed that the content of Ru3+ in the Ru catalysts was related to the catalytic activity.
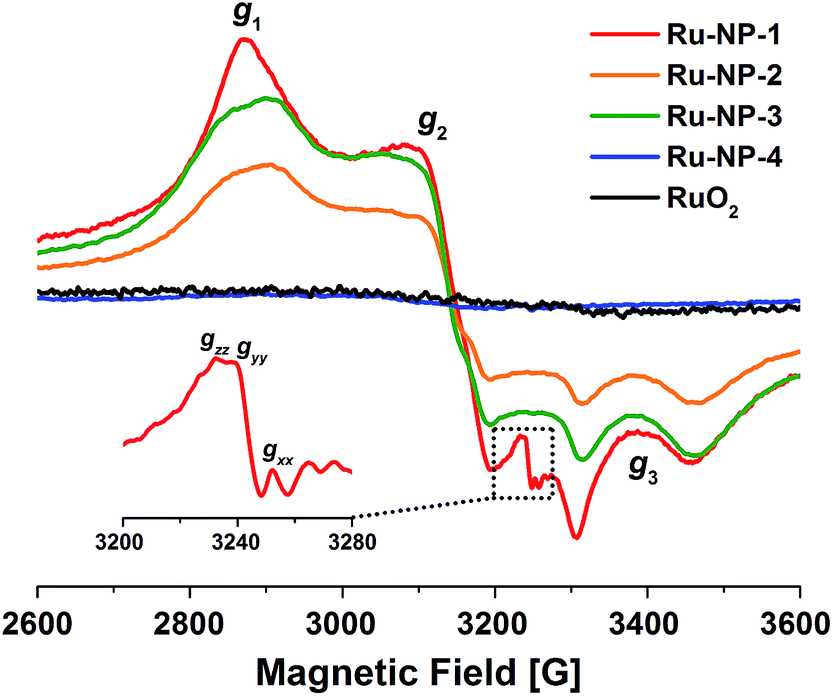 | ||
| Fig. 4 EPR spectra of Ru-NP-1 (red curve), Ru-NP-2 (orange curve), Ru-NP-3 (green curve) and Ru-NP-4 (blue curve) all as solid powders and RuO2 powders (black curve) of the same mass collected at 123 K. For Ru-NP-1, a signal was superimposed on the signal of Ru3+ species (g1 = 2.23, g2 = 2.04, g3 = 1.86), which was ascribed to the superoxo- or peroxo-like species (*O2˙−) adsorbed on the Ru catalyst, as shown in the inset. | ||
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Equivalentb [mol%] | Time [h] | Conversion [%] | Yieldc [%] | TOFd [s−1] |
a Reaction conditions: 0.10 mmol of 1a, 4 equiv. of 2a, 48 μL of 0.025 M AcOH, solvent: H2O/CH3OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), room temperature.b Molar ratio of Ru element to reagent 1a. ICP-AES was used for the quantification of the Ru element.c Isolated yield after column chromatography.d The TOF was obtained with the conversion of substrate 1a being lower than 15%. The methods to calculate the number of catalytic sites are shown in the ESI.e Reaction temperature was 60 °C. :1), room temperature.b Molar ratio of Ru element to reagent 1a. ICP-AES was used for the quantification of the Ru element.c Isolated yield after column chromatography.d The TOF was obtained with the conversion of substrate 1a being lower than 15%. The methods to calculate the number of catalytic sites are shown in the ESI.e Reaction temperature was 60 °C. |
||||||
| 1 | Ru-NP-1 | 8 | 42 | 100 | 94 | 1.6 |
| 2 | Ru-NP-1 | 8 | 2 | 100 | 93 | 33.6e |
| 3 | Ru-NP-2 | 8 | 72 | 41 | 34 | 0.28 |
| 4 | Ru-NP-3 | 8 | 72 | 73 | 57 | 0.45 |
| 5 | Ru-NP-4 | 8 | 72 | 74 | 45 | 0.10 |
| 6 | RuO2 | 80 | 45 | 30 | 18 | 0.091 |
Ru NPs with different Ru3+ contents were synthesized by just prolonging the time of synthesis of Ru-NP-1 to 16 h and 24 h, denoted as Ru-NP-3 and Ru-NP-4, respectively. As shown in Fig. S1 (ESI) and Table S1 (ESI†), the thickness of the plate-shaped NPs in these two samples remained the same as that of Ru-NP-1 (1.4 nm), and the morphology selectivity of the plates were also similar (77 wt% for Ru-NP-3 and 78 wt% for Ru-NP-4). The EPR results showed that the Ru3+ content of Ru catalysts with the same mass was Ru-NP-1 > Ru-NP-3 > Ru-NP-2 > Ru-NP-4 > RuO2 (see the magnified detail in Fig. 4). Table 1 compares the apparent TOF of different catalysts, which is defined as the average number of reactant molecules consumed on one surface Ru site per second (see ESI† for the calculation process). Different Ru nanocatalysts exhibited quite different catalytic activities under the same ambient conditions. The higher the content of Ru3+ in the nanocatalysts is, the higher the catalytic activity toward CDC reactions is.
