
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA phase-reversible Pd containing sphere-to-bridge-shaped peptide nanostructure for cross-coupling reactions†
Seongsoo Kim‡
 a,
Hong-Jun Cho‡b,
Namhun Leea,
Yoon-Sik Lee*b,
Dong-Sik Shin*c and
Sang-Myung Lee*a
a,
Hong-Jun Cho‡b,
Namhun Leea,
Yoon-Sik Lee*b,
Dong-Sik Shin*c and
Sang-Myung Lee*a
aDepartment of Chemical Engineering, Kangwon National University, Gangwon-do 24341, Republic of Korea. E-mail: sangmyung@kangwon.ac.kr; Fax: +82 33 251 3658; Tel: +82 33 250 6335
bSchool of Chemical and Biological Engineering, Seoul National University, Seoul 08826, Republic of Korea. E-mail: yslee@snu.ac.kr; Fax: +82 2 880 1604; Tel: +82 2 880 7073
cDepartment of Chemical and Biological Engineering, Sookmyung Women's University, Seoul 04310, Republic of Korea. E-mail: dshin@sm.ac.kr; Fax: +82 2 2077 7450; Tel: +82 2 2077 7236
First published on 30th June 2017
Abstract
A sphere-to-bridge-shaped peptide nanostructure was constructed from a tyrosine-rich peptide (H-YYACAYY-OH) via mediating Pd2+ ions as well as changing temperature. This novel assembly technique provided a recyclable Pd nano-catalyst with a function of reversible thermal phase transition between the homogeneous and heterogeneous states for cross-coupling reactions.
Natural peptides coordinated with various metal ions via amide bonds and functional groups on side chains can form various self-assembled structures.1 For example, ferritin is a natural intracellular protein which takes a specific conformation through interactions between peptide building blocks and Fe2+ ions. Artificial peptides that provide natural or non-natural metal binding sites have been developed to construct metal ion-induced self-assemblies of nanostructures including tubular-, fibre-, vesicle-, spherical-, and rod-coil-type ones.2 Such metal coordination can initiate intermolecular self-assembly by bringing two or more peptides into close proximity.3 Furthermore, several metal ions coordinated with peptides have been involved in a redox reaction to induce irreversible peptide cross-linking via covalent bonding e.g., dityrosine formation.4–7
Based on a peptide template, various types of metal-peptide nanostructures have been designed and utilized as electrochemical sensors,8 biological scaffolds,9,10 and catalysts.11–13 In particular, significant effort has been devoted in developing metal nanoparticles (NPs)-incorporated peptide nanostructures which can then be used as catalysts in C–C coupling reaction. To facilitate the reaction, for instant, Knecht group synthesized various shapes of Pd nanostructures with a large surface area by using a self-assembling peptide template composed of R5 peptide.14 Moreover, peptide amphiphiles possessing both hydrophobic carbon-chain and ionic peptide sequences are promising as bio-inspired-templates for growing PdNPs-incorporated nanofibers. Recently, several groups have demonstrated nanofiber incorporated with PdNPs using a self-assembled peptide amphiphile. The peptide-templated PdNPs showed high catalytic activity for the Suzuki coupling reaction under environmentally-friendly conditions.15,16 Although these peptide-templated PdNPs can afford efficient catalytic activities for the C–C coupling reactions, drawbacks originated from the heterogeneous state limits its broad and practical application of the catalyst.
A recent report has revealed that a tyrosine-rich peptide, YYACAYY (YC7), can be self-assembled into a two-dimensional peptide nanostructure via interaction of tyrosines and cysteines leading to cross-linking at an air/water interface.17 Based on this finding, we have successfully designed Pd2+-ion-mediated sphere-to-bridge-shaped peptide nanostructures (YC7@Pd2+) through thermally induced phase transition. During the transition, the Pd2+ ions have interacted with the YC7 peptide molecules through coordination, which might be a crucial driving force for leading to a distinctive self-assembled sphere-to-bridge peptide nanostructure. More interestingly, the self-assembled YC7@Pd2+ is dissolved into the aqueous solution via thermal transition during cross-coupling process over phase transition temperature and followed by re-assembly under the critical temperature achieving the PdNP-captured astrocyte-shaped peptide nanostructures (YC7@PdNP). This provides a great opportunity to take advantage of YC7@Pd2+ as a versatile and recyclable catalyst, capitalizing both of its homogeneous characteristic over critical temperature and heterogeneous characteristic under the same temperature. Herein, we demonstrate the feasibility of YC7@Pd2+ as a new type of catalyst under environmentally friendly conditions: (a) as a nano-catalyst during C–C coupling reactions; and (b) as a heterogeneous catalyst for separation, especially in Suzuki and Sonogashira coupling reactions. YC7@Pd2+, this catalytic system via switchable phase enabled reusable catalyst. To the best of our knowledge, this kind of the reassembly process is first demonstrated in this paper.
YC7 is a random coil-dominant peptide, which self-assembles in an aqueous phase into a two-dimensional nanostructure.17 From this peptide, we synthesized sphere-to-bridge-type peptide nanostructures (YC7@Pd2+) via thermally controlled and Pd2+-ion-mediated ionic interactions (Scheme 1). During heating the peptide up to 90 °C for 1 h, it was allowed to completely dissolve in water. As the peptide solution was gradually cooled to 60 °C, YC7 underwent a rapid transition into a secondary, α-helix-dominant structure. At this stage, Pd2+ ions were injected to induce distinctive Pd-peptide secondary structures via ionic interactions between phenolate anions of tyrosyl residues (or C terminal carboxylate anions) and Pd2+ ions. Without Pd2+ ions, YC7 molecules were self-assembled into irregular shaped nanostructures and nanosheets during the thermodynamic transition (90 °C → 60 °C) via interactions between tyrosines and cysteine crosslinking (Fig. S1†). However, the YC7@Pd2+, which was self-assembled by interactions with Pd2+ ions, was uniform and spherical with an average diameter of 88 ± 31 nm (count: 100) and further formed linked networks of sphere-to-bridge shapes (Fig. 1a and b). Physicochemical characterization of the nanostructure was performed with several analytical methods. First, the constituent elements of YC7@Pd2+, including Pd, N, O, S and Cl, were analysed by energy-dispersive X-ray microanalysis mapping (Fig. S2†). The results demonstrated that YC7@Pd2+ was composed of Pd2+ ions, evenly distributed within the YC7 matrix.
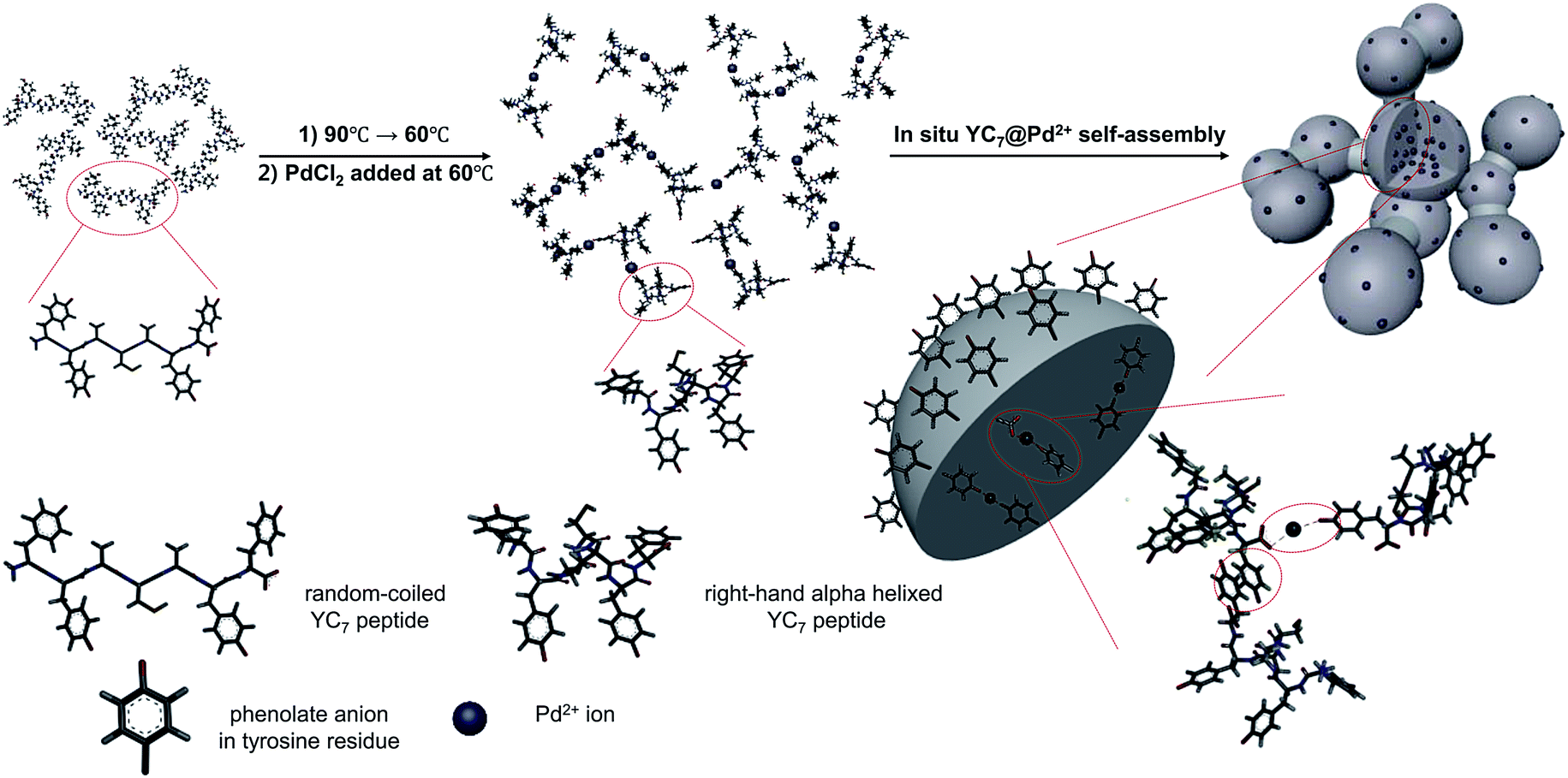 | ||
| Scheme 1 Proposed mechanism of YC7@Pd2+ synthesis by thermal phase transition of dissolved YC7 and Pd2+-ion-driven self-assembly. | ||
 | ||
| Fig. 1 (a) TEM image and (b) SEM image of YC7@Pd2+ nanostructure. | ||
To analyse the structure of YC7@Pd2+ and the interactions between Pd2+ ions and the peptides, it was further characterized by infrared (IR) spectroscopy (Fig. 2). Bands from ν(C–O), ν(CC), δ(COH) of Tyr in YC7 peptide were observed at 1070–1270 cm−1, which exhibited relatively strong intensity due to its polar character (Fig. 2). These peaks are unique in Tyr not in other residues of YC7 peptide. When the C–O stretching vibration bands of the self-assembled YC7 and YC7@Pd2+ were compared, YC7@Pd2+ gave strong peaks at 1232 cm−1 involving two stretching modes of ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and ν(C–O) in Tyr–OH, which are attributed to stronger polarity of phenolate (Tyr residue) in the presence of Pd2+.18–21 These results clearly show that there are strong interactions between Pd2+ ions and tyrosine groups of YC7@Pd2+. In addition, a new CO stretching vibration from the carboxylate of YC7@Pd2+ appeared at 1557 cm−1 which corresponds to the asymmetric stretching vibration of metal carboxylates.22
C) and ν(C–O) in Tyr–OH, which are attributed to stronger polarity of phenolate (Tyr residue) in the presence of Pd2+.18–21 These results clearly show that there are strong interactions between Pd2+ ions and tyrosine groups of YC7@Pd2+. In addition, a new CO stretching vibration from the carboxylate of YC7@Pd2+ appeared at 1557 cm−1 which corresponds to the asymmetric stretching vibration of metal carboxylates.22
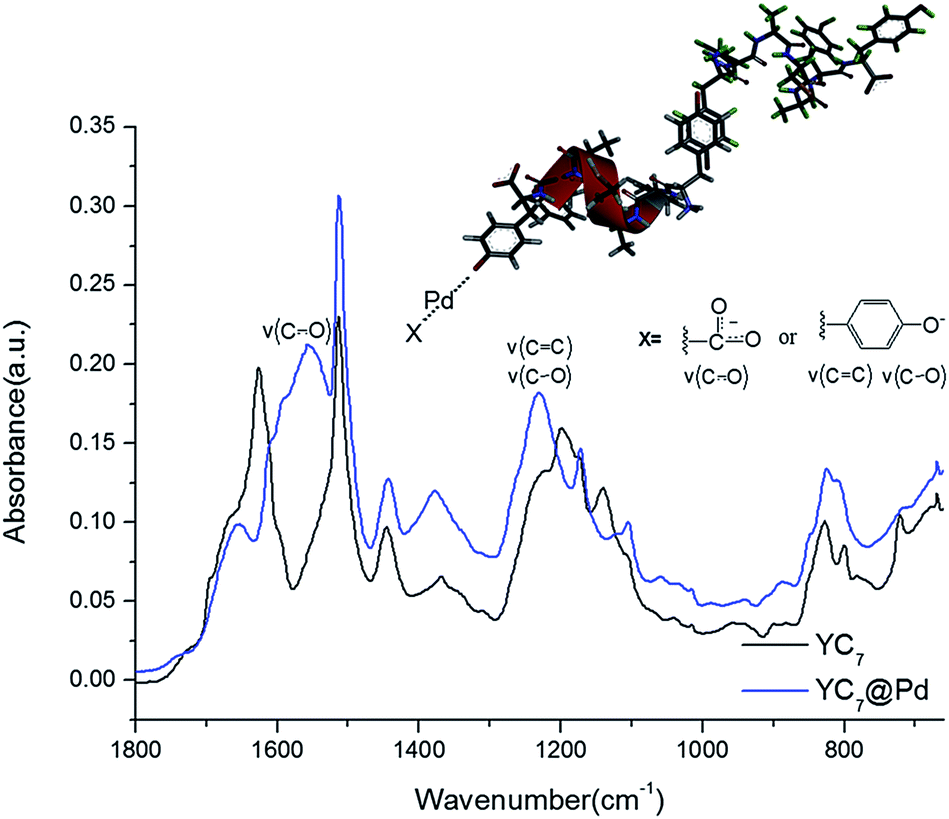 | ||
| Fig. 2 FT-IR spectra of YC7@Pd2+ and YC7 peptide. | ||
Additionally, the X-ray photoelectron spectroscopy (XPS) data of YC7@Pd2+ clearly revealed Pd peaks (3d2/5 and 3d3/5) at 343.20 and 337.95 eV which corresponded to Pd(II) (Fig. S3†).23 Taken together, YC7@Pd2+ is a non-crystalline peptide complex coordinated with Pd(II) at tyrosine residues, and has a potential as a Pd catalyst used in C–C coupling reactions. Consequently, Pd2+-ion-mediated peptide self-assembly was well characterized with IR and XPS.
Hence, this proves that the phenolate and carboxylate groups of YC7@Pd2+ were primarily involved in coordination with Pd2+ ions leading to the formation of a distinctive Pd-peptide nanostructure. Inductively coupled plasma atomic emission spectroscopy (ICP-OES) analysis revealed that the palladium content of YC7@Pd2+ was calculated to be 1.86 mmol Pd per g catalyst.
The X-ray diffraction (XRD) patterns of self-assembled YC7 exhibited distinctive peaks at 10° and 18° originating from the intersheet reflections of peptide sheets, respectively (Fig. S4†). In contrast, peptide sheet-originated regular sharp peaks of face-centered cubic Pd crystals did not appear in the XRD pattern of YC7@Pd2+ (Fig. S5†). This result supports that Pd2+ ions were able to collapse the intrinsic YC7 structure, leading to the irregular shaped peptide arrangement inside the sphere-to-bridge-shaped peptide nanostructure.
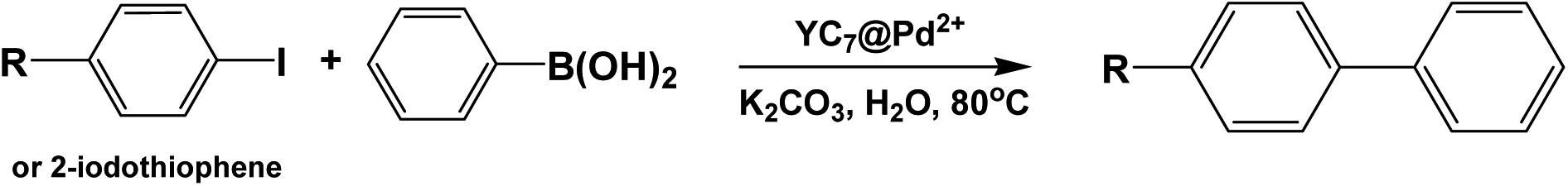
We investigated the catalytic activity of the YC7@Pd2+ by evaluating the efficiency of C–C coupling reactions including Suzuki, Heck and Sonogashira coupling reactions.24 Actually, YC7@Pd2+ nanostructures are decomposed into the solution at coupling reaction temperature, and then PdNPs instantly reduced from Pd2+ ions participate in the three kinds of coupling reactions. First, various bases for each coupling reaction were screened in aqueous solution.25 K2CO3 and pyrrolidine were selected as the best base in the Suzuki and the Sonogashira reactions, respectively. (Tables S1 and S2†). However, the catalytic activity of YC7@Pd2+ in the Heck coupling reactions gave unfavourable yields (<2%) even in the presence of cetyl trimethyl ammonium bromide (CTAB) (Table S3†).26 Therefore, YC7@Pd2+ was further used as a catalyst for the Suzuki and the Sonogashira coupling reactions of aryl iodides with phenylboronic acid or phenylacetylene in an aqueous solvent system at 80 °C, where YC7@Pd2+ can be transformed into the homogeneous phase.
An activated aryl iodide (4-iodoacetophenone) was well-converted to the corresponding biaryl compounds in the Suzuki coupling reaction with over 98% yield in only 1 h with 0.1 mol% of YC7@Pd2+ (Table 1, entry 1). Electron-donating substituents lowered the yields of the corresponding biaryl compounds (Table 1, entries 2–5). In particular, the oxygen-containing substrates (Table 1, entries 2 (OH) and 3 (OMe)) exhibited more efficient coupling than non-oxygen-containing substrates. These results demonstrated that the hydrogen bonding facilitated the access of YC7@Pd2+ to the substrate molecules under aqueous conditions providing a compatible coupling environment for the Suzuki coupling reaction. Furthermore, the use of CTAB as a phase-transfer catalyst promoted the interphase transfer of substrates, thereby boosting the coupling yields of the non-oxygen containing substrates (Table 1, entries 4 (H) and 5 (Me)). As a control experiment, compared with the coupling reaction catalyzed by YC7@Pd2+, the coupling yields by PdCl2 activation were lower than those of YC7@Pd2+ in same condition (Table S4†).
|
|||
|---|---|---|---|
| Entry | R | Time (h) | Yieldc (%) |
| a Conditions: aryl iodine and heterocyclic halides (0.155 mmol), phenylboronic acid (0.186 mmol), YC7@Pd2+ (0.1 mol%), K2CO3 (0.274 mmol) in water (1 mL) at 80 °C.b GC yields when 0.5 eq. of CTAB was used.c GC yields. | |||
| 1 | COCH3 | 1 | 98.7 |
| 2 | OH | 3 | 99.6 |
| 3 | OCH3 | 3 | 92.8 |
| 4 | H | 3/3b | 88.8/99.9b |
| 5 | CH3 | 6/3b | 70.8/96.0b |
| 6 | 2-Iodothiophene | 6/3b | 40.0/91.3b |
Given the successful catalytic activity of YC7@Pd2+ in the Suzuki coupling reactions, we further investigated the catalytic activity of YC7@Pd2+ in the Sonogashira coupling reactions with CuI under aqueous conditions.27 The Sonogashira coupling reaction catalysed by YC7@Pd2+ exhibited a high coupling yield with a strong electron-withdrawing substrate (Table S5,† entry 1). In contrast to the Suzuki coupling reaction, the hydrogen bonding derived from the electron-donating substrates (Table S5,† entries 3 and 6) was not relatively effective on the catalytic activity in the Sonogashira coupling reaction. CTAB strongly suppressed the cross-coupling reaction, and instead, accelerated homo-coupling reaction with activated aryl iodide. The results indicate that CTAB did not afford a compatible cross-coupling environment between the catalyst and the substrates in the Sonogashira coupling reaction, whereas it stabilized phenylacetylene with Cu+ ion to activate the homo-coupling pathway.
During the coupling reaction at 80 °C in an aqueous solvent, YC7@Pd2+ disassembled into a soluble Pd-peptide complex, which is reduced into the solubilized PdNP catalyst. After cooling down to room temperature, the PdNP-peptide complex was reassembled and transformed into a nanostructure which is similar with a shape of astrocyte (YC7@PdNP, Fig. S6b†). This reversible process between homogeneous and heterogeneous states facilitated easy isolation and reuse of YC7@PdNP from the reaction mixture. To evaluate the reusability of YC7@Pd2+, the catalyst was recycled in subsequent Suzuki coupling reactions with 4-iodophenol or 4-iodoacetophenone. While the coupling yields of the activated substrates (iodoacetophenone) decreased slightly in the fourth and fifth runs, 4-iodophenol which can form a hydrogen bonding with YC7@PdNP was converted to the corresponding biaryl compound in excellent yields (yield > 98%) even after 5th use (Fig. S7†).9 These results reconfirm that hydrogen bonding enabled the substrates for easy access to the Pd-peptide complex as well as possibly stabilizing the PdNPs for excellent catalytic activity during a series of Suzuki coupling reactions.
In conclusion, we developed a novel method for the construction of Pd2+-ion-mediated sphere-to-bridge-shaped peptide nanostructures, YC7@Pd2+, of which morphology can be controlled by temperature in an aqueous phase. Characterized by the switchable thermally-reversible phase transition, YC7@Pd2+ acted as a solubilized nano-catalyst during C–C coupling reactions and reassembled into a heterogeneous structure for isolation and reuse after each coupling reaction. YC7@Pd2+ showed an excellent activity as a catalyst for the Suzuki and the Sonogashira coupling reactions under aqueous conditions. Especially, in the Suzuki coupling reaction, hydrogen bonding capability of the substrates provided a favourable coupling environment, enhancing the coupling yield and reusability of the catalyst. The morphology controllable self-assembled Pd2+-ion-mediated peptide nanostructure can open a new avenue as a reusable catalyst for C–C coupling reactions under environmentally-friendly conditions.
Acknowledgements
This work was supported by the Ministry of Science, ICT & Future Planning and National Research Foundation of Korea through Basic Science Research Program (NRF-2014R1A1A1006711), the Ministry of Trade, Industry & Energy (MOTIE), Korea Institute for Advancement of Technology (KIAT) through the Encouragement Program for The Industries of Economic Cooperation Region (R0004027) and Kangwon National University through 2015 Research Grant (No. 520150081).Notes and references
- O. Yamauchi, A. Odani and M. Takani, J. Chem. Soc., Dalton Trans., 2002, 3411–3421 RSC.
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200–5219 RSC.
- M. M. Pires, D. E. Przybyla, C. M. R. Perez and J. Chmielewski, J. Am. Chem. Soc., 2011, 133, 14469–14471 CrossRef CAS PubMed.
- T. G. Huggins, M. C. W. Knecht, N. A. Detorie, J. W. Baynes and S. R. Thorpe, J. Biol. Chem., 1993, 268, 12341–12347 CAS.
- S. Takasaki, Y. Kato, M. Murata, S. Homma and S. Kawakishi, Biosci., Biotechnol., Biochem., 2005, 69, 1686–1692 CrossRef CAS PubMed.
- Y. K. A. Hilaly, T. L. Williams, M. S. Parker, L. Ford, E. Skaria, M. Cole, W. G. Bucher, K. L. Morris, A. A. Sada, J. R. Thorpe and L. C. Serpel, Acta Neuropathol. Commun., 2013, 1, 1–17 Search PubMed.
- K.-I. Min, G. Yun, Y. Jang, K.-R. Jim, Y. H. Ko, H.-S. Jang, Y.-S. Lee, K. Kim and D.-P. Kim, Angew. Chem., Int. Ed., 2016, 55, 6925–6928 CrossRef CAS PubMed.
- S. Kim, J. H. Kim, J. S. Lee and C. B. Park, Small, 2015, 11, 3623–3640 CrossRef CAS PubMed.
- T. Mizutaru, T. Sakuraba, T. Nakayama, G. Marzun, P. Wagener, C. Rehbock, S. Barcikowski, K. Murakami, J. Fujita, N. Ishiie and J. Yamamoto, J. Mater. Chem. A, 2015, 3, 17612–17619 CAS.
- Y.-X. Pan, H.-P. Cong, Y.-L. Men, S. Xin, Z.-Q. Sun, C.-J. Liu and S.-H. Yu, ACS Nano, 2015, 9, 11258–11265 CrossRef CAS PubMed.
- D. B. Pacardo, M. Sethi, S. E. Jones, R. R. Naik and M. R. Knecht, ACS Nano, 2009, 3, 1288–1296 CrossRef CAS PubMed.
- W. Wang, C. F. Anderson, Z. Wang, W. Wu, H. Cui and C.-J. Liu, Chem. Sci., 2017, 8, 3310–3324 RSC.
- W. Wang, Z. Wang, M. Yang, C.-J. Zhong and C.-J. Liu, Nano Energy, 2016, 25, 26–33 CrossRef CAS.
- A. Jakhmola, R. Bhandari, D. B. Pacardo and M. R. Knecht, J. Mater. Chem., 2010, 20, 1522–1531 RSC.
- I. Maity, D. B. Rasale and A. K. Das, RSC Adv., 2014, 4, 2984–2988 RSC.
- M. A. Khalily, O. Ustahuseyin, R. Garifullin, R. Genc and M. O. Guler, Chem. Commun., 2012, 48, 11358–11360 RSC.
- H.-S. Jang, J.-H. Lee, Y.-S. Park, Y.-O. Kim, J. Park, T.-Y. Yang and Y.-S. Lee, Nat. Commun., 2014, 5, 3665–3675 Search PubMed.
- M. S. Shongwe, C. H. Kaschula, M. S. Adsetts, E. W. Ainscough, A. M. Brodie and M. J. Morris, Inorg. Chem., 2005, 44, 3070–3079 CrossRef CAS PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS PubMed.
- L. I. Grace, R. Cohen, T. M. Dunn, D. M. Lubman and M. S. D. Vries, J. Mol. Spectrosc., 2002, 215, 204–219 CrossRef CAS.
- L. Rodriguez, E. Labisbal, A. S. Pedrares, J. A. G. Vazquez, J. Romero, M. L. Duran, J. A. Real and A. Sousa, Inorg. Chem., 2006, 45, 7903–7914 CrossRef CAS PubMed.
- V. Otero, D. Sanches, C. Montagner, M. Vilarigues, L. Carlyle, J. A. Lopes and M. J. Melo, J. Raman Spectrosc., 2014, 45, 1197–1206 CrossRef CAS.
- G. Kumar, J. R. Blackburn, R. G. Albridge, W. E. Moddeman and M. M. Jones, Inorg. Chem., 1972, 11, 296–300 CrossRef CAS.
- T. Suzuka, K. Kimura and T. Nagamine, Polymers, 2011, 3, 621–639 CrossRef CAS.
- I. Hoffmann, B. Blumenroder, S. O. N. Thumann, S. Dommer and J. Schatz, Green Chem., 2015, 17, 3844–3857 RSC.
- P. Qiu, J. Y. Zhao, X. Shi and X. H. Duan, New J. Chem., 2016, 40, 6568–6572 RSC.
- A. M. Thomas, A. Sujatha and G. Anilkumar, RSC Adv., 2014, 4, 21688–21698 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra04793b |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |