
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ir/C and Brφnsted acid functionalized ionic liquids: an efficient catalytic system for hydrogenation of nitrobenzene to p-aminophenol
Hong Wang,
Taotao Jiang,
Lei Ma *,
Hanbing Wang,
Xiaoliang Xu,
Chunshan Lu and
Xiaonian Li*
*,
Hanbing Wang,
Xiaoliang Xu,
Chunshan Lu and
Xiaonian Li*
Institute of Industrial Catalysis, Zhejiang University of Technology, State Key Laboratory Breeding Base of Green Chemistry Synthesis Technology, Hangzhou, 310014, P. R. China. E-mail: malei@zjut.edu.cn; xnli@zjut.edu.cn
First published on 20th June 2017
Abstract
In this study, we found that the phenylhydroxylamine intermediate could desorb more easily from an Ir surface than from a Pt surface, which is beneficial for inhibiting the over-hydrogenation of phenylhydroxylamine to aniline. On the other hand, the Brφnsted acid functionalized ionic liquids with sulfonic acid and bisulfate anions were acidic enough to catalyze the Bamberger rearrangement to form p-aminophenol from phenylhydroxylamine. On this basis, a new catalytic system constructed by Ir/C and Brφnsted acid functionalized ionic liquid was applied, for the first time, to the one-pot hydrogenation of nitrobenzene to p-aminophenol. Our results indicate that the PAP selectivity of Ir/C and [SO3H-bmim][HSO4] Brφnsted functionalized ionic liquid was far more than that of the traditional Pt/C and sulfuric acid catalyst system. Furthermore, the dually functionalized ionic liquid ([HSO3-b-N-Bu3][HSO4]) can be used simultaneously as an acid catalyst and also as a surfactant, due to its higher lipophilicity. Therefore, our new catalytic system has unique advantages in the hydrogenation of nitrobenzene to p-aminophenol.
Introduction
p-Aminophenol (PAP) is an important commercial chemical raw material, which is widely used in the field of pharmaceuticals, dyestuffs, polyurethanes and photographic chemicals.1–3 PAP is usually synthesized by the reduction of p-nitrochlorobenzene or p-nitrophenol,4–6 which has several drawbacks including multistep processes, scarcity of raw materials and environmental problems. Compared to the above synthetic methods, the one-pot synthesis of PAP from nitrobenzene (NB) is regarded as the most convenient, economical and challenging method at present. This process involves two steps: the first step is the hydrogenation of NB to an intermediate phenylhydroxylamine (PHA), and the second step is the conversion of PHA to PAP through the acid-catalysed Bamberger rearrangement.7 At the same time, the major by-product aniline (AN) can also be generated for the over hydrogenation of PHA in this transformation (Scheme 1). | ||
| Scheme 1 Hydrogenation of nitrobenzene to p-aminophenol. | ||
The one-pot hydrogenation of NB to PAP involves two kinds of catalysts: the hydrogenation catalyst and acid catalyst. The supported noble metal (e.g. Pt, Pd or Au) catalysts are usually used as hydrogenation catalysts for the reduction of the nitro group.8–12 In our previous study of the selective hydrogenation of halogenated nitrobenzene, we found that the adsorption properties of halogenated nitrobenzene on Ir were significantly different from those on Pd; Ir was more beneficial to the formation of PHA intermediates than Pd.13 Since PHA is the most important intermediate for the hydrogenation of NB to PAP, we supposed that the special adsorption properties of Ir might be favourable for the formation of PAP.
Sulfuric acid is usually used as the acid catalyst for the conversion of PHA to PAP through the Bamberger rearrangement, which causes some serious problems like considerable amounts of effluents and equipment corrosion.14,15 In the past few years, chemists have investigated some new acidic catalysts, including solid acid16–21 and the CO2/H2O system,22 to replace sulfuric acid for this transformation, but the results have not been ideal, due to the rapid deactivation or insufficient acidity of these acid catalysts.23–25
As a kind of environmentally friendly acid catalyst, Brφnsted acid functionalized ionic liquids (BFILs) have the advantages of solid acids in addition to the characteristics of liquid acids. BFILs also have unique properties like water-stability and structure-adjustability, which make them widely used in the field of esterification,26,27 hydrolysis,28,29 alkylation,30,31 the Biginelli reaction,32 hydrodeoxygenation,33 and Beckmann rearrangement,34 instead of traditional mineral acids. Recently, Cui et al. reported their work on the one-pot hydrogenation of NB to PAP using BFIL [HSO3-b-N-Me3][HSO4] as the acid catalyst and Pt/SiO2 as the hydrogenation catalyst.35
Based on our previous research results and the performance of BFILs in the hydrogenation of NB to PAP, we constructed a new catalytic system by the combination of Ir/C and BFILs to catalyze the one-pot hydrogenation of NB to PAP. The catalytic performance of this system was then studied and compared with that of the traditional Pt/C and sulfuric acid system. In addition, considering the structure-adjustability of BFILs, we also wanted to modify the structure of the BFILs to improve the lipophilicity, and found real dual functionalized ionic liquids with the functions of acid catalyst and surfactant. The dual functionalization of acidic ionic liquids might afford an opportunity for us to carry out the hydrogenation of NB to PAP without adding surfactant.
Experimental
Materials
N-Methylimidazole, triethylamine, tripropylamine, tributylamine, pyridine, 1,4-butanesultone and hexadecyl trimethyl ammonium bromide (CTAB) were purchased from Aladdin Reagent Co. Ltd. Sulfuric acid, nitrobenzene, aniline ethyl acetate, diethyl ether, acetonitrile, ethanol and isopropanol were purchased from Shanghai Lingfeng Chemical Reagent Co. Ltd. All reagents and raw materials were commercially available and used without any further purification.Preparation of catalysts
 | ||
| Scheme 2 Structures of the BFILs. | ||
The preparation of type B BFILs DF-A ([SO3H-bmim][HSO4]) was similar to that used in the literature37 and the detailed procedure is as follows: N-methylimidazole (16.42 g, 0.2 mol) and equimolar 1,4-butanesultone (27.23 g, 0.2 mol) were mixed in a 100 mL single-necked flask. Then, the mixture was stirred at 333 K under nitrogen until the white solid was completely generated. The solid was washed with ethyl acetate (20 mL × 3) and dried in vacuum at 353 K for 6 hours. A stoichiometric amount of sulphuric acid was added dropwise to the zwitterion and the mixture was stirred at 353 K for 6 hours, resulting the formation of DF-A. The product was washed with ethyl acetate (20 mL × 3) and dried under vacuum at 383 K for 6 hours. DF-B1 ([HSO3-b-N-Et3][HSO4]) and DF-C ([HSO3-bPy][HSO4]) were prepared according to the procedure described above for DF-A, with triethylamine and pyridine, respectively, instead of N-methylimidazole.
Type C BFILs DF-B2 ([HSO3-b-N-Pr3][HSO4]) and DF-B3 ([HSO3-b-N-Bu3][HSO4]) were prepared using the same procedure describe above, replacing N-methylimidazole with tripropylamine and tributylamine.38,39
Characterization of catalysts
The particle size of Ir or Pt on the active carbon surface was determined by transmission electron microscopy (TEM) using a Tecnai G2 F30 S-Twin microscope (Philips-FEI Co.). At least 200 individual Ir particles were counted for each catalyst. The Ir or Pt particle size of the catalysts, ds, was calculated using the following equation: ds = ∑nidi3/∑nidi2, where the visible particle size di on the micrographs was measured by a computerized system.
Spectral data for SF-A ([Hmim][HSO4]): 1H-NMR (500 MHz, D2O) δ 9.06 (s, 1H), 7.70 (t, 1H), 7.65 (t, 1H), 3.88 (s, 3H). 13C-NMR (125MHZ, D2O) δ 35.86, 120.41, 122.95, 135.86. IR (cm−1): 3149, 2967, 1639, 1586, 1451, 1048, 761.
Spectral data for SF-B ([Et3–NH][HSO4]): 1H-NMR (500 MHz, D2O) δ 3.09 (q, 6H), 1.18 (t, 9H). IR (cm−1): 2991, 2951, 1476, 1399, 1165, 1036.
Spectral data for SF-C ([HPy][HSO4]): 1H-NMR (500 MHz, D2O) δ 8.66 (d, 2H), 8.50 (m, 1H), 8.08–7.84 (m, 2H). IR (cm−1): 3071, 1639, 1545, 1488.
Spectral data for DF-A ([SO3H-bmim][HSO4]): 1H-NMR (500 MHz, D2O) δ 8.59 (s, 1H), 7.32 (m, 2H), 4.10 (t, 2H), 3.80–3.72 (m, 3H), 2.83–2.62 (m, 2H), 1.88 (m, 2H), 1.76–1.41 (m, 2H). 13C-NMR (125 MHz, D2O) δ 21.50, 28.43, 35.87, 49.28, 51.07, 122.64, 123.55, 136.56. IR (cm−1): 3156, 3115, 2960, 1575, 1169, 1031, 794, 747, 579.
Spectral data for DF-B1 ([HSO3-b-N-Et3][HSO4]): 1H-NMR (500 MHz, D2O) δ 3.08 (q, 6H), 2.99 (dd, 2H), 2.76 (dd, 2H), 1.76–1.52 (m, 4H), 1.06 (t, 9H). 13C-NMR (125 MHz, D2O) δ 7.36, 19.98, 21.90, 50.26, 52.28, 56.10. IR (cm−1): 2991, 1488, 1396, 1189, 730, 587.
Spectral data for DF-B2 ([HSO3-b-N-Pr3][HSO4]): 1HNMR (500 MHz, D2O) δ 3.19–3.08 (m, 2H), 3.07–2.98 (m, 6H), 2.88–2.74 (m, 2H), 1.77–1.67 (m, 2H), 1.64 (dd, 2H), 1.61–1.46 (m, 6H), 0.80 (t, 9H). IR (cm−1): 2976, 2884, 1487, 1473, 1169, 1054, 720, 579.
Spectral data for DF-B3 ([HSO3-b-N-Bu3][HSO4]): 1H NMR (500 MHz, D2O) δ 3.01–2.89 (m, 6H), 2.89–2.80 (m, 2H), 2.71–2.60 (m, 2H), 1.60–1.45 (m, 4H), 1.43–1.32 (m, 6H), 1.15–1.02 (m, 6H), 0.66 (q, 9H). IR (cm−1): 2963, 2876, 1472, 1168, 1054, 726, 579.
Spectral data for DF-C ([HSO3-bPy][HSO4]): 1H-NMR (500 MHz, D2O) δ 8.73 (d, 2H), 8.42 (m, 1H), 7.95 (t, 2H), 4.53 (t, 2H), 2.95–2.72 (m, 2H), 2.15–1.86 (m, 2H), 1.75–1.42 (m, 2H). 13C-NMR (125 MHz, D2O) δ 20.92, 29.56, 49.92, 61.28, 128.58, 144.38, 146.10. IR (cm−1): 3070, 2949, 1635, 1489, 1456, 1173, 728, 579.
Acidity of ionic liquids
In order to explore the relationship between the catalytic performance and the acidity of BFILs, the acidity of BFILs was measured by an acid–base titration method.43 Phenolphthalein was used as an indicator, and the concentration of NaOH aqueous solution was 2 mol L−1.
