
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versatile magnetic resonance singlet tags compatible with biological conditions†
Stefan
Glöggler
 *,
Stuart J.
Elliott
,
Gabriele
Stevanato
,
Richard C. D.
Brown
and
Malcolm H.
Levitt
*,
Stuart J.
Elliott
,
Gabriele
Stevanato
,
Richard C. D.
Brown
and
Malcolm H.
Levitt
School of Chemistry, University of Southampton, Southampton SO171BJ, UK. E-mail: stefan.gloeggler@mpibpc.mpg.de
First published on 10th July 2017
Abstract
The long lifetime of nuclear singlet states holds promise for the development of molecular tracers to study motional processes in proteins with increased precision or to act as imaging contrast agents. We introduce a singlet tag (STAG) based on bromoacrylate that is readily attached to a variety of biomolecules, compatible with biological conditions, and is relatively insensitive to the presence of molecular oxygen. The generality of this approach is demonstrated by tagging buildings blocks for biomolecules using a simple labelling route based on commercially available starting materials. Additionally, we demonstrate a singlet NMR experiment on the endogenous molecule phosphoenolpyruvate.
Introduction
Nuclear singlet states are NMR “silent” states that do not give rise directly to NMR signals but which may be populated and detected indirectly.1–30 Such a state is created e.g. if the spins of two I = 1/2 nuclei couple antiparallel to each other to form a total spin of 0. The singlet spin state is immune to the motional modulation of the in pair dipole–dipole interaction, with other relaxation mechanisms strongly attenuated. The time constant (TS) for the equilibration of the singlet state population with the triplet populations can exceed the longitudinal relaxation time (T1) by more than one order of magnitude, with a TS value exceeding 1 hour being observed in one case.13 Various applications of singlet states have been proposed, including the utilization of long TS times to probe slow dynamic processes in biomolecules and the binding of drugs,6,10,15–17 and as a means to support nuclear hyperpolarization in the context of molecular imaging.18,21,23,28,31–43 In some cases, the protons of biomolecules support nuclear singlet states. Examples include molecules relevant to neuroscience such as aspartate and glutamine, peptides, proteins and adenosinediphosphate (ADP).15–17,28,29 Proton singlet states with lifetimes TS up to 4.5 s have been reported under biologically compatible conditions (pH 7.0–7.4 and 25–37 °C) even in the presence of dissolved molecular oxygen and without deuteration of the solvent.28A more versatile approach is offered by artificial prosthetic groups supporting long-lived nuclear singlet states. In this paper we call such entities singlet tags (STAGs). Previously described examples include bromothiophene and fluorinated phenylacetyl moieties linked to tripeptides.28,29 The measured TS values were 11.7 s in D2O for the bromothiophene tag, and 2.6 s for the fluorinated phenylacetyl tag.28,29 However, in order to be applicable to in vitro and in vivo experiments, the singlet NMR phenomena should be insensitive to oxygen and functional in non-deuterated solvents (especially H2O rather than D2O, which is toxic for cell experiments and may change protein conformations). Ideally, the singlet states should be sustainable at high magnetic fields, and their 1H NMR resonances should be in a spectral region that does not overlap with the signal from water (4.7 ppm in a proton spectrum), signals from amino acids in proteins (aliphatic and aromatic functionalities, ranging from 0–5 ppm and 7–9 ppm in a proton spectrum) and typical signals from metabolites (0–5.3 ppm and 6.8–8.1 ppm in a proton spectrum).44,45
In this article we explore the use of substituted acrylates as STAGs. The acrylate moiety (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CO2−)) contains a pair of protons with similar chemical shifts and which are very close in space (internuclear distance ∼171.2 pm).46 The acrylate proton signals appear in a favourable chemical shift region around 6–7 ppm which does not typically overlap with other common proton signals. Acrylates have found a wide range of applications and are mostly used as monomers for polymerization reactions.47 Other important fields are their utilization as Michael acceptors, with particular use in click chemistry reactions48,49 and as precursors for imaging contrast agents.31–38,50–52 In the present case, the close proximity of the acrylate protons reduces the influence of relaxation agents such as paramagnetic species on nuclear singlet order, except at very short range. The relatively small shift difference of about 0.6 ppm between acrylate protons makes it feasible to spin-lock singlet states at high magnetic field by applying on resonant radiofrequency irradiation.4 We demonstrate a general synthetic route for bromoacrylate STAGs which utilizes inexpensive and commercially available reactants. The route allowed us to label several amino acids as well as a glucose derivative. We show that the bromoacrylate singlet lifetime in all of these compounds significantly exceeds T1 under conditions suitable for biological applications, i.e. in an aqueous phosphate buffer in the presence of molecular oxygen and without solvent deuteration. Furthermore, we have identified a metabolite, phosphoenolpyruvate, which contains an acrylate functionality and which supports a nuclear singlet state with a decay time constant TS more than four times longer than T1. Phosphoenolpyruvate becomes converted into pyruvate during glycolysis which converts into lactate in cancer cells.53
C(CO2−)) contains a pair of protons with similar chemical shifts and which are very close in space (internuclear distance ∼171.2 pm).46 The acrylate proton signals appear in a favourable chemical shift region around 6–7 ppm which does not typically overlap with other common proton signals. Acrylates have found a wide range of applications and are mostly used as monomers for polymerization reactions.47 Other important fields are their utilization as Michael acceptors, with particular use in click chemistry reactions48,49 and as precursors for imaging contrast agents.31–38,50–52 In the present case, the close proximity of the acrylate protons reduces the influence of relaxation agents such as paramagnetic species on nuclear singlet order, except at very short range. The relatively small shift difference of about 0.6 ppm between acrylate protons makes it feasible to spin-lock singlet states at high magnetic field by applying on resonant radiofrequency irradiation.4 We demonstrate a general synthetic route for bromoacrylate STAGs which utilizes inexpensive and commercially available reactants. The route allowed us to label several amino acids as well as a glucose derivative. We show that the bromoacrylate singlet lifetime in all of these compounds significantly exceeds T1 under conditions suitable for biological applications, i.e. in an aqueous phosphate buffer in the presence of molecular oxygen and without solvent deuteration. Furthermore, we have identified a metabolite, phosphoenolpyruvate, which contains an acrylate functionality and which supports a nuclear singlet state with a decay time constant TS more than four times longer than T1. Phosphoenolpyruvate becomes converted into pyruvate during glycolysis which converts into lactate in cancer cells.53
Results and discussion
The 1H NMR spectrum of bromoacrylic acid (BrAc, Fig. 1A), shows a distinct AX pattern in the range of 6–7 ppm. The shift difference between the acrylate protons is 0.6 ppm in H2O. The measured values of T1 and TS for BrAc solutions at two different magnetic fields and a selection of solvent preparations are shown in Table 1. The singlet relaxation times TS were estimated by using the pulse sequence of Sarkar et al. [ref. 6] as shown in Fig. 1C (see Table 1 of the ESI† for pulse sequence timings). Experiments exploited singlet spin-locking by applying an on resonant radiofrequency field with a nutation frequency of 3 kHz (at magnetic fields of 11.7 T and 22.2 T). Note that the values of TS are approximately four times larger than the values of T1 under the same conditions. The weak dependence of the relaxation times on the magnetic field strength indicate that chemical shift anisotropy is not a highly significant relaxation mechanism for this system. | ||
| Fig. 1 Investigation of the BrAc-STAG. (A) 1H NMR spectrum of bromoacrylic acid (BrAc) in H2O (PBS) at 11.7 T and 25 °C. The spectrum displays two doublets (AX-pattern) of the indicated protons in red, which are used to support singlet order. The NMR signal is in a region downfield of typical aliphatic protons and upfield of most aromatic protons found in metabolites. (B) Decay of proton singlet order in BrAc, observed using the pulse sequence in (C) in the presence of a spin-locking field (nutation frequency = 3 kHz). The observed singlet decay time constant TS is 5.7 times longer than T1 (TS = 14.7 ± 0.2 s, T1 = 2.8 ± 0.1 s). (C) Pulse sequence to generate singlet order, spin-lock it, and back-convert it into observable magnetization with subsequent acquisition of the signal [ref. 6 and 16]. A pulse field gradient is utilized as a filter to remove coherences that are not originating from the singlet state after back-conversion into observable magnetization. The timings τ1 and τ2 depend on the J-couplings and chemical shift difference of the protons, a table of which is displayed in the (ESI Table 1†). | ||
| Molecule | Magnetic field | Solvent | Oxygen condition | T 1/s | T S/s |
|---|---|---|---|---|---|
| BrAc | 11.7 T | D2O | Degassed | 3.0 ± 0.1 | 44.6 ± 1.0 |
| BrAc | 11.7 T | D2O | Non-degassed | 3.3 ± 0.1 s | 22.8 ± 2.0 |
| BrAc | 11.7 T | H2O (PBS) | Non-degassed | 2.8 ± 0.1 | 14.7 ± 0.2 |
| BrAc | 22.2 T | H2O (PBS) | Non-degassed | 2.9 ± 0.1 | 14.1 ± 0.5 |
| PEP | 11.7 T | H2O (PBS) | Non-degassed | 1.4 ± 0.1 | 12.5 ± 1.5 |
| PEP | 22.2 T | H2O (PBS) | Non-degassed | 1.6 ± 0.1 | 12.1 ± 0.1 |
Phosphoenolpyruvate (PEP, Fig. 2), is a metabolite of the glycolysis pathway which contains an acrylic moiety. Since glycolysis is up-regulated in a variety of cancer cells, PEP (as well as glucose derivatised STAGs) might serve as molecular imaging agents for cancer targeting. The 1H NMR spectrum of PEP is displayed in Fig. 2A. The shift difference between the acrylate protons is 0.4 ppm in H2O. The spectrum shows the characteristic J-splitting of the acrylic protons which is further split by the 4J-coupling of the protons to the 31P phosphorus nucleus (100% natural abundance, spin I = 1/2).
 | ||
| Fig. 2 Investigation of PEP. (A) 1H NMR spectrum of the two indicated protons in phosphoenol pyruvate in H2O (PBS) at a field of 11.7 T. The triplet-like multiplet structure is due to a superposition of the 2J coupling between the protons with an additional 4J coupling to the 31P nucleus (I = 1/2, natural abundance of close to 100%). (B) Decay of singlet order in PEP (the data points are within the standard error of 12%). The observed value of TS is 3.6 longer than T1. | ||
The measured values of T1 and TS for PEP solutions at two different magnetic fields in PBS (phosphate buffered saline, 360 mg NaH2PO4 and 266 mg Na2HPO4 in 300 mL ultrapure H2O) buffer are shown in Table 1. The nutation frequencies of the spin-locking fields for the TS measurements were 3 kHz at magnetic fields of 11.7 T and 22.2 T. The results are similar to those obtained for BrAc. The dependence of the relaxation times on magnetic field strength is again found to be weak, indicating that chemical shift anisotropy is not a strong relaxation mechanism for this system. We developed a general and robust route for coupling the bromoacrylate singlet tag to a variety of molecules. Since the hydrophilicity of bromoacrylic acid itself causes handling difficulties and promotes unwanted side reactions, we developed an alternative procedure (Fig. 3) starting from 2,3-dibromopropionic acid (or its acid chloride), which is subsequently converted into BrAc upon the addition of bases.51,54 2,3-dibromopropionic acid or its acid chloride underwent reactions with amines or alcohols, which are functional moieties found in a variety of metabolites, amino acids or peptides. In the presence of coupling reagents and a base we postulate that an intermediate product is formed based on the 2,3-dibromopropionic acid. However, the base slowly eliminates HBr from the intermediate structure so that in most cases the acrylic acid derivative is yielded in a one-pot process. Specific bases that we have utilized are 4-dimethylaminopyridine (DMAP), triethylamine (TEA) and diisopropylethylamine (DIPEA). The tagged molecules that we have synthesized and investigated are esters or amides, based on the following compounds: ethyl alcohol, propargyl alcohol, serine, alanine and acetylglucosylamine. To verify that different sites in amino acids can be tagged, alanine was labelled at the N-terminus and serine via its alcohol moiety. The labelling of propargylalcohol was chosen since the BrAc-STAG carrying an alkyne moiety from propargylalcohol may serve as a precursor for click reactions.55 The tagged structures are presented in Table 2. Singlet lifetimes in non-degassed PBS (unless indicated otherwise) are shown in Table 3. All of the demonstrated molecules show a significantly longer TS than T1, proving that singlet states with a significant larger TS/T1 ratio can be maintained in conditions that are compatible with biological investigations. Moreover, the presented STAGs may be utilized to label a variety of biomolecules or derivatives thereof and maintain their longevity. The STAGs described here require the application of a relatively strong radiofrequency field to quench singlet–triplet transitions. Applications which exploit the long singlet lifetime of the spin-locked singlet order may therefore be restricted to in vitro applications. To further improve the STAGs we intend to reduce the chemical shift difference of the protons, allowing operation with a very small or absent spin-locking field, leading to applicability in vivo without heating effects.
 | ||
| Fig. 3 General synthetic route for labelling molecules with BrAc-STAGs. For the labelling of alcohols or amines, 2,3-dibromopropionic acid or its acid chloride are utilized as a reagent. During the reaction a dibromo-intermediate is formed from which hydrobromic acid is eliminated upon reaction with a base. The intermediate is typically not isolated since the reactions are performed in the presence of bases (4-dimethylaminopyridine, triethylamine and diisopropylethylamine) and thus yield the final acrylate in a one-pot procedure. | ||
| Compound | Structure | Residue |
|---|---|---|
| 1 |
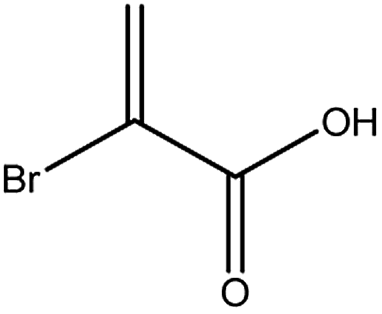
|
Free acid |
| 2 |

|
Ethyl |
| 3 |

|
Propargyl |
| 4 |
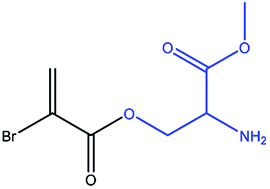
|
Serine |
| 5 |

|
Alanine |
| 6 |
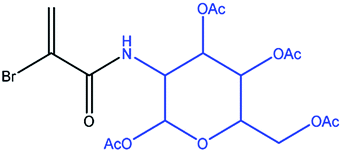
|
Glucose |
| 7 |
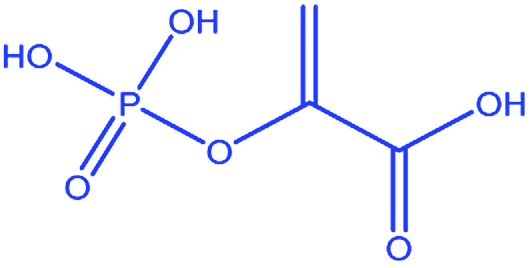
|
Phosphoenol pyruvate |
| Compound | T 1/s | T S/s | T S/T1 | B 0/T | c/mM | Spin-lock/kHz | Solvent |
|---|---|---|---|---|---|---|---|
| a Measured in MeOH due to limited solubility in PBS buffer. | |||||||
| 1 | 2.8 ± 0.1 | 14.7 ± 0.2 | 5.3 ± 0.2 | 11.7 | 10 | 3 | PBS |
| 2 | 2.4 ± 0.1 | 9.9 ± 0.3 | 4.1 ± 0.3 | 11.7 | 10 | 3 | PBS |
| 3 | 2.4 ± 0.1 | 13 ± 2 | 5 ± 2 | 11.7 | 10 | 3 | PBS |
| 4 | 1.4 ± 0.1 | 13.5 ± 0.5 | 9.6 ± 0.5 | 11.7 | 10 | 3 | PBS |
| 5 | 1.5 ± 0.1 | 11.9 ± 0.2 | 7.9 ± 0.2 | 11.7 | 10 | 3 | PBS |
| 6 a | 1.3 ± 0.1 | 8.3 ± 0.5a | 6.4 ± 0.5 | 11.7 | 10 | 3 | MeOH |
| 7 | 1.4 ± 0.1 | 13 ± 2 | 9 ± 2 | 11.7 | 10 | 3 | PBS |
Conclusions
We have identified haloacrylate STAGs that can be used as molecular labels for singlet NMR experiments in a variety of biological relevant molecules and which can be used under conditions relevant for biological investigations. The bromoacrylate STAG may be introduced by an efficient one-pot synthetic procedure that is readily performed with commercially available products. The metabolite phosphoenolpyruvate (PEP) contains an acrylate entity which also supports long-lived nuclear singlet order. This is a step towards metabolic imaging of PEP in living systems. Acrylate tags may also have applications in areas outside singlet NMR. Possible applications include their role as precursors for imaging contrast agents, including PET-tracers and hyperpolarized MRI tracers upon the hydrogenation with tritium or para-hydrogen respectively.31–38,50,51 The BrAc tag molecules may serve as new building blocks for Michael additions or thio–bromo reactions in click chemistry.49,56 Furthermore, we envision utilization of the haloacrylates as building blocks for polymerization reactions.Experimental details
NMR experiments were conducted on Bruker Avance III 500 or 950 MHz spectrometers with a Bruker TBO probe or a Bruker TCI cryo probe running TopSpin 3.2. software. Substrates 1–5 and 7 were dissolved in PBS buffer (360 mg NaH2PO4 and 266 mg Na2HPO4 in 300 mL ultrapure H2O) at pH = 7.0 at a concentration of 10 mM. 6 was dissolved in methanol (CH3OH). The pulse sequence displayed in Fig. 1C was used to perform singlet experiments for which the coupling parameters and respective timings are listed in the ESI.† The pulsed field gradient in the sequence had a sine shape with 5 Gcm-1 maximal amplitude and a duration of 3 ms. During the singlet sustaining period a continuous wave decoupling pulse with 3 kHz amplitude was applied, centred in between the two protons resonances of interest. A recycle delay of 80 s was used in protonated solvents and 300 s in D2O after sample degassing. 4 averages were acquired for each data point and experiments were repeated 6 times. Longitudinal relaxation times were estimated by the inversion recovery pulse sequence with 4 averages whereby each experiment was repeated 6 times. The recycle delay was set to 20 s.Acknowledgements
This research was supported by the Engineering and Physical Sciences Research Council (UK), grant codes EP/N002482 and EP/L505067/1, the Marie-Skłodowska-Curie program of the European Community (655864-SingMet), the Wolfson Foundation and Bruker Biospin (UK). We would like to acknowledge helpful support and discussion from L. J. Brown and G. Pileio. We would also like to acknowledge C. Griesinger and M. Zweckstetter (Max-Planck-Institute for Biophysical Chemistry and Deutsches Zentrum für Neurodegenerative Erkrankungen) for 950 MHz spectrometer access.Notes and references
- M. Carravetta and M. H. Levitt, J. Am. Chem. Soc., 2004, 126, 6228–6229 CrossRef CAS PubMed.
- M. Carravetta, O. G. Johannessen and M. H. Levitt, Phys. Rev. Lett., 2004, 92, 153003 CrossRef PubMed.
- M. Carravetta and M. H. Levitt, J. Chem. Phys., 2005, 122, 214505 CrossRef PubMed.
- G. Pileio and M. H. Levitt, J. Chem. Phys., 2009, 130, 214501 CrossRef PubMed.
- G. Pileio, M. Carravetta and M. H. Levitt, Proc. Natl. Acad. Sci. U. S. A., 2011, 107, 17135–17139 CrossRef PubMed.
- R. Sarkar, P. R. Vasos and G. Bodenhausen, J. Am. Chem. Soc., 2007, 129, 328–334 CrossRef CAS PubMed.
- A. S. Kiryutin, A. N. Pravdivtsev, A. V. Yurkovskaya, H.-M. Vieth and K. L. Ivanov, J. Phys. Chem. B, 2016, 120, 11978–11986 CrossRef CAS PubMed.
- G. Pileio, M. Carravetta, E. Hughes and M. H. Levitt, J. Am. Chem. Soc., 2008, 130, 12582–12583 CrossRef CAS PubMed.
- M. C. D. Tayler and M. H. Levitt, Phys. Chem. Chem. Phys., 2011, 13, 5556–5560 RSC.
- P. Ahuja, R. Sarkar, P. R. Vasos and G. Bodenhausen, J. Chem. Phys., 2007, 127, 134112 CrossRef PubMed.
- G. Pileio, M. Concistre, M. Carravetta and M. H. Levitt, J. Magn. Reson., 2006, 182, 353–357 CrossRef CAS PubMed.
- T. Theis, Y. Feng, T. Wu and W. S. Warren, J. Chem. Phys., 2014, 140, 014201 CrossRef CAS PubMed.
- G. Stevanato, J. T. Hill-Cousins, P. Håkansson, S. S. Roy, L. J. Brown, R. C. D. Brown, G. Pileio and M. H. Levitt, Angew. Chem., Int. Ed., 2015, 54, 3740–3743 CrossRef CAS PubMed.
- S. J. Elliott, L. J. Brown, J.-N. Dumez and M. H. Levitt, Phys. Chem. Chem. Phys., 2016, 18, 17965–17972 RSC.
- R. Buratto, D. Mammoli, E. Chiarparin, G. Williams and G. Bodenhausen, Angew. Chem., Int. Ed., 2014, 53, 11376–11380 CrossRef CAS PubMed.
- N. Salvi, R. Buratto, A. Bornet, S. Ulzega, I. R. Rebollo, A. Angelini, C. Heinis and G. Bodenhausen, J. Am. Chem. Soc., 2012, 134, 11076–11079 CrossRef CAS PubMed.
- R. Buratto, A. Bornet, J. Milani, D. Mammoli, B. Vuichoud, N. Salvi, M. Singh, A. Laguerre, S. Passemard, S. Gerber-Lemaire, S. Jannin and G. Bodenhausen, ChemMedChem, 2014, 9, 2509–2515 CrossRef CAS PubMed.
- Y. Feng, R. M. Davis and W. S. Warren, Nat. Phys., 2012, 8, 831–837 CrossRef CAS PubMed.
- A. K. Grant and E. Vinogradov, J. Magn. Reson., 2008, 194, 46–57 CrossRef PubMed.
- W. S. Warren, E. Jensita, R. T. Branca and X. Chen, Science, 2009, 323, 1711–1714 CrossRef CAS PubMed.
- G. Pileio, S. Bowen, C. Laustsen, M. C. D. Tayler, J. T. Hill-Cousins, L. J. Brown, R. C. D. Brown, J. H. Ardenkjaer-Larsen and M. H. Levitt, J. Am. Chem. Soc., 2013, 135, 5084–5088 CrossRef CAS PubMed.
- Y. Feng, T. Theis, X. Liang, Q. Wang and W. S. Warren, J. Am. Chem. Soc., 2013, 135, 9632–9635 CrossRef CAS PubMed.
- T. Theis, G. X. Ortiz Jr, A. W. J. Logan, K. E. Claytor, Y. Feng, W. P. Huhn, V. Blum, S. J. Malcolmson, E. Y. Chekmenev, Q. Wang and W. S. Warren, Sci. Adv., 2016, 2, e1501438 Search PubMed.
- Y. Zhang, X. Duan, P. C. Soon, V. Sychrovsky, J. W. Canary and A. Jerschow, ChemPhysChem, 2016, 17, 2967–2971 CrossRef CAS PubMed.
- Y. Zhang, P. C. Soon, A. Jerschow and J. W. Canary, Angew. Chem., Int. Ed., 2014, 53, 3396–3399 CrossRef CAS PubMed.
- S. Cavadini, J. Dittmer, S. Antonijevi and G. Bodenhausen, J. Am. Chem. Soc., 2005, 127, 15744–15748 CrossRef CAS PubMed.
- P. Ahuja, R. Sarkar, P. R. Vasos and G. Bodenhausen, J. Am. Chem. Soc., 2009, 131, 7498–7499 CrossRef CAS PubMed.
- S. J. DeVience, R. Walsworth and M. S. Rosen, NMR Biomed., 2013, 26, 1204–1212 CrossRef CAS PubMed.
- R. Buratto, D. Mammoli, E. Canet and G. Bodenhausen, J. Med. Chem., 2016, 59, 1960–1966 CrossRef CAS PubMed.
- M. C. D. Tayler and M. H. Levitt, Phys. Chem. Chem. Phys., 2011, 13, 9128–9130 RSC.
- M. Goldman, H. Johannesson, O. Axelsson and M. Karlsson, C. R. Chim., 2006, 9, 357–363 CrossRef CAS.
- J.-B. Hövener, E. Y. Chekmenev, K. C. Harris, W. H. Perman, T. T. Tran, B. D. Ross and P. Bhattacharya, Magn. Reson. Mater. Phys., Biol. Med., 2009, 22, 123–134 CrossRef PubMed.
- K. W. Waddell, A. M. Coffey and E. Y. Chekmenev, J. Am. Chem. Soc., 2011, 133, 97–101 CrossRef CAS PubMed.
- R. Reineri, T. Boi and S. Aime, Nat. Commun., 2015, 6, 5858 CrossRef PubMed.
- K. Münnemann, M. Kölzer, I. Blakey, A. K. Whittaker and K. J. Thurecht, Chem. Commun., 2012, 46, 1583–1585 RSC.
- J. A. Tang, F. Gruppi, R. Fleysher, D. K. Sodickson, J. W. Canary and A. Jerschow, Chem. Commun., 2011, 47, 958–960 RSC.
- S. Glöggler, S. Wagner and L.-S. Bouchard, Chem. Sci., 2015, 6, 4261–4266 RSC.
- T. Gutmann, M. Sellin, H. Breitzke, A. Stark and G. Buntkowsky, Phys. Chem. Chem. Phys., 2009, 11, 9170–9175 RSC.
- J. H. Ardenkjaer-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10158–10163 CrossRef CAS PubMed.
- K. Golman, R. in ‘t Zandt and M. Thaning, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11270–11275 CrossRef CAS PubMed.
- S. J. Nelson, J. Kurhanewicz, D. B. Vigneron, P. E. Z. Larson, A. L. Harzstark, M. Ferrone, M. can Criekinge, J. W. Chang, R. Bok, I. Park, G. Reed, L. Carvajal, E. J. Small, P. Munster, V. K. Weinberg, J. H. Ardenkjaer-Larsen, A. P. Chen, R. E. Hurd, L.-I. Odegardstuen, F. J. Robb, J. Tropp and J. A. Murray, Sci. Transl. Med., 2013, 5, 198ra108 Search PubMed.
- J. Kurhanewicz, D. B. Vigneron, K. Brindle, E. Y. Chekmenev, A. Comment, C. H. Cunningham, R. J. DeBerardinis, G. G. Green, M. O. Leach, S. S. Rajan, R. R. Rizi, B. D. Ross, W. S. Warren and C. R. Malloy, Neoplasia, 2011, 13, 81–97 CrossRef CAS PubMed.
- A. Comment, J. Magn. Reson., 2016, 264, 39–48 CrossRef CAS PubMed.
- D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges and B. D. Sykes, J. Biomol. NMR, 1995, 5, 67–81 CrossRef CAS PubMed.
- V. Govindaraju, K. Young and A. A. Maudsley, NMR Biomed., 2000, 13, 129–153 CrossRef CAS PubMed.
- M. A. Higgs and R. L. Sass, Acta Crystallogr., 1963, 16, 657–661 CrossRef CAS.
- J. M. G. Cowie and V. Arrighi, Polymers: Chemistry and Physics of Modern Materials, 3rd edn, CRC Press, Taylor and Francis Group, Boca Raton, FL, 2007 Search PubMed.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- M. M. Herth, M. Barz, D. Moderegger, M. Allmeroth, M. Jahn, O. Thews, R. Zentel and F. Rösch, Biomacromolecules, 2009, 10, 1697–1703 CrossRef CAS PubMed.
- S. W. Rhee and M. J. Tanga, J. Labelled Compd. Radiopharm., 2000, 43, 925–932 CrossRef CAS.
- J. L. Turner, D. Pan, R. Plummer, Z. Chen, A. K. Whittaker and K. L. Wooley, Adv. Funct. Mater., 2005, 15, 1248–1254 CrossRef CAS.
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS PubMed.
- V. Pace, L. Castoldi, A. R. Alcantara and W. Holzer, Green Chem., 2012, 14, 1859–1863 RSC.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3921–3939 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical synthesis, relaxation analysis, details of the NMR sequence timings. See DOI: 10.1039/c7ra05196d |
| This journal is © The Royal Society of Chemistry 2017 |