More importantly, an obvious signal ascribed to adsorbed superoxo- or peroxo-like species (*O2˙−)40–42 was observed in the EPR spectrum of Ru-NP-1, the sample with highest content of Ru3+ species. The *O2˙− species might form because of the electron transfer from the Ru sites on the surface of Ru NPs to oxygen molecules, as shown by:
| (Run+)surf + O2 → (Ru(n+1)+)surf − *O2˙− |
This process is more likely to occur on Ru3+ sites on the surface of Ru NPs. The signals of the *O2˙− species were not detected for Ru-NP-2 and RuO2, which had a lower Ru3+ content. If benzoquinone was added to the reaction as a radical quencher of *O2˙− species, no reactions occurred on Ru-NP-1 (entry 4 in Table S7, ESI†), indicating that the *O2˙− species mediated the SET process. Based on the previous characterizations, it is proposed that the oxidation level of Ru nanocatalysts determines the content of Ru3+ sites on which *O2˙− species can form and mediate the SET process. Therefore, Ru nanocatalysts with a moderate oxidation level may trigger the CDC reactions of tetrahydroisoquinolines and achieve excellent catalytic performances.
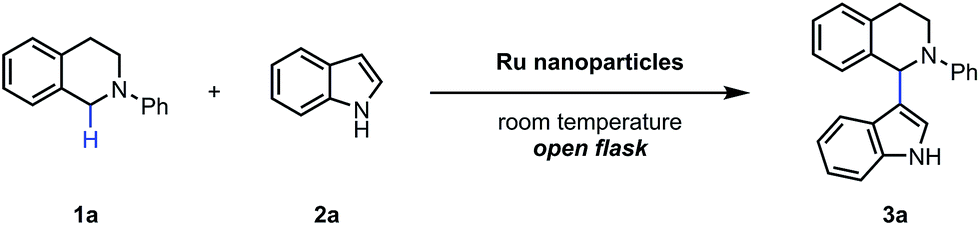
The content of Ru3+ depends on the oxidation level of Ru NPs. To investigate the relationship between the catalytic activity and oxidation levels of Ru NPs, an X-ray adsorption fine structure (XAFS) analysis of different Ru nanocatalysts was performed. Fig. 5a shows the X-ray absorption near edge structure (XANES) spectra of Ru-NP-1, Ru-NP-2, Ru-NP-3, Ru-NP-4, Ru foil and RuO2. The higher energy of the Ru K-edge reflects its higher oxidation level. Thus, the oxidation levels of different Ru NPs were in the sequence of Ru-NP-3 > Ru-NP-1 > Ru-NP-2 > Ru-NP-4. This result was consistent with the R-space extended X-ray absorption fine structure (EXAFS) data shown in Fig. 5b. Fourier transformation (F.T.) of the k2-weighted extended EXAFS data showed two main peaks around 2.0 Å and 2.7 Å, attributed to Ru–O and Ru–Ru scattering, respectively.43 The ratio of the peak intensity of Ru–O scattering to Ru–Ru scattering reflected the sequence of oxidation levels, which showed the same trend as XANES. Combined with the activity data of the SET reactions, Ru-NP-1 with a moderate oxidation level showed the highest catalytic activity.
 | ||
| Fig. 5 (a) XANES spectra and (b) R-space EXAFS spectra and the curve fitting for Ru K-edge of Ru foil (red curve), Ru-NP-1 (blue curve), Ru-NP-2 (green curve), Ru-NP-3 (pink curve), Ru-NP-4 (orange curve) and RuO2 (black curve). | ||
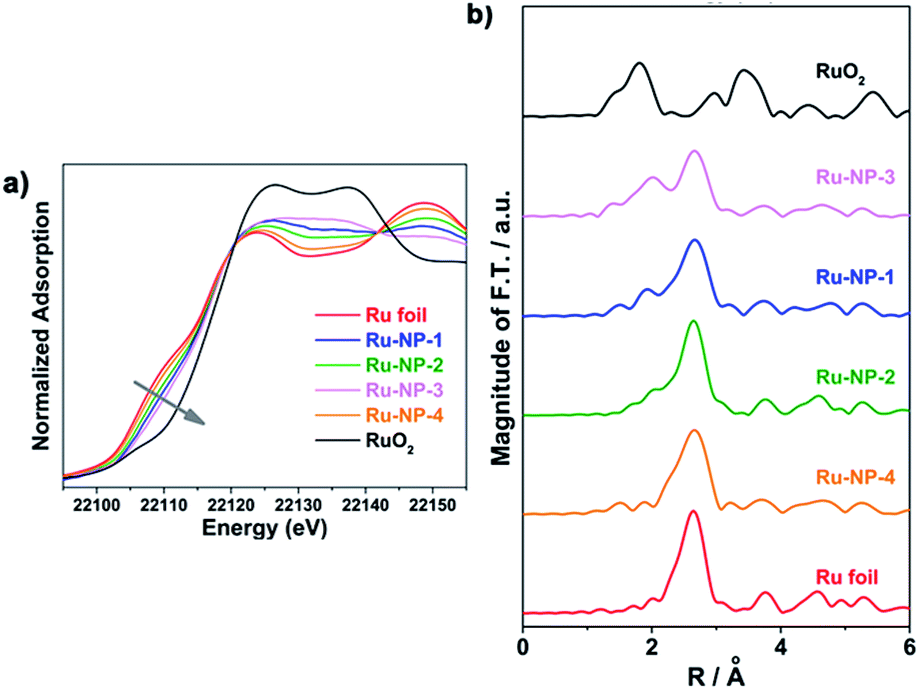
Next, XPS was used to investigate the surface oxidation level of the Ru NPs. Considering the probing depth of XPS and the ultra-thin nature of the Ru NPs, most Ru atoms in the sample could be detected in the analysis. As shown in Fig. 6a, the C-1s and Ru-3d regions of the samples were deconvoluted into three C-1s peaks at 284.80 eV (C–H), 285.80 eV (C–N) and 287.63 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and three Ru-3d doublets (3d5/2 and 3d3/2, Δ = 4.17 eV) with the binding energy (BE) of Ru 3d5/2 peak at 279.5–279.9 eV, 281.2–281.4 eV and 282.6–282.8 eV, assigned to the Ru(0), Ru(IV) and Ru(VI) states, respectively. The fitting results of the XPS spectra of Ru-3d electrons are listed in Table 2. Compared with Ru-NP-1, the post-reduced sample (Ru-NP-2) showed a significantly lower oxidation level. The oxidation level of Ru-NP-3 was higher than that of Ru-NP-1, probably because of the oxidizing effect of the oxygen sealed in the reactor, on the Ru NPs from 8 h to 16 h. As the reaction time was extended to 24 h, the Ru-NP-4 obtained showed greatly enhanced crystallinity (Fig. S2, ESI†) and became inert and were unable to be oxidized.44 Thus, Ru-NP-4 represented the lowest oxidation level. The XPS spectra in the Ru-3p regions showed the same trend of the oxidation levels of different Ru catalysts, as shown in Fig. 6b.
O), and three Ru-3d doublets (3d5/2 and 3d3/2, Δ = 4.17 eV) with the binding energy (BE) of Ru 3d5/2 peak at 279.5–279.9 eV, 281.2–281.4 eV and 282.6–282.8 eV, assigned to the Ru(0), Ru(IV) and Ru(VI) states, respectively. The fitting results of the XPS spectra of Ru-3d electrons are listed in Table 2. Compared with Ru-NP-1, the post-reduced sample (Ru-NP-2) showed a significantly lower oxidation level. The oxidation level of Ru-NP-3 was higher than that of Ru-NP-1, probably because of the oxidizing effect of the oxygen sealed in the reactor, on the Ru NPs from 8 h to 16 h. As the reaction time was extended to 24 h, the Ru-NP-4 obtained showed greatly enhanced crystallinity (Fig. S2, ESI†) and became inert and were unable to be oxidized.44 Thus, Ru-NP-4 represented the lowest oxidation level. The XPS spectra in the Ru-3p regions showed the same trend of the oxidation levels of different Ru catalysts, as shown in Fig. 6b.
 | ||
| Fig. 6 XPS spectra of Ru-NP-1, Ru-NP-2, Ru-NP-3, Ru-NP-4 and RuO2 powders. (a) The spectra in the C-1s and Ru-3d regions. Each spectrum was deconvoluted into C-1s peaks at 284.8 eV, 285.8 eV and 287.6 eV (violet curves), which were assigned to C atoms in the C–H, C–N and CO forms, respectively, and Ru-3d doublets (3d5/2 and 3d3/2, Δ = 4.17 eV) with the binding energy of Ru 3d5/2 peak at 279.5–279.9 eV, 281.2–281.4 eV and 282.6–282.8 eV which were assigned to Ru(0) (red curves), Ru(IV) (green curves) and Ru(VI) (blue curves) states, respectively.45–47 The fitting results of the Ru-3d spectra are shown in Table 2. (b) The spectra in the Ru-3p3/2 region. The red, green and blue vertical lines indicate the binding energy of Ru(0), Ru(IV) and Ru(VI) states, respectively.48,49 | ||
| Sample | Ru(0) | Ru(IV) | Ru(VI) | |||
|---|---|---|---|---|---|---|
| BE (eV) a | Ratio (%) | BE (eV) | Ratio (%) | BE (eV) | Ratio (%) | |
| a BE: binding energy of Ru 3d5/2 electrons. | ||||||
| Ru-NP-1 | 279.8 | 30 | 281.2 | 50 | 282.7 | 20 |
| Ru-NP-2 | 279.7 | 50 | 281.1 | 33 | 282.7 | 17 |
| Ru-NP-3 | 279.7 | 18 | 281.2 | 63 | 282.7 | 18 |
| Ru-NP-4 | 279.5 | 69 | 281.1 | 31 | — | 0 |
| RuO2 powders | — | 0 | 281.3 | 64 | 282.8 | 36 |
| Ru-NP-1 after catalysis | 279.9 | 30 | 281.4 | 50 | 282.9 | 20 |
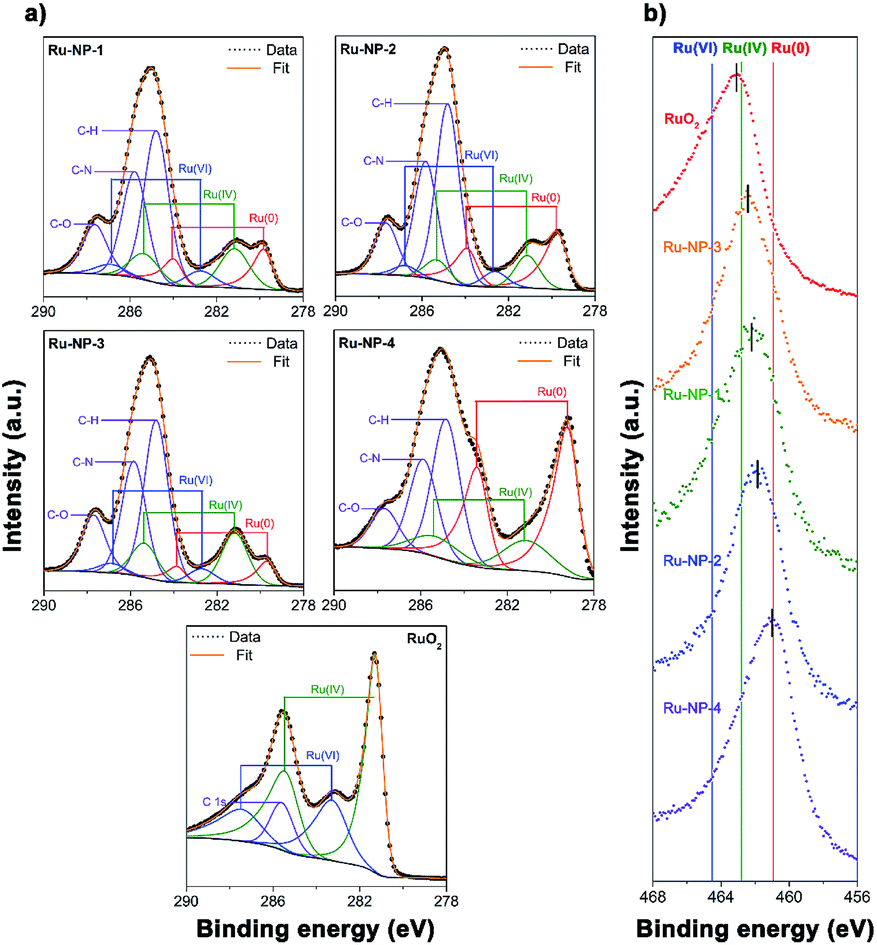
When the TOF values of all the Ru catalysts were plotted against their oxidation level (quantified as the percentage of surface Ru atoms with positive valences among all the surface Ru atoms deduced from the Ru-3d XPS spectra), a volcano shaped relationship was found, as shown in Fig. 7. The highest catalytic activity was achieved at a moderate oxidation level. The catalytic performances and the oxidation level of Ru NPs with other morphologies synthesized by hydrothermal methods were also investigated, and the results obtained agreed well with the volcano shaped relationship, as discussed in Fig. S5 and S6 (ESI) and Tables S2 and S6 (ESI†).
 | ||
| Fig. 7 Plot of the TOF values of all the catalysts against their oxidation level quantified as the percentage of surface Ru atoms with positive valencies among all the surface Ru atoms deduced from the Ru-3d XPS spectra. A volcano shaped relationship between the TOF values and the oxidation level is indicated by the red line. | ||
In this work, Ru nanocatalysts were synthesized using a hydrothermal method. With the understanding of the growth mechanism of the Ru NPs33 (Fig. S11–S13, ESI†), after the nucleation and growth steps, Ru NPs were oxidized mainly using subcritical water in hydrothermal synthesis,50–53 together with the generation of active *O2˙− species at the surfaces of the Ru NPs as revealed by the EPR measurements. Ru(0) was still the body constituent to support the structure and morphology of these Ru nanocatalysts with different oxidation levels. In the interface, the oxidized Ru nanocatalysts triggered the CDC reaction of tetrahydroisoquinoline derivatives via the SET process. The volcano shaped relationship between the catalytic activity and the oxidation level confirmed the as-proposed process on how Ru NPs catalyze the CDC reactions: the *O2˙− species formed on Ru3+ sites on the surface of Ru NPs were the active sites to mediate the SET process and connected the inorganic nanocatalysts and organic substrates. For the completely reduced Ru NPs, a few Ru3+ sites existed on the surface, whereas the Ru3+ sites first increased with increasing degree of oxidation of Ru NPs, and then decreased as the Ru4+ sites became dominant on the surface. The content of Ru3+ sites was highest on Ru NPs with a moderate oxidation level, which consequently generated more *O2˙− species by the electron transfer from oxygen molecules to the Ru3+ sites and the highest catalytic activity for the CDC reaction was exhibited with the sample of Ru-NP-1.
Based on the previous discussion, it is now possible to systematically clarify the mechanism as shown in Scheme 1. In the SET process, Ru NPs with superoxo- or peroxo-like species captured one single electron from tetrahydroisoquinoline derivative 1a with lone pair electrons to generate aminium radical cation A and transferred it to oxygen from the air to form the superoxide anion radical (O2˙−), which is the impetus of this reaction. Then radical intermediate B could be achieved by several conversions following the formation of aminium radical ion A and attack by nucleophiles yielding the final complex molecules. In this heterogeneous catalysis, it is supposed that a large amount of reactive oxygen radicals on the surface of Ru NPs, that are associated with their oxidation level, could drive the catalytic cycle and enhance the activities of Ru NPs for the CDC reactions. The detailed explanations of the organic mechanism including control experiments and proposed pathways are shown in Table S7 (ESI) and Fig. S14 (ESI†).
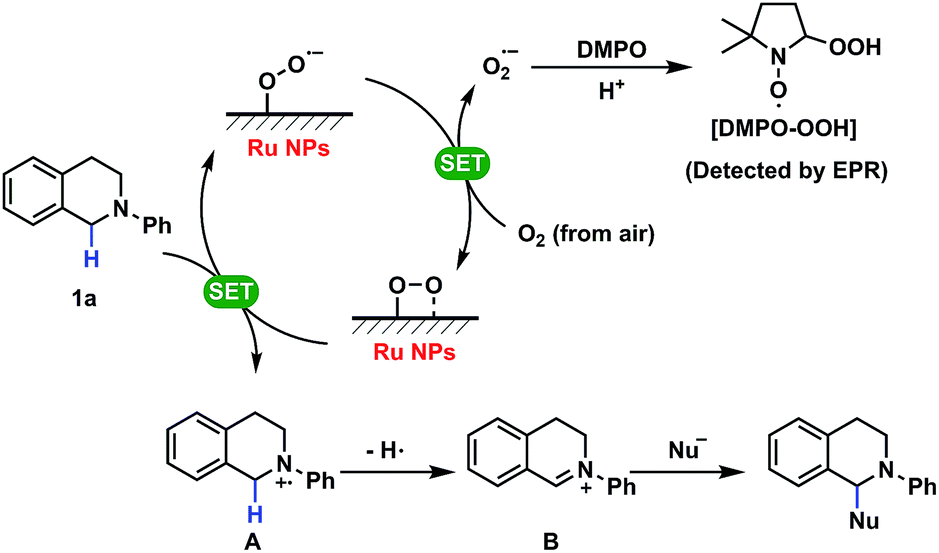 | ||
| Scheme 1 Mechanism of SET reactions catalyzed by Ru NPs. | ||
The general mechanism of the Ru-nanocatalyzed CDC reactions was further explored using the standard conditions [8 mol% Ru-NP-1, 0.10 mmol of N-phenyl tetrahydroisoquinoline derivatives, 4 equiv. of indoles, 10–48 μL of AcOH, H2O/CH3OH (1:1) as solvent, room temperature, concentration of 0.025 M (ESI†)] in the optimization studies. Significantly, all the positions of the substituted nucleophilic indoles were reactive and the yields were generally high (Fig. 8 3b–3j). The product (3d) that substitutes the indole at the 4-position has not been discovered before. In addition, the substrates substituted N-phenyl tetrahydroisoquinolines at all positions of N-aryl group perform high reactivity and the obtained products (3k–3o) are also first discovered. The reaction proceeds satisfactorily resulting in the formation of the crossing-coupling products with excellent yields (4a and 5). All the experimental results mentioned previously, shows that Ru-NP-1 with a moderate oxidation level presented excellent catalytic performance and a broad applicability, by virtue of the powerful single electron grabbing capacity under ambient conditions.
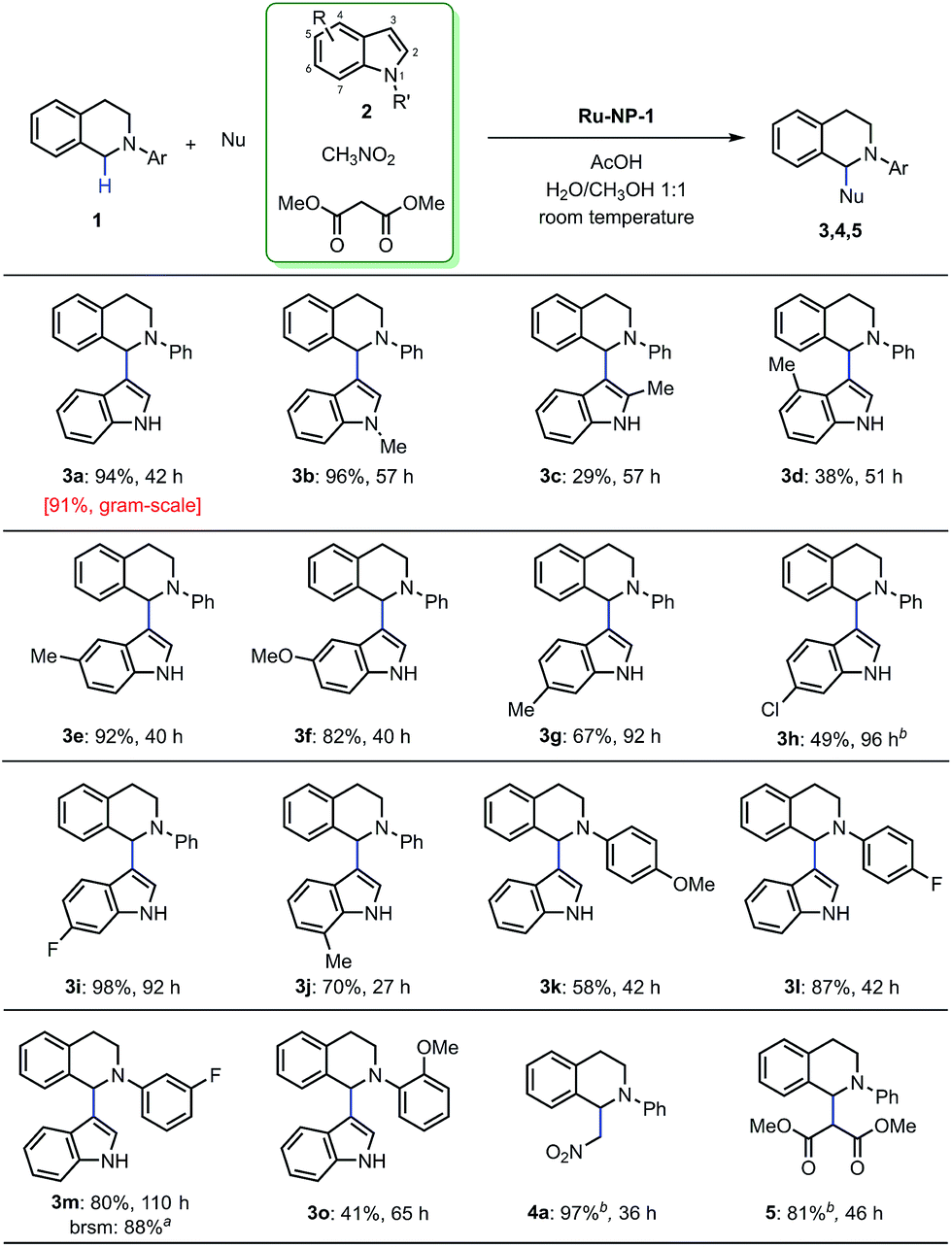 | ||
| Fig. 8 Ru NP catalyzed CDC reactions. Reaction conditions: 8 mol% of Ru nanocatalyst, 1 equiv. of 1, 4 equiv. of 2, 10–48 μL of AcOH, 0.025 M in concentration. Isolated yields were obtained after column chromatography, except that the yields of products 4a and 5 were determined using 1H-NMR using 4-chloroacetophenone as an internal standard. a Recovered 10% of 1d, 88% of yield based on the recovered starting material (brsm). b Solvent of 3h, 4a and 5 is CH3OH. | ||
Conclusions
A novel Ru nanocatalyst with a different oxidation level was synthesized using a hydrothermal strategy. The Ru NPs with a moderate oxidation level exhibited excellent reactivity and selectivity for the CDC reaction of tetrahydroisoquinolines under ambient conditions. This heterogeneous catalytic process was free of energy supplement, sacrificial reductants or sacrificial oxidants compared to homogeneous catalysis, and can be conducted at the gram scale. It was proposed and confirmed that there was a heterogeneous catalytic mechanism of Ru-nanocatalyzed CDC reactions. Ru NPs firstly capture one electron from amine substrates to generate an aminium radical cation and subsequently transfer the electron to the oxygen. Then, the active aminium radical cation could react with nucleophiles to form complex structures. The active aminium radical cation can be obtained for a series of available substrates. The catalytic behaviors present a volcano shaped correlation to the oxidation level of the NPs. The Ru3+ content was the highest for Ru NPs with a moderate oxidation level, which consequently generated more *O2˙− species by electron transfer from Ru3+ sites to oxygen molecules and exhibited a higher catalytic activity to trigger this SET process.By introducing catalytically active NPs into the SET process of CDC reactions, not only were the underlying principles and the associated dominant factors of nanocatalysis for the development of efficient and versatile approaches to heterogeneous catalysis under ambient conditions studied, but the results also provided a thorough insight into the relationship between the inorganic nanocatalysts and organic methodologies. Extended research with other NPs is necessary to facilitate the development of heterogeneous catalysts with high reactivity and selectivity in an even larger array of chemical reactions.
Experiments
Hydrothermal synthesis of Ru nanocatalysts
In a typical synthesis, 100 mg of PVP and 80 mg of Na2C2O4 were first dissolved in 10 mL of water, followed by addition 0.24 mmol RuCl3·xH2O, 0.062 mL of hydrochloric acid (1 M) and 0.1 mL of formaldehyde solution (40 wt%). The solution was then diluted to 15 mL, loaded in to a 25 mL Teflon-lined container and sealed in a matched steel autoclave. The autoclave was then heated in an oven kept at 160 °C for 8 h, 16 h or 24 h for the synthesis of Ru-NP-1, Ru-NP-3 and Ru-NP-4, respectively. After the reaction, the autoclave was cooled naturally to 25 °C and 45 mL of acetone was added and the product was then collected by centrifugation at 7800 rpm for 10 min. The yield of Ru NPs using the hydrothermal method was measured using ICP-AES and found to be about 85%.A post-reduction process generating Ru-NP-2 was also used to lower the oxidation level of Ru-NP-1. The as-obtained Ru-NP-1 was dispersed in 15 mL of water containing 100 mg of PVP. The dispersion was loaded into a 50 mL Teflon-lined reactor which was then filled with 2.0 MPa of H2. The reduction process was kept at 150 °C for 4 h. After the reaction, 45 mL acetone was added and the NPs were collected by centrifugation at 7800 rpm for 10 min.
General procedure of Ru-nanocatalyzed CDC reactions
The concentration of a Ru NP colloidal CH3OH solution was determined using ICP-AES. Ru nanocatalysts with the same Ru content (8 mol% of the substrate content) were added into the solvents [H2O/CH3OH (1:1)] with 0.10 mmol of tetrahydroisoquinoline derivatives, 4 equiv. of indoles and 10–48 μL of AcOH. The mixture (concentration: 0.025 M) was stirred under room temperature. The reaction tube was immersed in water bath under room temperature. When the thin layer chromatography (TLC) indicated the disappearance of the starting material, the reaction mixture was filtered through a thin pad of silica gel. The filter cake was washed with petroleum ether/ethyl acetate, and the combined filtrate was concentrated. The crude product was purified using flash column chromatography on silica gel to give the final product.
Acknowledgements
The Authors wish to thank the National Natural Science Foundation of China (No. 21461162001, 21371011, 21621061, 21229101, 21573005 and 21331001), the National Basic Research Program of China (No. 2012CBA01204), the National Key Research and Development Program of China (no. 2016YFB0701100), the Beijing Natural Science Foundation (No. 2162019), and the 7th China Postdoctoral Science Foundation Funded Project (No. 2014T70009) for financial support. ML was also partly supported by a Postdoctoral Fellowship of Peking-Tsinghua Center for Life Sciences.Notes and references
- G. A. Somorjai, Chem. Rev., 1996, 96, 1223 CrossRef CAS PubMed.
- T. S. Ahmadi, Z. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924 CrossRef CAS PubMed.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed.
- C. Wang, H. Daimon, T. Onodera, T. Koda and S. H. Sun, Angew. Chem., Int. Ed., 2008, 47, 3588 CrossRef CAS PubMed.
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 437, 121 CrossRef CAS PubMed.
- Y. Wu, D. S. Wang and Y. D. Li, Chem. Soc. Rev., 2014, 43, 2112 RSC.
- X. Q. Huang, Z. P. Zhao, J. M. Fan, Y. M. Tan and N. F. Zheng, J. Am. Chem. Soc., 2011, 133, 4718 CrossRef CAS PubMed.
- A. R. Tao, S. Habas and P. D. Yang, Small, 2008, 4, 310 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. D. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097 CrossRef CAS PubMed.
- Y. Wu, S. F. Cai, D. S. Wang, W. He and Y. D. Li, J. Am. Chem. Soc., 2012, 134, 8975 CrossRef CAS PubMed.
- S. B. Wang, W. Zhu, J. Ke, M. Lin and Y. W. Zhang, ACS Catal., 2014, 4, 2298 CrossRef CAS.
- L. X. Dai, W. Zhu, M. Lin, Z. P. Zhang, J. Gu, Y. H. Wang and Y. W. Zhang, Inorg. Chem. Front., 2015, 2, 949 RSC.
- M. Lin, L. Q. Kang, J. Gu, L. X. Dai, S. B. Tang, T. Zhang, Y. H. Wang, L. D. Li, X. Y. Zheng, W. Zhu, R. Si, X. F. Fu, L. D. Sun, Y. W. Zhang and C. H. Yan, Nano Res., 2017, 10, 922 CrossRef CAS.
- L. J. Durndell, C. M. A. Parlett, N. S. Hondow, M. A. Isaacs, K. Wilson and A. F. Lee, Sci. Rep., 2015, 5, 9425 CrossRef CAS PubMed.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed.
- J. M. Brown and N. A. Cooley, Chem. Rev., 1988, 88, 1031 CrossRef CAS.
- Y. N. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem.–Asian J., 2014, 9, 26 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed.
- S. A. Girard, T. Knauber and C. J. Li, From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling, 2014 Search PubMed.
- X. Lu, B. Xiao, Z. Q. Zhang, T. J. Gong, W. Su, J. Yi, Y. Fu and L. Liu, Nat. Commun., 2016, 7, 11129 CrossRef PubMed.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed.
- W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761 RSC.
- A. Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2004 Search PubMed.
- C. A. Parrodi and P. J. Walsh, Angew. Chem., Int. Ed., 2009, 48, 467925 Search PubMed.
- Z. Shao and F. Peng, Angew. Chem., Int. Ed., 2010, 49, 9566 CrossRef CAS PubMed.
- S. Gupta, H. Joshi, N. Jain and A. K. Singh, J. Mol. Catal. A: Chem., 2016, 423, 135 CrossRef CAS.
- R. Hudson, S. lshikawa, C. J. Li and A. Moores, Synlett, 2013, 24, 1637 CrossRef CAS.
- T. Zeng, G. Song, A. Moores and C. J. Li, Synlett, 2010, 13, 2002 Search PubMed.
- Q. Y. Meng, Q. Liu, J. J. Zhong, H. H. Zhang, Z. J. Li, B. Chen, C. H. Tung and L. Z. Wu, Org. Lett., 2012, 14, 5993 Search PubMed.
- G. Ertl, Angew. Chem., Int. Ed. Engl., 1990, 29, 1219 CrossRef.
- H.-J. Freund, G. Meijer, M. Scheffler, R. Schlögl and M. Wolf, Angew. Chem., Int. Ed., 2011, 50, 10064 CrossRef CAS PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524 CrossRef CAS PubMed.
- A. X. Yin, W. C. Liu, J. Ke, W. Zhu, J. Gu, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2012, 134, 20479 CrossRef CAS PubMed.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed.
- Q. Y. Meng, J.-J. Zhong, Q. Liu, X.-W. Gao, H.-H. Zhang, T. Lei, Z.-J. Li, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2013, 135, 19052 CrossRef CAS PubMed.
- K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed.
- J.-L. Clément, N. Ferré, D. Siri, H. Karoui, A. Rockenbauer and P. Tordo, J. Org. Chem., 2005, 70, 1198 CrossRef PubMed.
- M. Matsushita, K. Kamata, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 6632 CrossRef CAS PubMed.
- D. Moonshiram, J. W. Jurss, J. J. Concepcion, T. Zakharova, I. Alperovich, T. J. Meyer and Y. Pushkar, J. Am. Chem. Soc., 2012, 134, 4625 CrossRef CAS PubMed.
- G.-D. Lei and L. Kevan, J. Phys. Chem., 1991, 95, 4506 CrossRef CAS.
- H. Tsunoyamo, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Soc. Chem., 2009, 131, 7086 CrossRef PubMed.
- M. D'Arienzo, J. Carbajo, A. Bahamonde, M. Crippa, S. Polizzi, R. Scotti, L. Wahba and F. Morazzoni, J. Am. Soc. Chem., 2011, 133, 17652 CrossRef PubMed.
- T. Mitsudome, Y. Takahashi, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2014, 53, 8348 CrossRef CAS PubMed.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, J. Am. Chem. Soc., 2013, 135, 8016 CrossRef CAS PubMed.
- A. Lewera, W. P. Zhou, C. Vericat, J. H. Chung, R. Haasch, A. Wieckowski and P. S. Bagus, Electrochim. Acta, 2006, 51, 3950 CrossRef CAS.
- V. Mazzieri, F. Coloma-Pascual, A. Arcoya, P. C. L'Argentière and N. S. Figoli, Appl. Surf. Sci., 2003, 210, 222 CrossRef CAS.
- R. Bavand, A. Yelon and E. Sacher, Appl. Surf. Sci., 2015, 355, 279 CrossRef CAS.
- R. Kotz, H. J. Lewerenz and S. Stucki, J. Electrochem. Soc., 1983, 130, 825 CrossRef.
- Z. L. Liu, J. Y. Lee, M. Han, W. X. Chen and L. M. Gan, J. Mater. Chem., 2002, 12, 2453 RSC.
- H. Hayashi, A. Ueda, A. Suino, K. Hiro and Y. Hakuta, J. Solid State Chem., 2009, 182, 2985 CrossRef CAS.
- S. A. Simakov and Y. Tsur, J. Nanopart. Res., 2007, 9, 403 CrossRef CAS.
- S. Yu, S. Li, X. Ge, M. Niu, H. Zhang, C. Xu and W. Li, Ind. Eng. Chem. Res., 2014, 53, 2238 CrossRef CAS.
- N. Lock, M. Bremholm, M. Christensen, J. Almer, Y.-S. Chen and B. B. Iversen, Chem.–Eur. J., 2009, 15, 13381 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra05726a |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2017 |