Solubility of NB in the reaction solution
The solubility of NB in the reaction solution was checked by HPLC, which was similar to that used in the literature.44 The method is as follows: the slight excess of 6 NB was sonicated in an ultrasonic washer for 30 minutes and then stirred at 353 K for 5 hours. After that, 1 mL of the upper homogeneous was transferred to the ethanol solution and diluted to a fixed volume, then determined by HPLC.Evaluation of catalyst
This reaction was carried out in a 75 mL stainless steel autoclave (Parr MRS5000). In a typical experiment, NB, catalyst, BFILs or 10% sulphuric acid solution and CTAB were introduced into the autoclave. After the air in the reactor was replaced with nitrogen and hydrogen, three times, respectively, the reactions were performed at 0.8 MPa H2, 353 K, the optimal reaction conditions that were previously determined. The final products were analysed by HPLC on an Agilent 1260 (column Agilent TC-C18, 5 μm) system with UV detector (λ = 254 nm). The column temperature was 298 K. CH3CN/H2O (70/30, v/v) was used as the mobile phase and the flow rate was 1 mL min−1.Recycling of Ir/C and ionic liquids
The Ir/C catalyst in the system was separated by vacuum filtration, and the aniline by-product was separated by ordinary extraction. After most of the water was removed by distillation under reduced pressure, PAP precipitated in the ice-water bath, due to its low solubility in aqueous solution. The remaining BFILs could then be reused.35Results and discussion
Hydrogenation catalyst
In our previous study of the hydrogenation of halogenated nitrobenzene, we had compared the catalytic performance of the Ir/C and the Pd/C catalysts.13 DFT calculations showed that adsorption properties of halogenated nitrobenzene on the Ir surface were different from that on the Pd surface. The distance between oxygen and Ir was much shorter than that between oxygen and Pd. Moreover, in our study of the hydrogenation of NB to AN, we also found that the amount of PHA intermediate detected in the solution of the Ir/C catalyst was much larger than that of the Pd/C catalyst. The adsorption properties of halogenated nitrobenzene on Ir aroused our interest. As we all know, for the hydrogenation of NB to PAP, NB was first hydrogenated to the PHA intermediate and then the PHA intermediate was desorbed from the surface of the hydrogenation catalyst and converted to PAP through the acid-catalysed Bamberger rearrangement. Therefore, the desorption properties of the PHA intermediate from the hydrogenation catalyst directly determined the amount of PHA involved in the next Bamberger rearrangement. Considering the above results, we conceived of Ir as the hydrogenation catalyst for the one-pot hydrogenation of NB to PAP. Thus, we first compared the catalytic performance of Ir with the traditional Pt catalyst.The XPS spectra of Ir/C and Pt/C are shown in Fig. 1. The Ir 4f7/2 could be deconvoluted into two peaks at about 61.1 eV and 62.2 eV, which were assigned to metallic Ir(0) and Ir(4+) respectively.45,46 The Pt 4f7/2 could be deconvoluted into two peaks at about 71.5 eV and 72.3 eV, which were assigned to metallic Pt(0) and Pt(2+) respectively.47,48 The presence of Ir(4+) and Pt(2+) might be related to the oxidation of Ir and Pt particles in air.46
 | ||
| Fig. 1 Ir/C (Pt/C) XPS spectra of the survey scan (a, c), and Ir(Pt) 4f high-resolution XPS spectra of Ir/C (Pt/C) (b, d). | ||
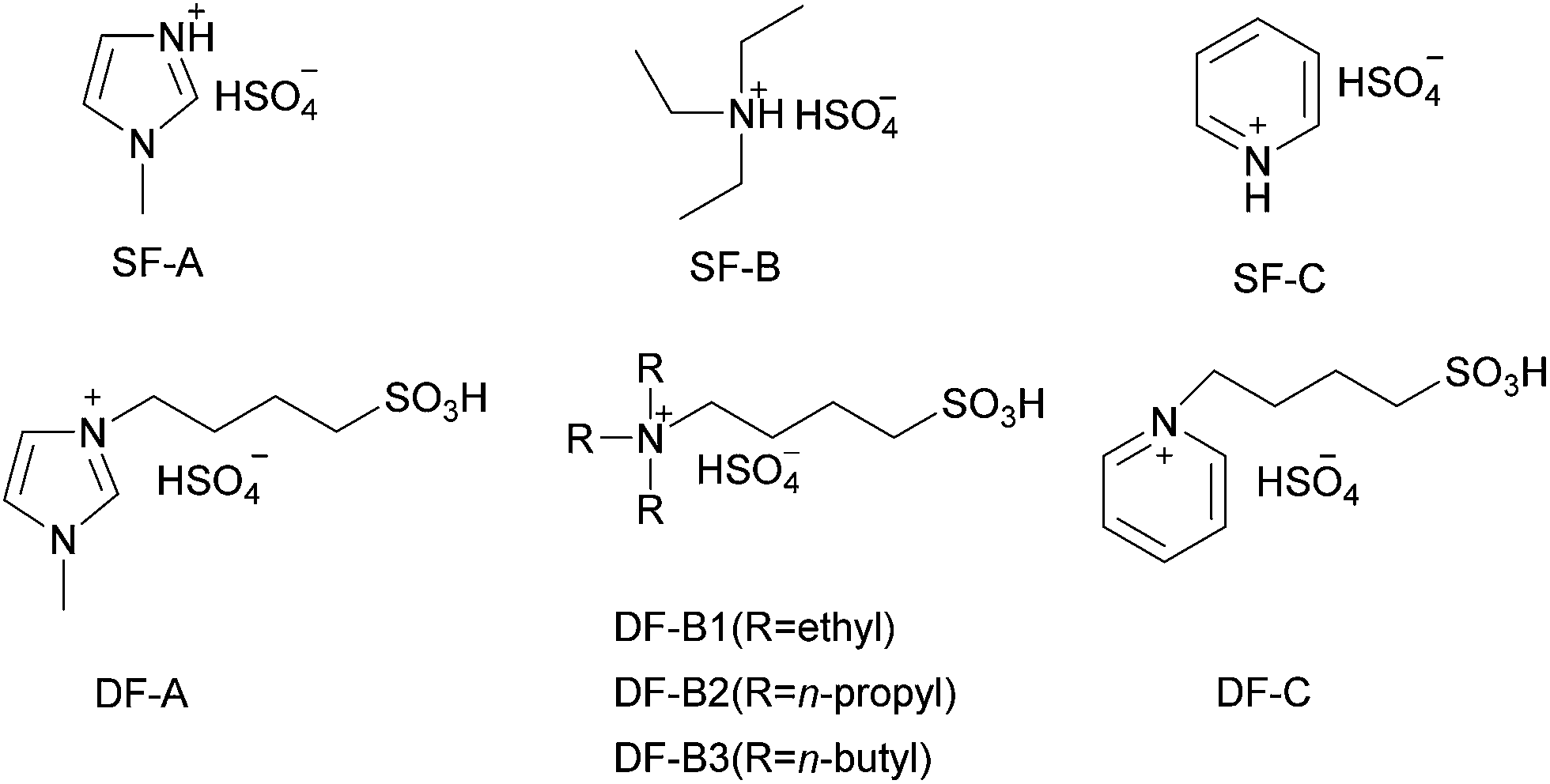
High resolution transmission electron microscopy (HR-TEM) images (Fig. 2a, b, e and f) revealed that Ir(Pt) particles were found on the surface of the activated carbon support. Moreover, the hydrogenation of NB to PAP did not cause any appreciable changes in the catalysts. At the same time, transmission electron microscopy (TEM) images and particle size distribution of the Ir(Pt) particles (Fig. 2c and d) demonstrated that the surface of metal particles were in a small and uniform state and the average particle size of the Ir/C catalyst was very close to that of the Pt/C catalyst. All these results indicated that the Ir and Pt should have similar amounts of active sites under the same metal load.
 | ||
| Fig. 2 HR-TEM (a, b, e, f) images, TEM images (c, d) and particle size distribution (c, d) of fresh and used Ir/C and Pt/C catalysts. | ||
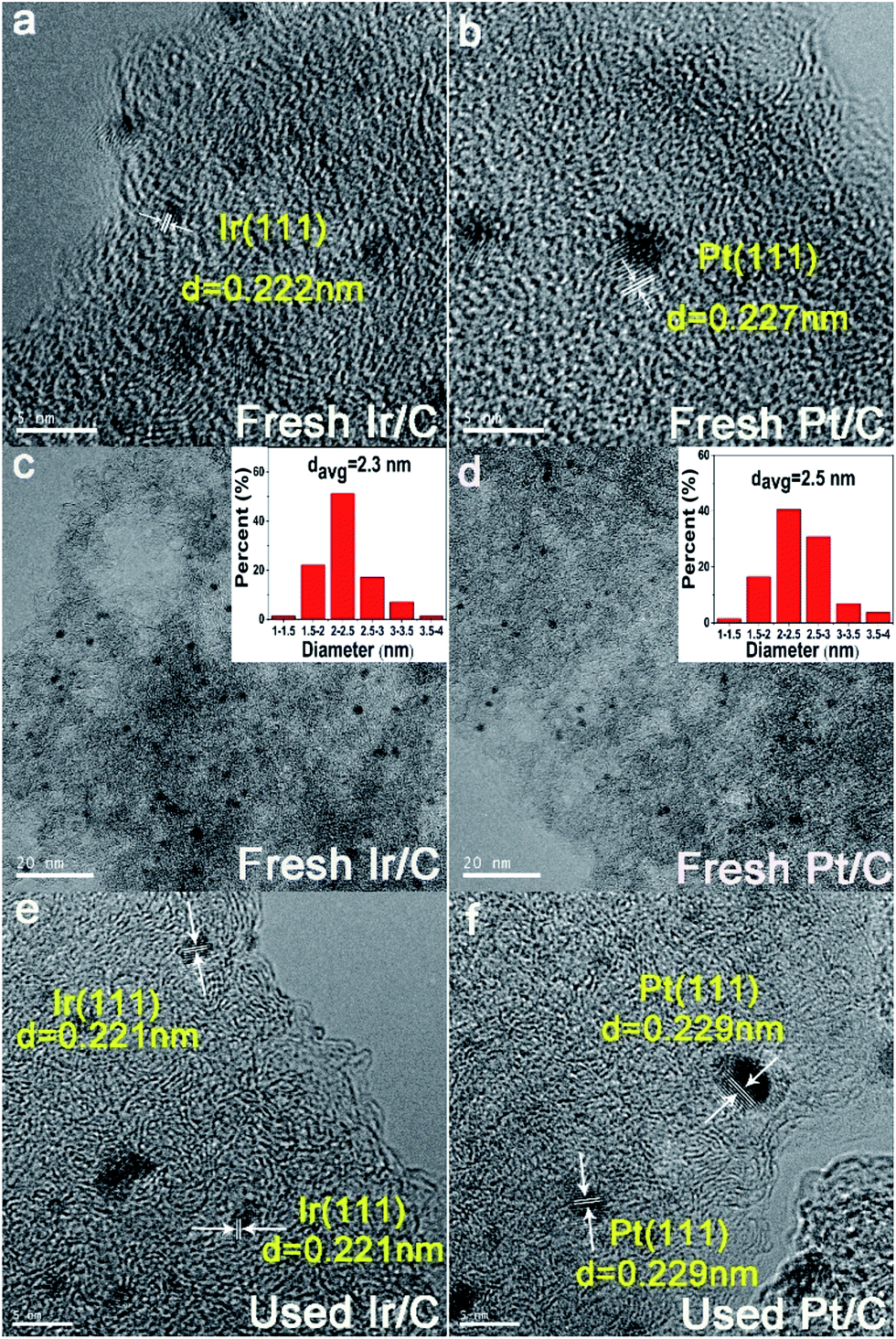
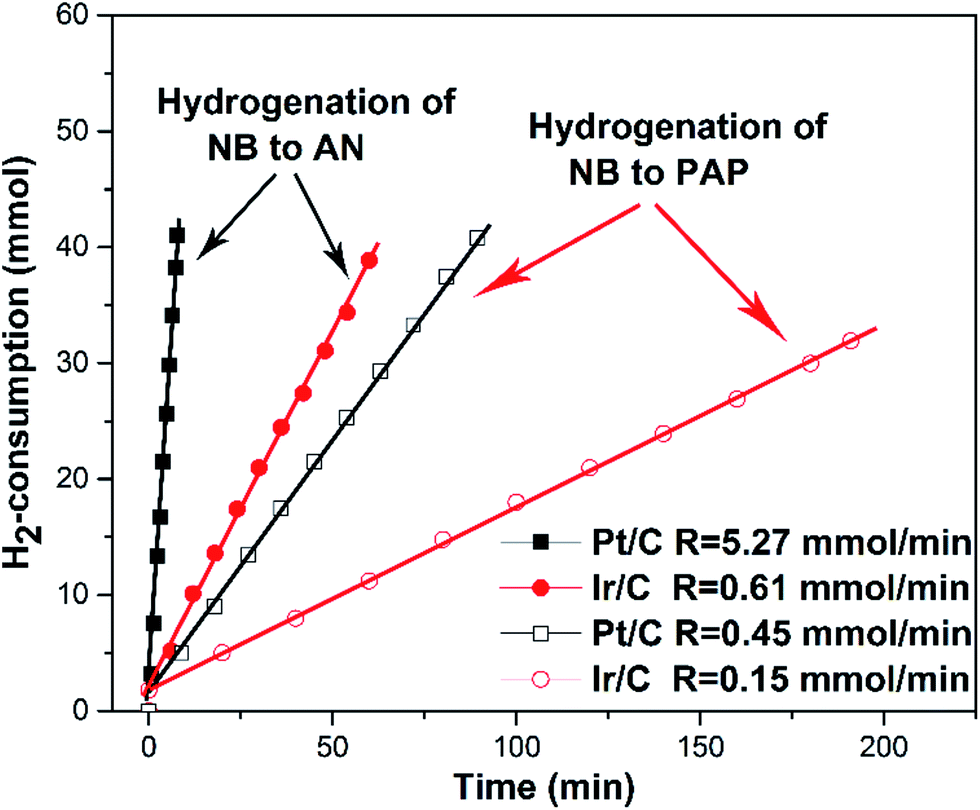
The hydrogen consumption rates for the hydrogenation of NB to AN were then studied and the results are shown in Fig. 3, where we can easily find that the hydrogen consumption rate of the Pt/C catalyst was much higher than that of the Ir/C catalyst. Obviously, this significant difference in the hydrogenation rate was not caused by the number of metal active sites. In order to find out the reason, the product distribution of the Pt/C catalyst for hydrogenation of NB to AN was then investigated and the results were demonstrated in Fig. 4. Compared to our previous study results,13 we found that the amount of the PHA intermediate distribution of Pt/C was much lower than that of Ir/C (about 21%). This indicated that the PHA intermediate desorbed more easily from the Ir surface than from the Pt surface. The more easily PHA was desorbed from the metal surface, the less PHA was left on the metal surface to be further hydrogenated to AN, which should be the main reason why the hydrogen consumption rate of Ir/C was much lower than that of Pt/C.
 | ||
| Fig. 3 Hydrogen consumption rate of the Ir/C and Pt/C catalysts for the hydrogenation of NB to ANa and hydrogenation of NB to PAPb. Reaction conditions: (a) 0.02 mol of NB, 0.1 g of catalyst, 25 mL of ethanol, PH2 = 1.0 MPa, T = 353 K, stirring rate = 1200 rpm. (b) 0.02 mol of NB, 0.1 g of catalyst, 25 mL of 10% sulphuric acid solution, 0.025 g of CTAB, PH2 = 0.8 MPa, T = 353 K, stirring rate = 1000 rpm. | ||
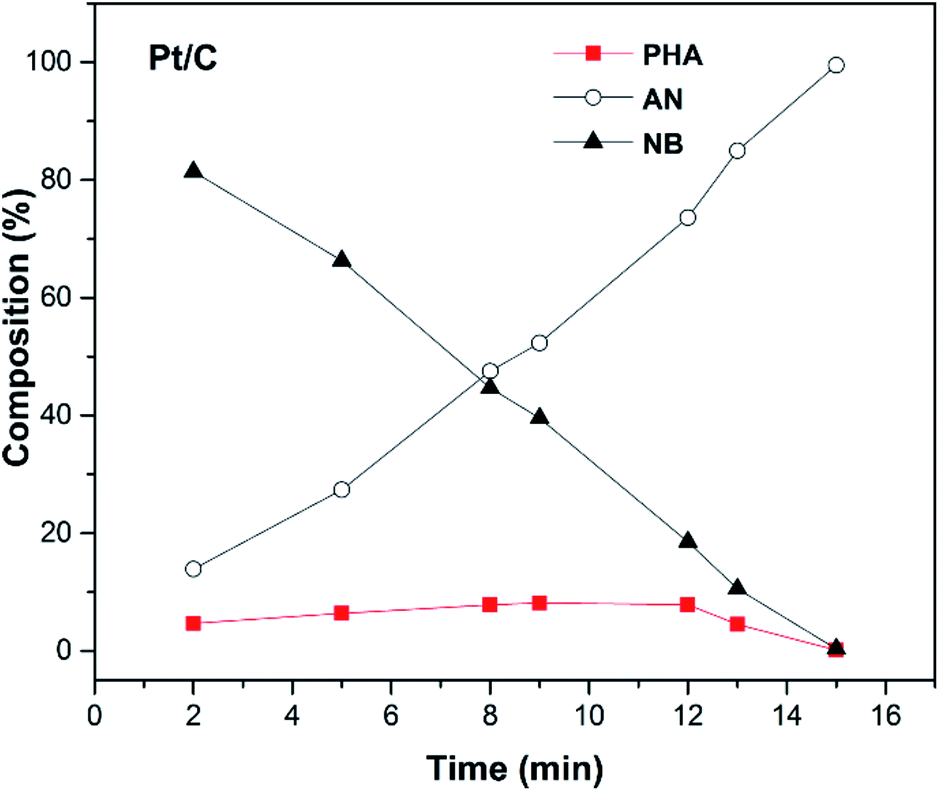 | ||
| Fig. 4 Product distribution of catalytic hydrogenation of NB to AN on the Pt/C catalyst. Reaction conditions: 0.05 g of catalyst, 25 mL of ethanol, 0.02 mol of NB, PH2 = 1.0 MPa; T = 353 K, stirring rate = 1200 rpm. | ||
As an intermediate of the hydrogenation of NB to PAP, the distinct adsorption properties of PHA on different metal catalysts could not only affect the hydrogen consumption rate, but also influence the PAP selectivity. Fig. 3 showed the hydrogen consumption rate of the hydrogenation of NB to PAP. As we had speculated, the hydrogen consumption rate of Ir and Pt catalysts all decreased sharply, compared to the hydrogenation of NB to AN, and the hydrogen consumption rate of the Ir/C catalyst was still much lower than that of the Pt/C. On the other hand, we further studied the catalytic performance of Ir/C and Pt/C for the hydrogenation of NB to PAP (Table 1). Our results indicated that the PAP selectivity of the Ir/C catalyst was significantly higher than that of the Pt/C catalyst. We thought the reason was that the easier desorption of PHA from Ir allowed more PHA to be involved in the following Bamberger rearrangement, which led to the lower hydrogen consumption rate and higher selectivity of PAP. Thus, we concluded that Ir was more suitable than Pt for the hydrogenation of NB to PAP.
| Catalyst | Conversion (%) | Selectivity (%) | |
|---|---|---|---|
| PAP | AN | ||
| a Reaction conditions: 0.1 g of catalyst, 25 mL of 10% sulphuric acid solution, 0.02 mol of NB, 0.025 g of CTAB, PH2 = 0.8 MPa, T = 353 K, stirring rate = 1000 rpm. | |||
| 1% Pt/C | 100 | 40.5 | 59.5 |
| 1% Ir/C | 100 | 60.4 | 39.5 |
Brφnsted acid functionalized ionic liquids
When the hydrogenation catalyst was determined, we selected BFILs as acid catalysts and constructed a new catalytic system with Ir/C for the hydrogenation of NB to PAP. In this regard, three types of BFILs were synthesized. The type A BFILs (SF-A, SF-B and SF-C) were single functionalized ionic liquids, which were characterized by the bisulfate ion as the anion and N-methylimidazole, triethylamine or pyridine as the cations, respectively. Based on the preparation of type A BFILs, type B BFILs (DF-A, DF-B1 and DF-C) were designed with butyl sulfonic acid functionalized to the N atom. The purpose of this reform was to increase the acid strength of the corresponding BFILs. In order to enhance the lipophilicity of DF-B1, type C BFILs (DF-B2 and DF-B3) were then prepared by replacing the ethyl group in DF-B1 with n-propyl (DF-B2) or n-butyl (DF-B3) respectively. Type C BFILs were more targeted to functionalization, compared to DF-B1. They were a type of dually functionalized ionic liquid and were expected to be used as acid catalyst and surfactant at the same time.The hydrogen consumption rates in the BFILs were firstly investigated in the presence of surfactant. As shown in Fig. 5, the hydrogen consumption rates in type A and type B BFILs were different. We could easily see that the hydrogen consumption rate in type A BFILs was much higher than that in type B BFILs. The hydrogen consumption rates in DF-A, DF-B1 and DF-C were basically within the same range. The above experimental results could be explained through the mechanism of the hydrogenation of NB to PAP. This reaction involved two competitive parallel reactions, and the acidity of BFILs affects the trend of the Bamberger rearrangement reaction. When the acidity of BFILs was stronger, the Bamberger rearrangement was dominant in the parallel reaction and the over hydrogenation side reaction was inhibited, which led to the lower hydrogen consumption rate. The acidity of type B BFILs stemmed not only from the bisulfate anion, but also the sulfonic acid, which made them more acidic than type A BFILs and resulted in the lower hydrogen consumption rate.
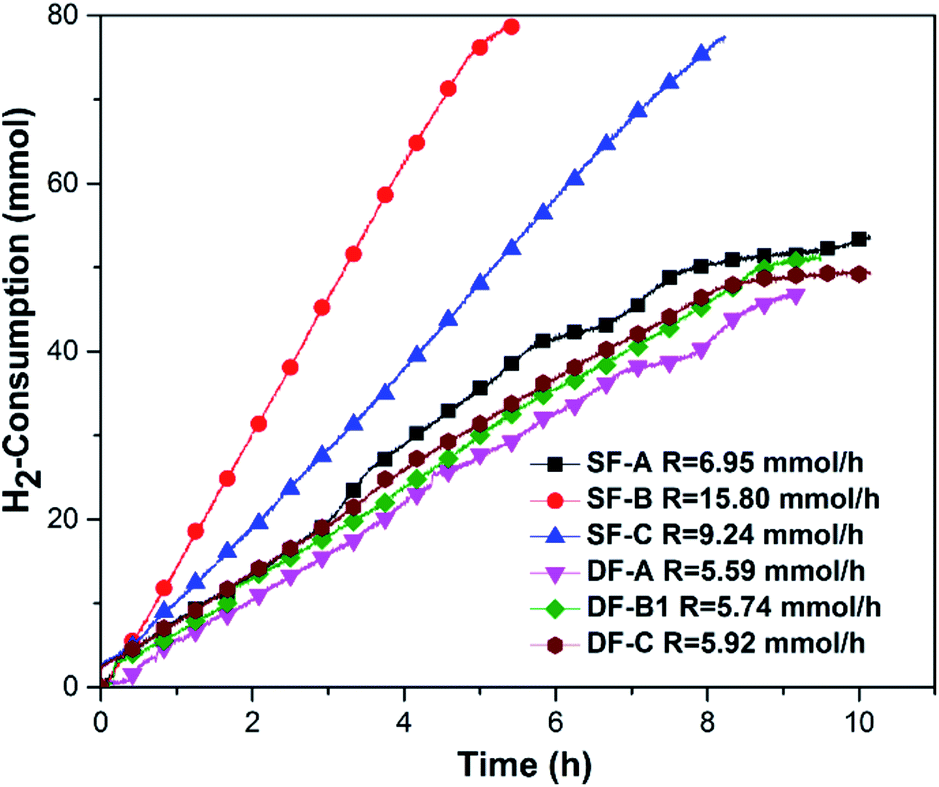 | ||
| Fig. 5 The hydrogen consumption rate of the reaction in different BFILs in the presence of CTAB. Reaction conditions: 0.02 mol of NB, 25 mL of H2O, 0.1 g of Ir/C, 0.025 g of CTAB, 353 K, 0.8 MPa, 1000 rpm, 0.05 mol BFILs of type A, 0.025 mol BFILs of type B. | ||
Considering the influence of the acidity on the Bamberger rearrangement reaction, we thought that it was necessary to further compare the acidity of the different BFILs quantitatively. Therefore, the acidity of the BFILs was quantitatively tested by sodium hydroxide titration. The relationship between the amount of acid and the catalytic performance of different kinds of BFILs was studied. The results are presented in Table 2, where it can be seen that the selectivity of PAP was proportional to the acidity of BFILs. The strong acidity of BFILs benefited the Bamberger rearrangement to form PAP and thus increased the selectivity of PAP; the relative acidity of the SO3H-functionalized ILs was stronger than that of the anion-functionalized ILs33 and thus, the type B BFILs should possess stronger acidity than that of the type A BFILs. Due to the acidity of type A BFILs being much weaker than that of type B, there was an obvious gap in the PAP selectivity between type A and type B BFILs. We could also see that the PAP selectivity in type B BFILs were very close to that in sulfuric acid. Among all the BFILs of type B, DF-A showed the best catalytic performance and the PAP selectivity of DF-A was even higher than that of sulfuric acid. We thought that this reason could be attributed to its weak steric hindrance49 and strong acidity. Our results indicate that BFILs could already replace sulfuric acid for the hydrogenation of NB to PAP in the presence of surfactant.
| BFILs | Amount of acid | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|---|
| PAP | AN | ||||
| a Reaction conditions: 0.02 mol of NB, 25 mL of H2O, 0.1 g of Ir/C, 0.025 g of CTAB, 353 K, 0.8 MPa, 1000 rpm, 0.05 mol of type A, 0.025 mol of type B. | |||||
| Type A | SF-A | 1.65 | 100 | 51.2 | 48.8 |
| SF-B | 0.83 | 100 | 29.3 | 70.7 | |
| SF-C | 1.06 | 100 | 32.7 | 67.3 | |
| Type B | DF-A | 1.94 | 100 | 62.4 | 37.6 |
| DF-B1 | 1.73 | 100 | 56.4 | 43.6 | |
| DF-C | 1.85 | 100 | 56.9 | 43.1 | |
Due to the lower solubility of NB in the reaction solution, surfactant must be added for the hydrogenation of NB to PAP. The addition of surfactant led to some problems such as a complicated system and troublesome operation. Considering the properties of the BFILs, we further explored the catalytic performance of different BFILs without the existence of surfactant. The result is shown in Table 3. As we predicted, the PAP selectivity greatly decreased both the type B BFILs and sulfuric acid, implying the non-negligible importance of surfactant for such a complex reaction system. However, the catalytic performance of DF-B1 caught our attention. Although DF-B1 was the weakest acid among all the type B BFILs, it showed the highest PAP selectivity without the existence of surfactant. We speculated that the catalytic performance of the BFILs was also related to lipophilicity in addition to acidity. The lipophilicity of the BFILs increased the solubility of the reactants, which would make the reaction occur more easily. This provided us with a good idea for modifying the DF-B1. In order to further enhance the lipophilicity of DF-B1, we increased the length of the carbon chain connected to the N atom by replacing the ethyl group in DF-B1 with the n-propyl or n-butyl carbon chain. As expected, the solubility of NB in type C BFILs improved significantly, and the selectivity of PAP increased with the elongation of the carbon chain. DF-B3 showed the best catalytic performance with 50.8% selectivity of PAP. Our results indicated that the DF-B3 dually functionalized ionic liquid could simultaneously be used as an acid catalyst and surfactant for the hydrogenation of NB to PAP.
| BFILs | Solubility (g L−1) | Conversion (%) | Selectivity (%) | ||
|---|---|---|---|---|---|
| PAP | AN | ||||
| a Reaction conditions: 0.02 mol NB, 0.025 mol BFILs, 25 mL of H2O, 0.1 g of Ir/C, 353 K, 0.8 MPa, 1000 rpm. | |||||
| H2SO4 | — | — | 100 | 43.9 | 56.1 |
| Type B | DF-A | 3.1 | 100 | 32.6 | 67.4 |
| DF-B1 | 3.4 | 100 | 37.8 | 62.2 | |
| DF-C | 2.8 | 100 | 30.8 | 69.2 | |
| Type C | DF-B2 | 7.6 | 100 | 44.3 | 55.7 |
| DF-B3 | 11.5 | 100 | 50.8 | 49.2 | |
Recyclability is one of the important properties of catalysts. Herein, we chose the catalytic system composed of Ir/C and DF-B3 to conduct the recycling experiments. As shown the Fig. 6, the conversion of NB remained stable and the selectivity of PAP decreased slightly after being recycled three times. This indicated that the new catalytic system of Ir/C and DF-B3 had good stability in the hydrogenation of NB to PAP.
 | ||
| Fig. 6 Recycling experiments of Ir/C and DF-B3 for hydrogenation of NB to PAP. Reaction conditions: 0.02 mol of NB, 0.025 mol of DF-B3, 25 mL of H2O, 0.1 g of Ir/C, 353 K, 0.8 MPa, 1000 rpm. | ||
Conclusions
The desorption properties of the PHA intermediate from the hydrogenation metal catalyst and the acidity of the acid catalyst were the key factors in the one-pot hydrogenation of NB to PAP. Our work included the following two aspects: the PHA intermediate was more easily desorbed from the surface of the Ir catalyst, compared to the traditional Pt catalyst; the BFILs with bisulfate ion and sulfonic acid exhibited similar acidity to sulfuric acid. A new catalyst system was constructed through the combination of Ir/C and BFILs and applied to the catalysis of the one-pot hydrogenation of NB to PAP. Our results indicate that the PAP selectivity of Ir/C and [SO3H-bmim][HSO4] was far greater than that of Pt/C and sulfuric acid. In addition, we found that the dually functionalized ionic liquid ([HSO3-b-N-Bu3][HSO4]) could be used simultaneously as acid catalyst and surfactant, due to its higher lipophilicity. Therefore, the great catalytic performance of the Ir/C and BFILs system could open new ground for the study of the catalyzed hydrogenation of NB to PAP.Acknowledgements
This work is financially supported by National Natural Science Foundation of China (Grant No. 21473159, 21476208) and Natural Science Foundation of Zhejiang Province (LY17B060008).Notes and references
- K. I. Min, J. S. Choi, Y. M. Chung, W. S. Ahn, R. Ryoo and P. K. Lim, Appl. Catal., A, 2008, 337, 97–104 CrossRef CAS.
- C. V. Rode, M. J. Vaidya, R. Jaganathan and R. V. Chaudhari, Chem. Eng. Sci., 2001, 56, 1299–1304 CrossRef CAS.
- Z. Dong, T. Wang, J. Zhao, T. Fu, X. Guo, L. Peng, B. Zhao, N. Xue, W. Ding and Z. Xie, Appl. Catal., A, 2016, 520, 151–156 CrossRef CAS.
- M. Blosi, S. Albonetti, A. L. Costa, N. Sangiorgi and A. Sanson, Chem. Eng. J., 2013, 215–216, 616–625 CrossRef CAS.
- Z. Ma, L. Zhang, R. Chen, W. Xing and N. Xu, Chem. Eng. J., 2008, 138, 517–522 CrossRef CAS.
- M. A. Bhosale, D. R. Chenna, J. P. Ahire and B. M. Bhanage, RSC Adv., 2015, 5, 52817–52823 RSC.
- E. A. Braude, Nature, 1952, 169, 80 CrossRef.
- S. K. Tanielyan, J. J. Nair, N. Marin, G. Alvez, R. J. McNair, D. Wang and R. L. Augustine, Org. Process Res. Dev., 2007, 11, 681–688 CrossRef CAS.
- Y. Gao, F. Wang, S. Liao and D. Yu, React. Kinet. Catal. Lett., 1998, 64, 351–357 CrossRef CAS.
- B. Zhang, Y. Yuan, K. Philippot and N. Yan, Catal. Sci. Technol., 2015, 5, 1683–1692 CAS.
- J. Li, R. R. Nasaruddin, Y. Feng, J. Yang, N. Yan and J. Xie, Chem.–Eur. J., 2016, 22, 14816–14820 CrossRef PubMed.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS PubMed.
- L. Ma, J. Wang, H. Wang, Q. Zhang, C. Lu, X. He and X. Li, Chin. J. Chem. Eng., 2017, 25, 306–312 CrossRef CAS.
- G. Quartarone, L. Ronchin, A. Tosetto and A. Vavasori, Appl. Catal., A, 2014, 475, 169–178 CrossRef CAS.
- S. Liu, Y. Hao and J. Jiang, Ind. Eng. Chem. Res., 2014, 53, 8372–8375 CrossRef CAS.
- S. Wang, Y. Ma, Y. Wang, W. Xue and X. Zhao, J. Chem. Technol. Biotechnol., 2008, 83, 1466–1471 CrossRef CAS.
- S. Wang, Y. Wang, Y. Gao and X. Zhao, Chin. J. Catal., 2010, 31, 637–644 CrossRef CAS.
- S. Wang, Y. Jin, B. He, Y. Wang and X. Zhao, Sci. China: Chem., 2010, 53, 1514–1519 CrossRef CAS.
- P. Liu, Y. Hu, M. Ni, K. You and H. Luo, Catal. Lett., 2010, 140, 65–68 CrossRef CAS.
- A. Deshpande, F. Figueras, M. L. Kantam, K. J. Ratnam, R. S. Reddy and N. S. Sekhar, J. Catal., 2010, 275, 250–256 CrossRef CAS.
- Y. Liu, Y. Fang, X. Lu, Z. Wei and X. Li, Chem. Eng. J., 2013, 229, 105–110 CrossRef CAS.
- T. Zhang, J. Jiang and Y. Wang, Org. Process Res. Dev., 2015, 19, 2050–2054 CrossRef CAS.
- T. Komatsu and T. Hirose, Appl. Catal., A, 2004, 276, 95–102 CrossRef CAS.
- X. Liu, M. Liu, X. Guo and J. Zhou, Catal. Commun., 2008, 9, 1–7 CrossRef CAS.
- K. L. Toews, R. M. Shroll, C. M. Wai and N. G. Smart, Anal. Chem., 2002, 67, 4040–4043 CrossRef.
- E. Rafiee and F. Mirnezami, J. Mol. Struct., 2017, 1130, 296–302 CrossRef CAS.
- J. Lunagariya, A. Dhar and R. L. Vekariya, RSC Adv., 2017, 7, 5412–5420 RSC.
- Y. Liu, W. Xiao, S. Xia and P. Ma, Carbohydr. Polym., 2013, 92, 218–222 CrossRef CAS PubMed.
- F. Parveen, K. Gupta and S. Upadhyayula, Carbohydr. Polym., 2017, 159, 146–151 CrossRef CAS PubMed.
- H. Wang, X. Meng, G. Zhao and S. Zhang, Green Chem., 2017, 19, 1462–1489 RSC.
- P. Gan and S. Tang, Chin. J. Chem. Eng., 2016, 24, 1497–1504 CrossRef CAS.
- A. G. Khiratkar, P. N. Muskawar and P. R. Bhagat, RSC Adv., 2016, 6, 105087–105093 RSC.
- N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed. Engl., 2010, 49, 5549–5553 CrossRef CAS PubMed.
- R. Kore and R. Srivastava, J. Mol. Catal. A: Chem., 2013, 376, 90–97 CrossRef CAS.
- Y. Cui, D. Yuan, Y. Wang and X. Zhao, J. Ind. Eng. Chem., 2009, 60, 345–350 CAS.
- J. Weng, C. Wang, H. Li and Y. Wang, Green Chem., 2006, 8, 96–99 RSC.
- J. Sun, H. Li, H. Song, Q. Wu, Y. Zhao and Q. Jiao, RSC Adv., 2015, 5, 87200–87205 RSC.
- D. Fang, X. L. Zhou, A. Zhiwen Ye and Z. L. Liu, Ind. Eng. Chem. Res., 2006, 45, 7982–7984 CrossRef CAS.
- S. Liu, X. Liu and C. Wang, Ind. Eng. Chem. Res., 2013, 52, 16719–16723 CrossRef CAS.
- Y. Zhao, J. Long, F. Deng, X. Liu, Z. Li, C. Xia and J. Peng, Catal. Commun., 2009, 10, 732–736 CrossRef CAS.
- D. Fang, Q. R. Shi, J. Cheng, K. Gong and Z. L. Liu, Appl. Catal., A, 2008, 345, 158–163 CrossRef CAS.
- J. Gui, X. Cong, D. Liu, X. Zhang, Z. Hu and Z. Sun, Catal. Commun., 2004, 5, 473–477 CrossRef CAS.
- X. L. Zhang, H. L. An, H. Q. Zhang, X. Q. Zhao and Y. J. Wang, Ind. Eng. Chem. Res., 2014, 53, 16707–16714 CrossRef CAS.
- F. Yu, G. Liu, E. Liang, C. Gao and W. Zhong, Mil. Med. Sci., 2012, 36, 49–55 CAS.
- L. Yuan, Z. Yan, L. Jiang, E. Wang, S. Wang and G. Sun, J. Energy Chem., 2016, 25, 805–810 CrossRef.
- G. Y. Fan, L. Zhang, H. Y. Fu, M. L. Yuan, R.-X. Li, H. Chen and X. J. Li, Catal. Commun., 2010, 11, 451–455 CrossRef CAS.
- J. Fan, K. Qi, L. Zhang, H. Zhang, S. Yu and X. Cui, ACS Appl. Mater. Interfaces, 2017, 9, 18008–18014 CAS.
- L. Zhu, T. Zheng, C. Yu, J. Zheng, Z. Tang, N. Zhang, Q. Shu and B. H. Chen, Appl. Surf. Sci., 2017, 409, 29–34 CrossRef CAS.
- T. Patra, S. Ahamad and S. Upadhyayula, Appl. Catal., A, 2015, 506, 228–236 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |