DOI:
10.1039/C7RA04247G
(Paper)
RSC Adv., 2017,
7, 42519-42528
An amphipathic trans-acting phosphorothioate RNA element delivers an uncharged phosphorodiamidate morpholino sequence in mdx mouse myotubes†
Received
14th April 2017
, Accepted 22nd August 2017
First published on 4th September 2017
Abstract
An efficient method for the delivery of uncharged polyA-tailed phosphorodiamidate morpholino sequences (PMO) in mammalian cells consists of employing a synthetic 8-mer amphipathic trans-acting poly-2′-O-methyluridylic thiophosphate triester element (2′-OMeUtaPS) as a transfection reagent. Unlike the dTtaPS DNA-based element, this RNA element is potent at delivering polyA-tailed PMO sequences to HeLa pLuc 705 cells or to myotube muscle cells. However, much like dTtaPS, the 2′-OMeUtaPS-mediated internalization of PMO sequences occurs through an energy-dependent mechanism; macropinocytosis appears to be the predominant endocytic pathway used for cellular uptake. The transfected PMO sequences induce alternate splicing of either the pre-mRNA encoding luciferase in HeLa pLuc 705 cells or the excision of exon 23 from the pre-mRNA encoding dystrophin in myotube muscle cells of the mdx mouse model of muscular dystrophy with an efficiency comparable to that of commercial cationic lipid reagents but without detrimental cytotoxicity.
Introduction
Human genes undergo alternative splicing events, which are activated by a complex and highly regulated machinery requiring the sequence-specific binding of multiple proteins to nuclear pre-mRNAs.1,2 Alternative splicing events are crucial to the treatment of Duchenne muscular dystrophy (DMD), which is the most common form of muscular dystrophy. This disease leads to progressive deterioration of muscle function and is caused mainly by a frameshift deletion, non-sense or duplication mutations in the DMD gene encoding the protein dystrophin.3–6 Thus, skipping an exon that contains a nonsense coding mutation or an out-of-frame genomic deletion, would allow the production of a functional dystrophin protein upon deletion of the non-sense coding mutation(s) or restoration of the correct mRNA reading frame.7,8
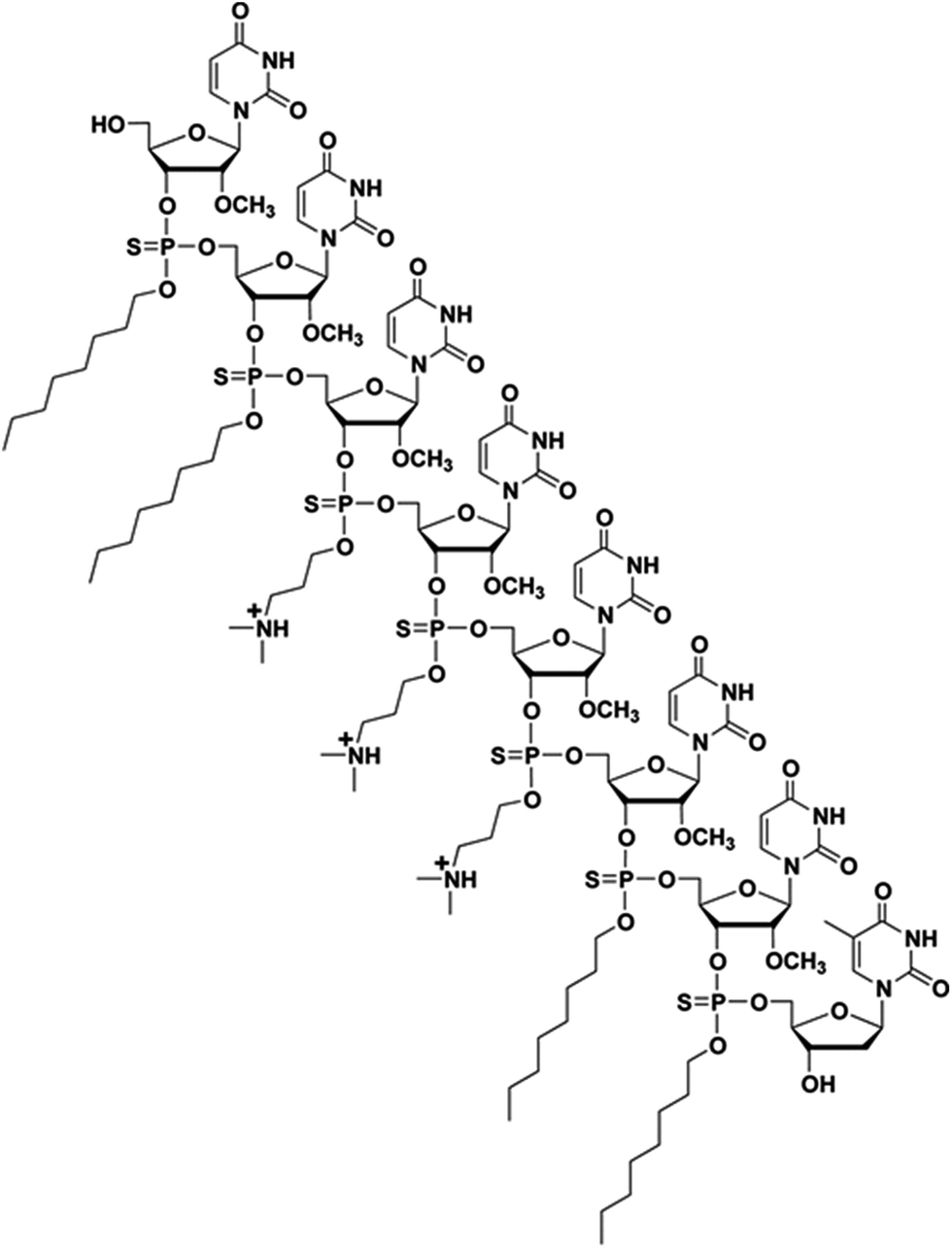
Steric interference imparted by RNase H-incompetent oligonucleotide analogues, complementary to specific pre-mRNA splice sites, has been shown to be efficient at redirecting exon splicing during assembly of mature mRNAs.1,2 Specifically, uncharged peptide nucleic acids (PNA), phosphorodiamidate morpholino (PMO) sequences and negatively charged 2′-O-methyl and 2′-O-methoxyethyl RNA phosphorothioates, locked nucleic acid (LNA), and tricyclo–DNA sequences have been found effective in exon skipping experiments.1,7 Out of these modified nucleic acid sequences, the 2′-O-methyl RNA phosphorothioate and PMO sequences have been extensively investigated in exon-skipping therapies for DMD; low levels of dystrophin production were reported in two independent clinical trials.7 Of particular relevance, the use of PMOs leading to the excision of exon 23 from dystrophin mRNA, produced superior results in the mdx mouse model of muscular dystrophy, when compared to treatment with 2′-O-methyl RNA phosphorothioate sequences.9 Although PMO sequences exhibit superior stability and safety relative to other nucleic acid sequences, the cellular internalization of PMOs is notoriously poor. The conjugation of cationic cell-penetrating peptides (CPP) or a tetraguanidinium-linked nonpeptidic transporter to PMOs led to improved cellular uptake of these sequences; CPP conjugates resulted in pre-mRNA splicing correction activities in mammalian cells and animal models.10 However, the positively charged CPP–PMO conjugates produced undesirable toxicity.11 A less laborious and more efficient approach to the delivery of PMO sequences is necessary to prompt an alternate splicing of exon 23 from dystrophin mRNA transcripts in mammalian muscle cells and/or mdx mice. In this context, we have recently reported the use of a chemically synthesized amphipathic trans-acting polythymidylic thiophosphate triester DNA element (dTtaPS) for the delivery of polyA-tailed PNA or PMO sequences in several mammalian cell lines.12,13 The recognition of the polyA-tailed PNA or PMO sequences by dTtaPS was found to be specific, necessary and sufficient for internalization of PNA and PMO sequences in HeLa pLuc 705 cells; restoration of luciferase activity in this cell line confirmed the correct splicing of luciferase pre-mRNA transcripts. The dTtaPS-mediated cellular internalization of PNA sequences was ∼10-fold greater than that of PMO sequences.12 Such a discrepancy in cellular internalization was tentatively rationalized on the basis of a presumably greater affinity of dTtaPS for polyA-tailed PNA than for polyA-tailed PMO sequences. Thus, the development of a trans-acting nucleic acid element that would exhibit a higher affinity for polyA-tailed PMO sequences became a matter of urgency. Given that PMO sequences have shown high binding affinity for complementary RNA sequences14 and given the fact that PMO–RNA duplexes have been found to be more stable than DNA–RNA duplexes,15 we hypothesized that a trans-acting 2′-OMe RNA element might exhibit higher affinity for polyA-tailed PMO sequences than dTtaPS. With the intent of substantiating our hypothesis, the chemical synthesis of 2′-OMeUtaPS, an octameric 2′-O-methyluridylic acid functionalized with three N,N-dimethylaminopropyl and four octyl thiophosphate triester functions (Fig. 1) was carried out. The purpose of this investigation is to: (i) validate the functionality of these sequences by restoring luciferase production in HeLa pLuc 705 cells through alternate splicing and, (ii) demonstrate efficient excision of exon 23 from dystrophin pre-mRNA in mdx mouse myotubes.
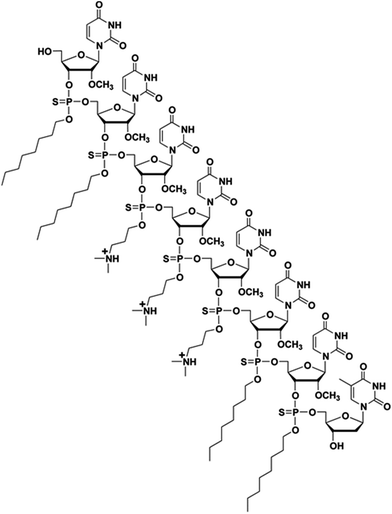 |
| | Fig. 1 Chemical structure of a chimeric 2′-OMeUtaPS transfection reagent. | |
Results and discussion
The chemical synthesis of 2′-OMeUtaPS
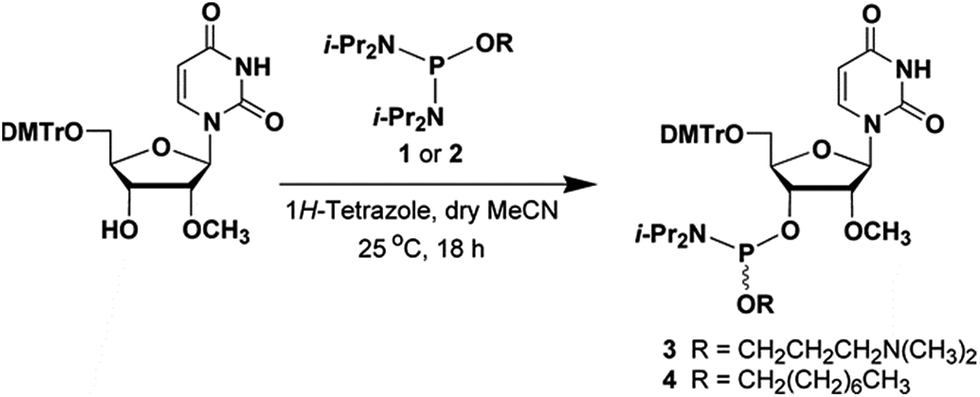
The phosphordiamidites 1 and 2 needed for the preparation of the ribonucleoside phosphoramidites 3 and 4 (Fig. 2) were prepared as reported earlier.12,13 Treatment of commercially available 5′-O-(4,4′-dimethoxytriyl)-2′-O-methyluridine with the phosphordiamidite 1 or 2 and 1H-tetrazole in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, respectively, in anhydrous MeCN afforded the ribonucleoside phosphoramidite 3 or 4, in yields of ca. 80–85% after purification by chromatography on silica gel and subsequent lyophilization from dry benzene. The identity of phosphoramidites 3 and 4 was confirmed by 31P-NMR spectroscopy and high-resolution mass spectrometry. (HRMS, data shown as ESI.†) The solid-phase synthesis of 2′-OMeUtaPS on an appropriately functionalized controlled-pore glass (CPG) support loaded with 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine as the leader nucleoside was easily achieved using the ribonucleoside phosphoramidites 3 and 4 as 0.1 M solutions in MeCN. The coupling time of the 1H-tetrazole-activated phosphoramidites 3 and 4 was extended to 10 min to maximize stepwise coupling efficiencies, which were determined to be in the order of 98–99% with an overall synthesis yield of 92 ± 5% based on the 4,4′-dimethoxytrityl cation assay.16 Upon exposure to pressurized gaseous amines and release from the CPG support, the purity of diastereomeric 2′-OMeUtaPS was assessed by C4-RP-HPLC analysis. Similar to the chromatographic profile of dTtaPS,13 2′-OMeUtaPS displayed numerous broad unresolved product peaks consistent with the diastereomeric and amphiphilic features of this class of compounds (data not shown). The purity of 2′-OMeUtaPS was nonetheless highly comparable to that of dTtaPS and was deemed adequate to support our investigations.
:2:1, respectively, in anhydrous MeCN afforded the ribonucleoside phosphoramidite 3 or 4, in yields of ca. 80–85% after purification by chromatography on silica gel and subsequent lyophilization from dry benzene. The identity of phosphoramidites 3 and 4 was confirmed by 31P-NMR spectroscopy and high-resolution mass spectrometry. (HRMS, data shown as ESI.†) The solid-phase synthesis of 2′-OMeUtaPS on an appropriately functionalized controlled-pore glass (CPG) support loaded with 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine as the leader nucleoside was easily achieved using the ribonucleoside phosphoramidites 3 and 4 as 0.1 M solutions in MeCN. The coupling time of the 1H-tetrazole-activated phosphoramidites 3 and 4 was extended to 10 min to maximize stepwise coupling efficiencies, which were determined to be in the order of 98–99% with an overall synthesis yield of 92 ± 5% based on the 4,4′-dimethoxytrityl cation assay.16 Upon exposure to pressurized gaseous amines and release from the CPG support, the purity of diastereomeric 2′-OMeUtaPS was assessed by C4-RP-HPLC analysis. Similar to the chromatographic profile of dTtaPS,13 2′-OMeUtaPS displayed numerous broad unresolved product peaks consistent with the diastereomeric and amphiphilic features of this class of compounds (data not shown). The purity of 2′-OMeUtaPS was nonetheless highly comparable to that of dTtaPS and was deemed adequate to support our investigations.
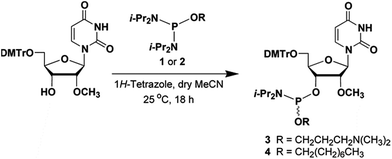 |
| | Fig. 2 Chemical illustrations of the phosphordiamidites 1 and 2 and ribonucleoside phosphoramidites 3 and 4 needed for the solid-phase synthesis of 2′-OMeUtaPS. DMTr, 4,4′-dimethoxytrityl; i-Pr, isopropyl. | |
Concentration-dependence of the 2′-OMeUtaPS-assisted internalization of PMO sequences in live HeLa pLuc 705 cells for restoration of luciferase activity
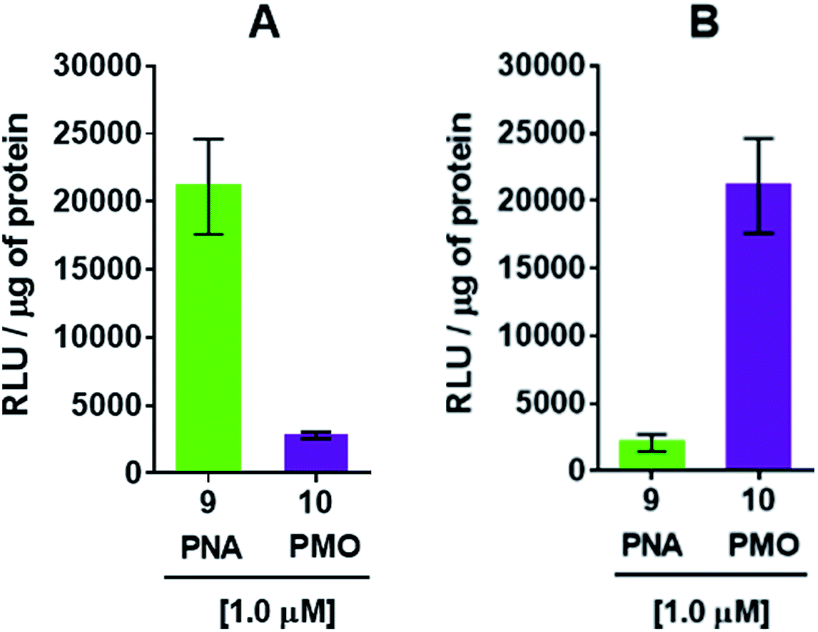
The efficiency of dTtaPS in mediating the cellular uptake of polyA-tailed PNA and polyA-tailed PMO sequences into various mammalian cell lines was assessed earlier by flow cytometry and through a splice correction assay performed in HeLa pLuc 705 cells.12,13 While the cellular internalization of polyA-tailed PMO sequences 5 and 6 (Table 1) exceeded 60% of the total number of cells in three out of the four cell lines investigated, the cellular uptake of PMO sequence 7, lacking the polyA tail, occurred in only 10–15% of the total number of cells in all cell lines under study.12,13 When assayed in HeLa pLuc 705 cells, the dTtaPS-assisted delivery of the PNA sequence 9 was found more efficient (8–10-fold) than that of PMO sequence 10 on the basis of luciferase activity production (Fig. 3A). However, when 2′-OMeUtaPS was used for the delivery of each sequence in HeLa pLuc 705 cells, uptake of the PMO sequence 10 (Table 1) was more efficient (8–10 fold) than that of the PNA sequence 9 based on restoration of luciferase activity (Fig. 3B).
Table 1 Commercial PMO and PNA sequences
| Sequence |
Type |
5′-Sequence-3′a |
| Fl, fluorescein; 16 is a fully phosphorothioated RNA sequence. |
| 5 |
PMO |
CCTCTTACCTCAGTTACA-AAAAAA-Fl |
| 6 |
PMO |
GTGGCCGTTTACGTCGCC-AAAAAA-Fl |
| 7 |
PMO |
CTTCTTACCTCAGTTACA-Fl |
| 8 |
PNA |
Fl-GTGGCCGTTTACGTCGCC-AAAAAA-NH2 |
| 9 |
PNA |
H-CCTCTTACCTCAGTTACA-AAAAAA-NH2 |
| 10 |
PMO |
CCTCTTACCTCAGTTACA-AAAAAA |
| 11 |
PMO |
CCTCTTACCTCAGTTACA-AATAAA |
| 12 |
PMO |
CCTCTTACCTCAGTTACA-TTTTTT-Fl |
| 13 |
PMO |
CCTCTTACCTCAGTTACA-CCCCCC-Fl |
| 14 |
PMO |
GGCCAAACCTCGGCTTACCTG-AAAAAA |
| 15 |
PNA |
H-GGCCAAACCTCGGCTTACCTG-AAAAAA-NH2 |
| 16 |
2′-OMe |
GGCCAAACCUCGGCUUACCUGAAAU |
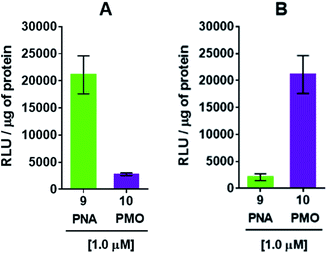 |
| | Fig. 3 (A) dTtaPS-mediated delivery of polyA-tailed PNA and PMO sequences 9 and 10, respectively, in HeLa pLuc 705 cells cultured in serum-containing medium. (B) 2′-OMeUtaPS-mediated delivery of polyA-tailed PNA and PMO sequences 9 and 10, respectively, under conditions identical to those used in (A). In all experiments, the concentration of each sequence was kept at 1.0 μM whereas the concentration of dTtaPS or 2′-OMeUtaPS was kept at 2.0 μM. Error bars represent the mean ± SD of three independent experiments. RLU, relative light units. | |
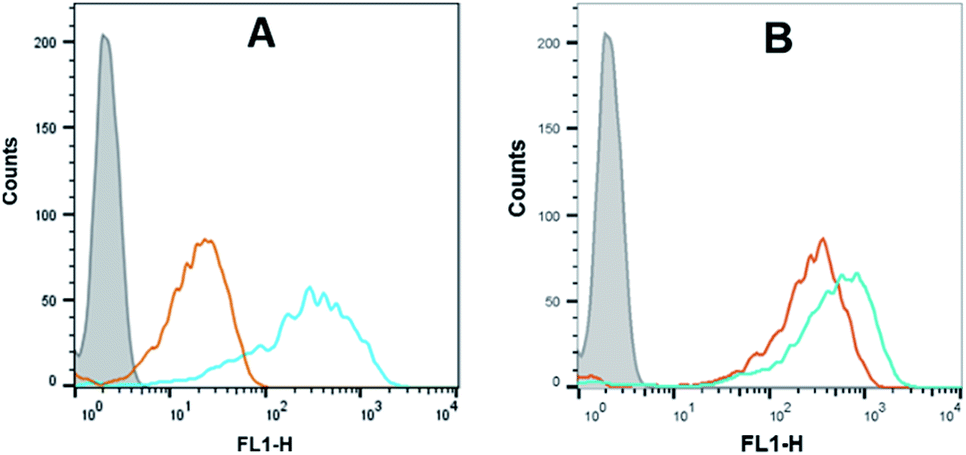
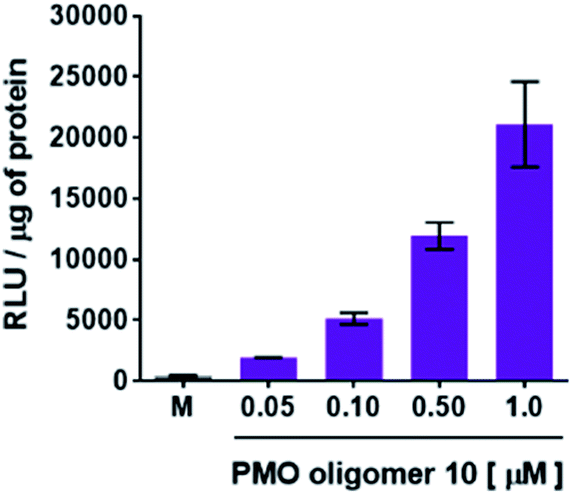
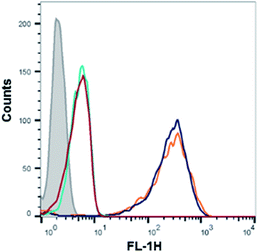
Flow cytometry analysis of the dTtaPS-mediated cellular uptake of the control PMO sequence 6 is significantly less than that of the control PNA sequence 8 as determined by the level of fluorescence per cell (Fig. 4A); this is consistent with the luciferase assay results obtained from the delivery of the PNA and PMO sequences 9 and 10, respectively (Fig. 3A). However, flow cytometry analysis of the 2′-OMeUtaPS-assisted delivery of polyA-tailed PMO and PNA sequences 6 and 8, respectively, revealed a marginally higher level of fluorescence per cell for the PNA sequence 8 when compared to that of the PMO sequence 6 (Fig. 4B). The results of this experiment suggest that the 2′-OMeUtaPS-assisted delivery of polyA-tailed PNA sequence 8 is comparably efficient to that of the PMO sequence 6 but the affinity of 2′-OMeUtaPS for the polyA-tail of PNA sequence 9 appears to interfere with either endosomal release of the sequence or nuclear membrane penetration; either outcome can lead to poor exon skipping activity in HeLa pLuc 705 cells when compared to that produced from the 2′-OMeUtaPS-mediated delivery of polyA-tailed PMO sequence 10 (Fig. 3B). The 2′-OMeUtaPS-assisted internalization of PMO sequence 10 in HeLa pLuc 705 cells is concentration-dependent. Fig. 5, clearly shows that the PMO sequence induced splice correction of the pre-mRNA encoding luciferase; the production of luciferase activity increased commensurately with the concentration of PMO sequence 10.
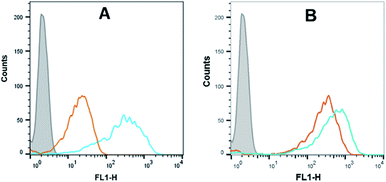 |
| | Fig. 4 (A) Flow cytometry analysis of the dTtaPS-assisted cellular uptake of the control PMO sequence 6 (peak area with an orange border) and PNA sequence 8 (peak area with a cyan border) in HeLa pLuc 705 cells. (B) Same as in (A) but using 2′-OMeUtaPS as the delivery reagent. Gray peak area, untreated HeLa pLuc 705 cells. Experimental conditions are described in the Experimental section. | |
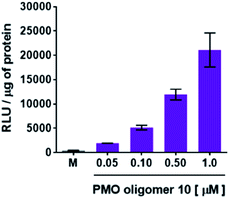 |
| | Fig. 5 Concentration-dependence of the 2′-OMeUtaPS-mediated delivery of PMO sequence 10 on luciferase activity production in serum-containing media. The concentration of 2′-OMeUtaPS was kept at 2.0 μM in all experiments. Error bars represent the mean ± SD of three independent experiments. RLU, relative light units. | |
Recognition of the PMO-polyA stretch by 2′-OMeUtaPS is sequence specific
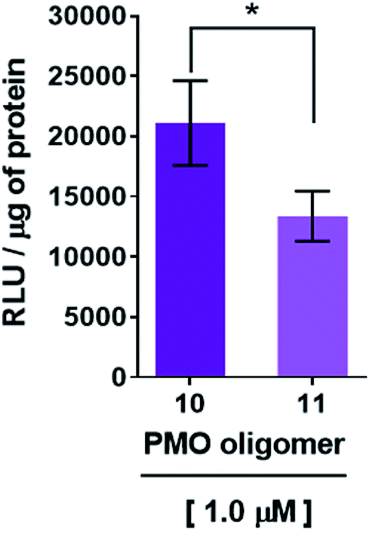
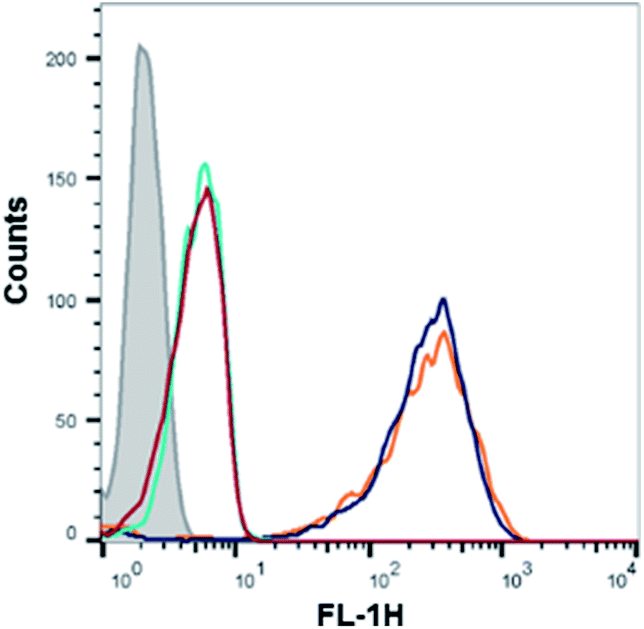
Recognition of the PMO-polyA tail of sequence 10 by either dTtaPS or 2′-OMeUtaPS is necessary for cellular internalization in mammalian cells. This was first demonstrated by replacing one PMO-A with a PMO-T in the polyA stretch of sequence 10. As shown in Fig. 6, the presence of a PMO-T in the middle of the polyA tail of PMO sequence 11 resulted in a statistically significant decrease in luciferase activity that is consistent with a weaker binding of 2′-OMeUtaPS to the modified polyA stretch of PMO sequence 11. Moreover, replacing the polyA stretch of 5 with a PMO polyT or a polyC tail led to dismal uptake of PMO sequence 12 or 13 in HeLa pLuc 705 cells, as demonstrated by flow cytometry analyses (Fig. 7). The results of this experiment show that the 2′-OMeUtaPS-mediated internalization of PMO sequence 12 or 13 is comparable to that of PMO sequence 7, lacking the polyA tail, or that of PMO sequence 5 in the absence of 2′-OMeUtaPS.
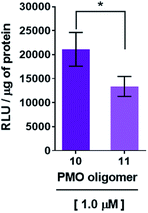 |
| | Fig. 6 Decreased recognition and internalization of PMO sequence 11 by 2′-OMeUtaPS in HeLa pLuc 705 cells resulting from an A → T substitution in the polyA tail of PMO sequence 10. The concentrations of 2′-OMeUtaPS was kept at 2.0 μM. Error bars represent the mean ± SD of three independent experiments. RLU, relative light units. | |
 |
| | Fig. 7 Flow cytometry analysis of the 2′-OMeUtaPS-assisted cellular uptake of the PMO sequence 5 (peak area with a black border), control PMO sequence 6 (peak area with an orange border), PMO sequence 12 (peak area with a red border) and PMO sequence 13 (peak area with a cyan border) in HeLa pLuc 705 cells. Gray peak area accounts for untreated HeLa pLuc 705 cells. | |
The 2′-OMeUtaPS-mediated internalization of polyA-tailed PMO sequence 10 in HeLa pLuc 705 cells is inhibited by urea
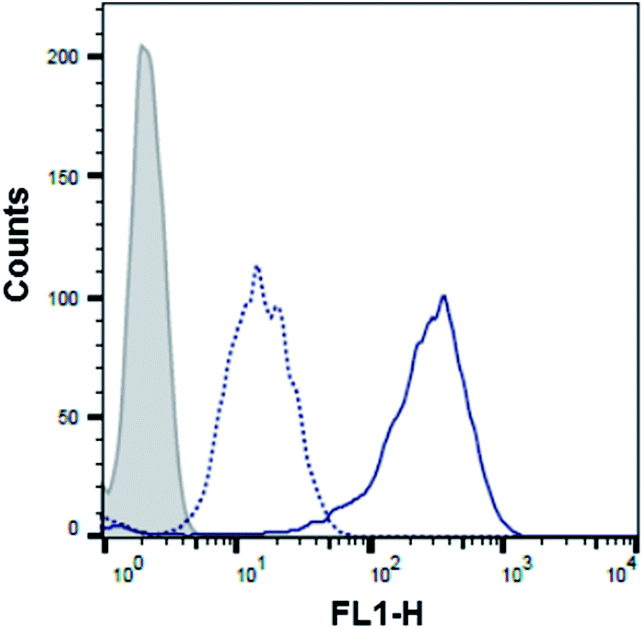
With the aim of further demonstrating that the polyA tail of PMO sequence 5 is required for its 2′-OMeUtaPS-mediated internalization in HeLa pLuc 705 cells, a cellular uptake experiment was conducted in the presence of urea, which by virtue of its denaturing properties was expected to interfere with the ability of 2′-OMeUtaPS to recognize the polyA tail of the PMO sequence and lead to a significant decrease in cellular uptake. As anticipated, flow cytometry analysis revealed (Fig. 8) that the 2′-OMeUtaPS-assisted internalization of the PMO sequence 5, when performed in the presence of 2.0 M urea, resulted in a decreased cellular uptake in the order of ∼80–90% relative to that of the control experiment performed in the absence of urea. These results suggest that the recognition of polyA-tailed PMO sequences by 2′-OMeUtaPS is sequence-specific, necessary and sufficient for efficient cellular uptake in live HeLa pLuc 705 cells.
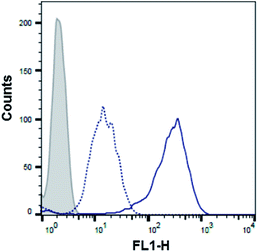 |
| | Fig. 8 Flow cytometry analysis of the 2′-OMeUtaPS-mediated internalization of PMO sequence 5 (peak area with a solid line border) in the presence of urea (peak area with a dotted line border) in HeLa pLuc 705 cells. Gray peak area accounts for untreated HeLa pLuc 705 cells. The concentration of 5 and 2′-OMeUtaPS was 1.0 μM and 2.0 μM, respectively, while the concentration of urea is 2.0 M during the course of complex formation. | |
Mechanistic authentication of the 2′-OMeUtaPS-assisted internalization of PMO sequences
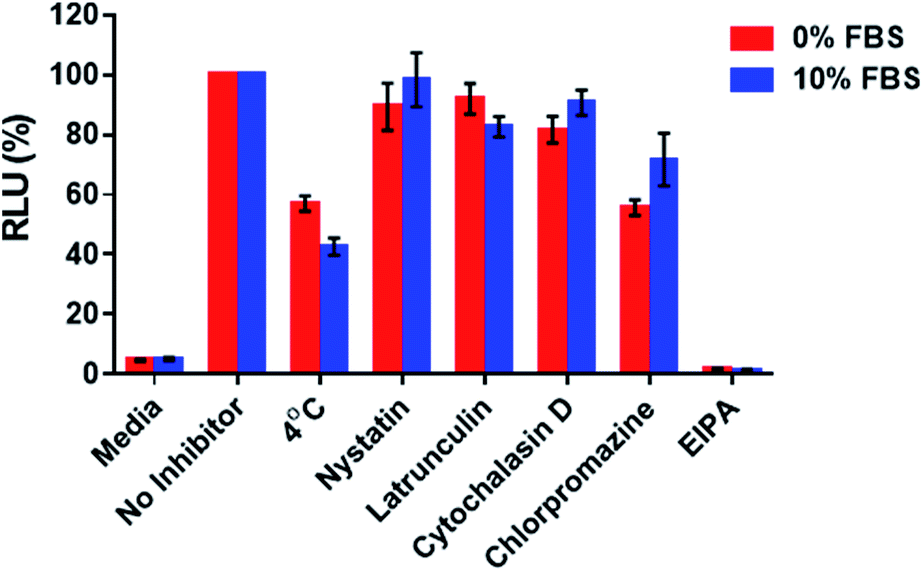
With the aim of assessing whether the 2′-OMeUtaPS-mediated internalization of PMO sequence 10 in HeLa pLuc 705 cells proceeded through an energy dependent mechanism, a cellular uptake experiment was carried out at 37 °C while another experiment was similarly conducted at 4 °C (Fig. 9). The results of this investigation show that when the cellular uptake experiment was carried out at 4 °C, restoration of luciferase activity decreased by more than 40% relative to that measured when the experiment was carried out at 37 °C. These findings are in agreement with an endocytic uptake mechanism.17 Identification of the most probable endocytic pathway leading to the 2′-OMeUtaPS-mediated uptake of PMO sequence 10 was initiated. For this purpose, chlorpromazine for inhibition of clathrin-coated pits-mediated endocytosis,17,18 nystatin and cytochalasin D for inhibition of caveolae-mediated endocytosis,17,19 latrunculin for inhibition of both clathrin-coated pits- and caveolae-mediated endocytosis19 and 5-(N-ethyl-N-isopropyl)amiloride (EIPA) for inhibition of macropinocytosis17,18 were employed at concentrations that are known to not significantly cause cell cytotoxicity.18 Although, the clathrin-coated pits-mediated endocytosis pathway appears to moderately inhibit the 2′-OMeUtaPS-assisted internalization of PMO sequence 10 in HeLa pLuc 705 cells, macropinocytosis is clearly the prevailing endocytic pathway used for this internalization process (Fig. 9).
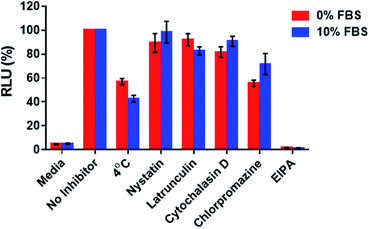 |
| | Fig. 9 Inhibition of luciferase activity production at 4 °C or 37 °C by endocytic pathway inhibitors upon 2′-OMeUtaPS-mediated internalization of PMO sequence 10 in both serum-containing and serum-free media. For each experiment, the extracellular concentrations of 2′-OMeUtaPS and sequence 10 are 2.0 μM and 1.0 μM, respectively, whereas the extracellular concentrations of nystatin, latrunculin, cytochalasin D, chlorpromazine and EIPA are 30 μM, 2 μM, 2 μM, 50 μM and 0.5 mM, respectively. Error bars represent the mean ± SD of three independent experiments. EIPA, 5-(N-ethyl-N-isopropyl)amiloride; RLU, relative light units; FBS, fetal bovine serum. | |
The 2′-OMeUtaPS-mediated internalization of polyA-tailed PMO sequence 14 in mdx mouse myotubes induced efficient excision of exon 23 from dystrophin pre-mRNA
Total RNA was extracted from myotubes using TRIzol (Life Technologies) as per the manufacturer's recommendations and was isolated by precipitation in cold (−20 °C) isopropanol. The total RNA was then subjected to reverse-transcription using qScript cDNA SuperMix; the cDNA was amplified using mouse-specific TaqMan probes designed to amplify the splice junction at exon 22–24 and the region spanning exons 23–24 of the non-skipped exon product.
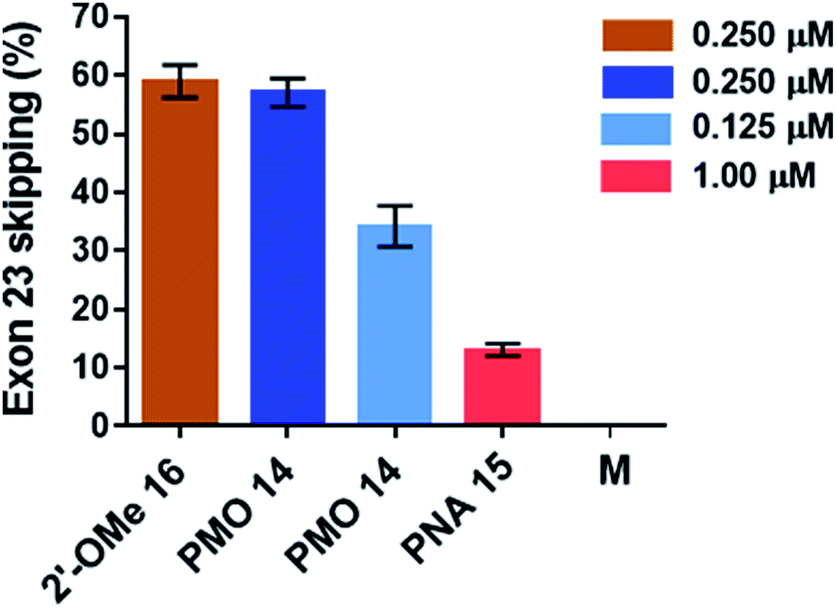
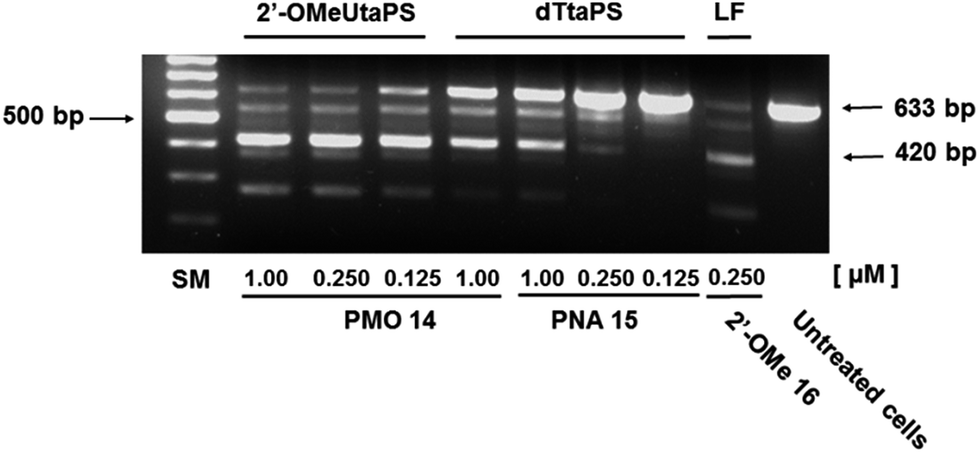
The percentage of exon 23 skipping was calculated as described in the Experimental section of this report. Fig. 10 demonstrates that the 2′-OMeUtaPS-mediated internalization of PMO sequence 14 in mdx mouse myotubes appears to be dose-dependent, leading to the excision of exon 23 from 60% of the pre-mRNA transcripts at a PMO sequence concentration of 250 nM in serum-containing medium. The result of this exon skipping experiment is vastly superior to that obtained with the transfection of PNA sequence 15 by 2′-OMeUtaPS, at a sequence concentration of 1.00 μM in serum-containing medium, while being comparable to that obtained with the Lipofectamine® 2000-mediated transfection of the positive control 2′-OMe RNA sequence 16 at a concentration of 250 nM in serum-free medium (Fig. 10). The efficiency of 2′-OMeUtaPS at skipping exon 23 from the mdx mouse dystrophin pre-mRNA was also demonstrated by performing a nested RT-PCR assay consisting of reverse-transcribing total RNA, isolated from mdx mouse myotubes, and amplifying the cDNA encoding exons 20–26 with appropriate DNA primers. Re-amplification of the primary PCR product encoding exons 20 to 24 was then initiated using another set of DNA primers; the secondary PCR products were separated by electrophoresis on a 1.5% agarose gel (Fig. 11). The 633 bp secondary PCR product corresponds to the unspliced pre-mRNA exon 23, whereas the shorter 420 bp PCR product corresponds to the correctly spliced exon 23 in agreement with the 213 bp difference between the secondary PCR products reported by others.20 As expected, the dTtaPS-mediated transfection of PMO sequence 14 in mdx mouse myotubes was considerably less efficient at correctly splicing exon 23 even at a PMO concentration of 1.00 μM. Contrary to expectations, the dTtaPS-mediated transfection of PNA sequence 15 in mdx mouse myotubes was found relatively inefficient at correctly splicing exon 23 even at a PNA concentration of 1.00 μM.
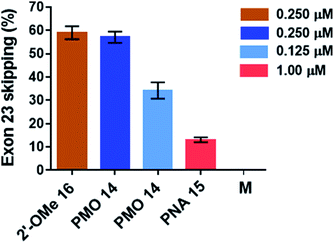 |
| | Fig. 10 Efficiency of 2′-OMeUtaPS at inducing the excision of the exon 23 from the mdx mouse dystrophin pre-mRNA upon transfection of PMO and PNA sequences 14 and 15, respectively, in mdx mouse myotubes. The concentration of 2′-OMeUtaPS was kept at 2 μM. All experiments were performed in serum-containing medium with the exception of the transfection of 2′-OMe RNA sequence 16, which was carried out using Lipofectamine® 2000 as the transfection reagent in serum-free-containing medium. Error bars represent the mean ± SD of three independent experiments. M, medium. | |
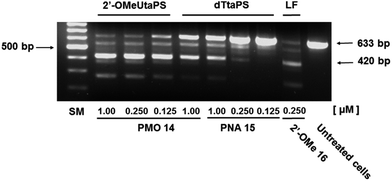 |
| | Fig. 11 Efficiency of 2′-OMeUtaPS and dTtaPS at inducing the excision of exon 23 from the mdx mouse dystrophin pre-mRNA upon transfection of the PMO or PNA sequence 14 or 15, respectively, in mdx mouse myotubes. The concentration of 2′-OMeUtaPS or dTtaPS was kept at 2 μM in serum containing medium. Lipofectamine® 2000 (LF) was used as a transfection reagent in serum free-medium at a concentration recommended by the supplier. Total RNA was extracted from transfected myotubes and amplified by nested RT-PCR using appropriate sets of DNA primers (see Experimental section). Two major PCR products were separated by electrophoresis on an agarose gel; the larger 633 bp and shorter 420 bp secondary PCR products correspond to the unspliced and correctly spliced pre-mRNA exon 23, respectively. SM, size marker. | |
Cytotoxicity studies
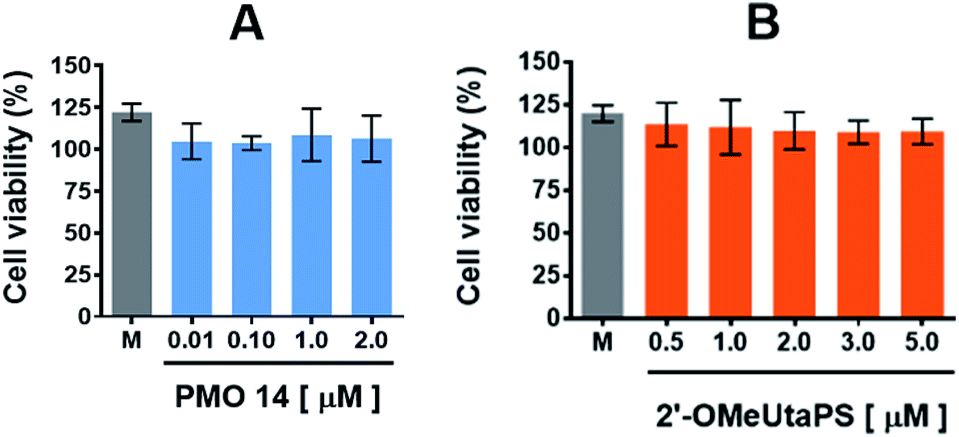
The cytotoxicity of PMO sequence 14 and 2′-OMeUtaPS in HeLa pLuc 705 cells was evaluated in serum containing (10% FBS) DMEM medium over a period of 18 h using a commercial cell-counting kit. Increasing the concentration of PMO 14 from 100 nM to 2.0 μM (Fig. 12A) or 2′-OMeUtaPS from 0.5 μM to 5.0 μM (Fig. 12B) did not induce significant cytotoxicity at concentrations optimal for transfection experiments.
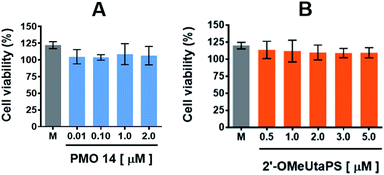 |
| | Fig. 12 Cytotoxicity of PMO sequence 14 (A) and 2′-OMeUtaPS (B) in HeLa pLuc 705 cells when increasing their respective concentrations. Each cytotoxicity study was evaluated over a period of 18 h in serum-containing (10% FBS) DMEM medium, using a commercial cell-counting kit, and expressed as a percentage of the viable cells. Error bars represent the mean ± SD of three independent experiments. M, medium. | |
Experimental
Materials and methods
Common chemicals and solvents including acetonitrile, benzene, triethylamine, dichloromethane, hexane, acetone, DMSO, 3-(N,N-dimethylamino)propan-1-ol, 1-octanol, bis(N,N-diisopropylamino)chlorophosphine, anhydrous solvents (MeCN, CH2Cl2, C6H6) and deuterated solvents (C6D6, DMSO-d6) were all purchased from commercial sources and used without further purification. 5′-O-(4,4′-Dimethoxytrityl)-2′-O-methyluridine was commercially available and used as received. All ancillary reagents commonly used in solid-phase DNA/RNA synthesis including 3-(dimethylaminomethylidene)amino-3H-1,2,4-dithiazole-3-thione or 3H-1,2-benzodithiol-3-one 1,1-dioxide and the succinylated long chain alkylamine controlled-pore glass (CPG) support, loaded with 2′-deoxythymidine as the leader nucleoside, were commercially available and used without further purification. The PNA and PMO sequences listed in Table 1 or mentioned anywhere in the text were purchased from PNA Bio Inc. and Gene Tools LLC, respectively, and used as received.
Cell culture media and reagents including OptiMEM, DMEM, DMEM high glucose, L-glutamine, sodium pyruvate, penicillin, GlutaMAX, streptomycin, hygromycin, monensin, trypan blue, 5% horse serum, mouse-specific TaqMan probes and pMAT vectors were acquired from Life Technologies; 20% heat-inactivated FBS was bought from Atlanta Biologicals, whereas 2% chick embryo extract and IFN-γ were obtained from United States Biological and R&D Systems, respectively. Cell lysis and Bright glow reagents needed for the luciferase assay protocol were acquired from Promega. Trypsin, TriZol and Platinum TaqPCRx DNA Polymerase were purchased from Thermo Fisher and qScript cDNA SuperMix from Quanta Bio. A High Capacity cDNA Reverse Transcription Kit was obtained from Applied Biosystems; forward and reverse DNA primers were acquired from Integrated DNA Technologies.
Flash chromatography purifications were performed on glass columns (6.0 cm or 2.5 cm I.D.) packed with silica gel 60 (EMD, 230–400 mesh), whereas analytical thin-layer chromatography (TLC) analyses were conducted on 2.5 cm × 7.5 cm glass plates coated with a 0.25 mm thick layer of silica gel 60 F254 (EMD). Analytical RP-HPLC analysis of 2′-OMeUtaPS was performed using a 5 μm Vydac C-4 wide pore (300 Å) column (25 cm × 4.6 mm). A linear gradient running from MeCN:H2O:AcOH (199:799:2, v/v/v to 799:199:2, v/v/v) was pumped at a flow rate of 1 mL min−1 for 20 min and was found adequate for its intended purpose. 1H-Decoupled 31P NMR analyses of the deoxyribonucleoside phosphoramidites 3 and 4 were performed using an NMR spectrometer operating at 121.5 MHz (300 MHz for 1H). Samples were maintained at a temperature of 298 K; all spectra were recorded in deuterated solvents and chemical shifts δ were reported in parts per million (ppm) relative to appropriate internal references. High resolution mass analyses of 3 and 4 and low-resolution MS analysis of 2′-OMeUtaPS were carried out under contract at a reputable mass spectrometry core facility.
Synthesis of N,N,N′,N′-tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (1) and N,N,N′,N′-tetraisopropyl-O-[octan-1-yl]phosphordiamidite (2)
To a stirred solution of 3-(N,N-dimethylamino)propan-1-ol (436 μL, 3.70 mmol) and bis(N,N-diisopropylamino) chlorophosphine (1.00 g, 3.70 mmol) in anhydrous benzene (20 mL) was added, under inert atmosphere, Et3N (1.00 mL, 7.17 mmol). Progress of the reaction was monitored by 31P-NMR spectroscopy; formation of the product (δP 122.4 ppm) was found to be complete within 2 h at ∼25 °C. The suspension was passed through a glass column packed with silica gel (∼15 g), which had been pre-equilibrated in benzene:Et3N (9:1 v/v); the filtrate was evaporated to an oil under reduced pressure. The oily material was dissolved in dry benzene (10 mL) and the resulting solution was swirled in a dry ice-acetone bath. The frozen material was lyophilized under high vacuum to give the phosphordiamidite 1 as a viscous oil (1.01 g, 3.03 mmol, 82%). 1H NMR (300 MHz, C6D6): δ 3.70 (dt, J = 7.3, 6.4 Hz, 2H), 3.55 (sept, J = 6.8 Hz, 2H), 3.52 (sept, J = 6.8 Hz, 2H), 2.36 (t, J = 7.1 Hz, 2H), 2.11 (s, 6H), 1.77 (quint, J = 6.8 Hz, 2H), 1.25 (d, J = 6.8 Hz, 12H), 1.20 (d, J = 6.8 Hz, 12H). 13C NMR (75 MHz, C6D6): δ 62.8 (d, 2JCP = 21.8 Hz), 56.9, 45.6, 44.6 (d, 2JCP = 12.6 Hz), 30.4 (d, JCP = 8.6 Hz), 24.8 (d, JCP = 8.6 Hz), 24.1 (d, JCP = 5.6 Hz). 31P NMR (121 MHz, C6D6): δ 123.3.
The preparation of phosphordiamidite 2 was performed at the same scale and under conditions similar to those employed for the synthesis of 1 with the exception of using 1-octanol (583 μL, 3.70 mmol) instead of 3-(N,N-dimethylamino)propan-1-ol. The phosphordiamidite 2 was isolated as a viscous oil, (1.17 g, 3.24 mmol, 88%). 1H NMR (300 MHz, C6D6): δ 3.65 (dt, J = 7.1, 6.4 Hz, 2H), 3.57 (sept, J = 6.8 Hz, 2H), 3.53 (sept, J = 6.8 Hz, 2H), 1.65 (dq, J = 6.6, 6.4 Hz, 2H), 1.43 (m, 2H), 1.34–1.18 (br m, 8H), 1.27 (d, J = 6.8 Hz, 12H), 1.22 (d, J = 6.8 Hz, 12H), 0.89 (dd, J = 6.8, 6.4 Hz, 3H). 13C NMR (75 MHz, C6D6): δ 64.7 (d, 2JCP = 21.8 Hz), 44.6 (d, 2JCP = 12.6 Hz), 32.2, 32.1 (d, JCP = 8.6 Hz), 29.8, 29.7, 26.6, 24.8 (d, JCP = 8.5 Hz), 24.1 (d, JCP = 5.6 Hz), 23.1, 14.3. 31P NMR (121 MHz, C6D6): δ 123.2.
General procedure for the preparation of deoxyribonucleoside phosphoramidites 3 and 4
To a stirred solution of N,N,N′,N′-tetraisopropyl-O-[3-(N,N-dimethylamino)prop-1-yl]phosphordiamidite (1, 667 mg, 2.00 mmol) or N,N,N′,N′-tetraisopropyl-O-[octan-1-yl]phosphordiamidite (2, 721 mg, 2.00 mmol) in MeCN (20 mL) was added 4,4′-dimethoxytrityl 2′-O-methyluridine (561 mg, 1.00 mmol) along with 0.45 M 1H-tetrazole in MeCN (2.2 mL, 1.0 mmol). The reaction mixture was stirred for 18 h at ∼25 °C. The reaction mixture was then concentrated under reduced pressure to a gummy material; the crude ribonucleoside phosphoramidite was purified by chromatography on silica gel (∼25 g), which was equilibrated in a solution of hexane:Et3N (95:5 v/v). The product was eluted from the column using a gradient of CH2Cl2 (0 → 95%) in hexane:Et3N (95:5 v/v). Fractions containing the pure ribonucleoside phosphoramidite, as detected by TLC, were pooled together and were evaporated to dryness under low pressure. The foamy material was dissolved in dry benzene (5 mL) and the resulting solution was manually stirred in a dry ice-acetone bath. The frozen material was then lyophilized under high vacuum to give the ribonucleoside phosphoramidite 3 or 4, each as a white powder in yields of 85% or 80%, respectively. 3: 31P NMR (121 mHz, C6D6): δ 149.5, 149.1 ppm, +HRMS: calcd for C42ClH57N4O9P [M + Cl]− 827.355, found 827.351. 4: 31P NMR (121 mHz, C6D6): δ 148.6, 148.3 ppm, +HRMS: calcd for C45ClH62N3O9P [M + Cl]− 854.391, found 854.393.
Solid-phase synthesis of chimeric 2′-OMeUtaPS
The solid-phase synthesis of 2′-OMeUtaPS was performed on a 0.2 μmole scale using a succinylated long chain alkylamine controlled-pore glass (CPG) support loaded with 5′-O-(4,4′-dimethoxytrityl)-2′-deoxythymidine as the leader nucleoside. The synthesis of the trans-acting RNA element was carried out using a DNA/RNA synthesizer and the appropriate phosphoramidites (3 or 4) as 0.1 M solutions in dry MeCN. The synthetic cycle recommended for the preparation of 2′-OMeUtaPS differs from the conventional cycle used for synthesis of unmodified oligonucleotides in that the “capping step” is performed after the oxidative sulfuration reaction.21 The reaction time of each of the following synthesis cycle steps has been extended to ensure optimal production of 2′-OMeUtaPS: (i) 5′-deblocking reaction (3% TCA in CH2Cl2, 60 s); (ii) 1H-tetrazole-mediated phosphoramidite coupling reaction (600 s); (iii) oxidation reaction (sulfuration solution, 600 s); and (iv) capping reaction (Cap A and Cap B solutions, 120 s). Upon complete assembly of 2′-OMeUtaPS, spectrophotometric (498 nm) measurements of the DMTr cation, released from the final synthesis cycle, revealed an overall synthesis yield of 92 ± 5%. The synthesis column was placed into a stainless steel pressure vessel and exposed to pressurized methylamine gas (∼2.5 bar, 25 °C, 3 min). Upon removal of residual methylamine gas from the pressure container, 2′-OMeUtaPS was eluted off the synthesis column using 500 μL of a solution consisting of Et3N:MeCN:H2O (1:60:39 v/v/v). The eluate was then evaporated to dryness using a stream of air. Air-dried 2′-OMeUtaPS can be stored indefinitely at −20 °C. 2′-OMeUtaPS was characterized by mass spectrometry; +ESI-MS: calcd for C127H205N19O54P7S7 [M + 2H]2+ corresponding to a calculated mass of 3301.0, found 3300.9.
Cell culture
The HeLa pLuc 705 cell line was exponentially grown in Dulbecco's Minimum Essential Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1.5 mM L-glutamine, 1.0 mM sodium pyruvate, 100 U penicillin, 100 μg mL−1 streptomycin and 200 μg mL−1 hygromycin; the cell culture was maintained at 37 °C in a humidified incubator under 5% CO2.
Complex formation between PNA or PMO sequences and dTtaPS or 2′-OMeUtaPS
An appropriate volume (1 μL) of a 100 μM PNA or PMO sequence stock solution was added to serum-free OptiMEM (20 μL). To this solution was added 2 μL of a 100 μM dTtaPS or 2′-OMeUtaPS stock solution and OptiMEM (27 μL) to produce a 2× stock solution of the complexes. The solution was incubated at 37 °C for 30 min and then stored at 4 °C for 15 min prior to immediate use in all cellular uptake experiments. Under these conditions, the extracellular concentration of the PNA/PMO sequence and 2′-OMeUtaPS/dTtaPS is 1.0 μM and 2.0 μM, respectively.
Flow cytometry analysis of cellular uptake in HeLa pLuc 705 cells
The internalization of fluorescently-labelled PNA or PMO sequence 8 or 6 in HeLa pLuc 705 cells using either dTtaPS or 2′-OMeUtaPS, as the delivery reagent, was assessed by flow cytometry. Cells were seeded in a 96-well plate (104 cells per well) and allowed to grow at 37 °C for 24 h in 10% FBS-MEM. The culture medium of each well was then replaced with fresh medium (100 μL) containing the fluorescently-labelled PNA or PMO sequence 8 or 6, each at a final concentration of 1 μM. After an 18 h-incubation at 37 °C, the medium was removed from each well by suction and 0.25% trypsin (50 μL) was added. Upon complete cell detachment, a solution (100 μL) of ice-cold 2% FBS in phosphate-buffered saline (PBS, pH 7.4) was added to each well. The cells were collected into tubes, to which was added 0.4% trypan blue (50 μL) and 200 μM monensin (20 μL), and analyzed by fluorescence-activated cell sorting (FACS) using a BD FACScan™ flow cytometer. A total of 5000 events were counted; histograms demonstrating cellular internalization of the fluorescently-labelled PNA and PMO sequences are presented in Fig. 4A and B, 7 and 8.
Luciferase assay protocol
HeLa pLuc 705 cells were seeded in a 96-well plate (2 × 104 cells per well) and allowed to grow at 37 °C for 18 h in 10% FBS-DMEM. The culture medium of each well was then replaced with either fresh serum-free or 20% serum-containing OptiMEM (50 μL) for experiments intended to be performed in serum-free or in 10% serum-containing media. A 2× solution of PNA or PMO sequence:2′-OMeUtaPS or dTtaPS complexes (50 μL) was added to the cells in order to achieve pre-determined complex concentrations, as indicated in the figure captions. After a 4 hour incubation at 37 °C, 20% FBS in OptiMEM (100 μL) was added to serum-free experiments and cells were incubated for an additional 18 h at 37 °C. The media was then removed by suction and the cells were mixed with a cell lysis reagent (50 μL) and agitated at ambient temperature for 10 minutes. The cell lysate (30 μL) was placed in a white 96-well plate and Bright-Glow reagent (20 μL) was added. Luciferase activity was measured using a microplate reader. For each well, luminescence was integrated over a period of 1 s and recorded as relative light units (RLU). Luminescence measurements were normalized to the amount of protein present in the test sample.
Protein concentration measurements
Protein concentration is measured, employing the Pierce Coomassie (Bradford) protein assay kit and a fraction (5 μL) of cell lysate prepared above for the luciferase assay, as per the manufacturer's instructions.
Energy-dependence of the 2′-OMeUtaPS-assisted internalization of PMO sequence 10 in HeLa pLuc 705 cells
HeLa pLuc 705 cells were seeded in a 96-well plate (2 × 104 cells per well) and allowed to grow at 37 °C for 18 h in 10% FBS-DMEM. While the cell culture was maintained at 37 °C or 4 °C, a 2× solution of a PMO sequence 10:2′-OMeUtaPS complex was prepared as described above. The cell culture medium was replaced with the PMO sequence 10:2′-OMeUtaPS complex solution to provide a final concentration of 1.0 μM and 2.0 μM, in PMO sequence and 2′-OMeUtaPS, respectively. After an incubation of 1 h at 37 °C or 4 °C, the medium was removed by suction and luciferase activity was measured as per the above luciferase assay protocol. Luminescence measurements were normalized to the amounts of protein present in the test samples and reported as a percentage of the luminescence produced relative to that measured from the experiment performed at 37 °C. The results of these experiments performed in the presence or absence of FBS are depicted in Fig. 9.
Endocytic pathways associated with the 2′-OMeUtaPS-mediated internalization of the PMO sequence 10 in HeLa pLuc 705 cells
HeLa pLuc 705 cells were seeded in a 96-well plate and grown in 10% FBS-DMEM, as described above. The cell culture was then pre-incubated for 30 min at 37 °C in the presence of nystatin or latrunculin, cytochalasin D, chlorpromazine, EIPA (5-(N-ethyl-N-isopropyl)amiloride), the final concentration of which was 30 μM or 2 μM, 2 μM, 50 μM, 0.5 mM, respectively. A 2× solution of PMO sequence 10:2′-OMeUtaPS complex was added to the cells to produce a final concentration of 1.0 μM and 2.0 μM in PMO sequence 10 and 2′-OMeUtaPS, respectively. The cells were then incubated for an additional 30 min at 37 °C, whereupon luciferase activity was measured as per the above luciferase assay protocol. Luminescence measurements were normalized to the amounts of protein present in the test samples and reported as the percentage of the luminescence produced relative to that measured in the absence of endocytic pathway inhibitors. The results of these experiments performed in the presence or absence of FBS are depicted in Fig. 9.
Transfection inhibition assay
An arbitrary amount of PMO sequence 5 was solubilized in serum-free OptiMEM (10 μL); 2′-OMeUtaPS S was added to the solution to achieve a final concentration that is twice that of the sequence. A solution of 4 M urea in serum-free OptiMEM (10 μL) was added to produce a final urea concentration of 2 M. After a 30 min incubation at 37 °C, the PMO sequence 5:2′-OMeUtaPS complexes were stored at 4 °C for 15 minutes or until used. Stock solutions (2×) of the complexes were made by adding OptiMEM to appropriate volumes (300 μL). HeLa pLuc 705 cells were seeded in a 96-well plate (2 × 104 cells per well) and allowed to grow at 37 °C for 18 h in 10% FBS-DMEM, as described above. The cell culture of each well was replaced with fresh 20% serum-containing OptiMEM (50 μL) to which was added 50 μL of the 2× stock solution of the PMO sequence 5:2′-OMeUtaPS complexes to produce an extracellular urea concentration of 67 mM. After a 4 hour incubation at 37 °C, the medium was replaced with fresh 10% FBS-DMEM; cells were allowed to grow for 18 h at 37 °C. The medium was removed from each well by suction and 0.25% trypsin (50 μL) was added. Upon complete cell detachment, the cells were processed as described above in the protocol for flow cytometry analysis. A total of 5000 events were counted. The FACS histogram presented as Fig. 8 illustrates the outcome of this experiment.
Cytotoxicity studies
The cytotoxicity of PMO sequence 14 and 2′-OMeUtaPS in HeLa pLuc 705 cells was evaluated over a period of 18 h in serum-containing (10% FBS) DMEM medium, using a commercial cell-counting kit. Increasing the concentration of 14 from 0.01 μM to 2.0 μM or 2′-OMeUtaPS from 0.5 μM to 5.0 μM did not induce significant cytotoxicity based on cell viability measurements (Fig. 12A and B) when compared to that of the medium.
Procedure for the culture and differentiation of myoblasts
H-2Kb-tsA58 (H2K) mdx myoblasts20,22 were maintained in DMEM, high glucose (4500 mg L−1), pyruvate (100 mg L−1) and GlutaMAX supplemented with 20% heat inactivated FBS, 2% chick embryo extract and 1% penicillin/streptomycin (100 U mL−1) on 0.4% gelatin coated plates. Myoblasts proliferation (5000 cells per cm2) was carried out at 33 °C in the presence of IFN-γ (20 ng mL−1) and 10% CO2. Myoblasts (26000 cells per cm2) were plated on 24-well 0.4% gelatin coated plates. Within 24 h, the media was aspirated from the cells and replaced with DMEM, high glucose (4500 mg L−1), pyruvate (100 mg L−1) and GlutaMAX supplemented with 5% horse serum and 1% penicillin/streptomycin (100 U mL−1). The cells were allowed to differentiate over three days at 37 °C under 5% CO2.
Myotube transfection assays
Myotubes were transfected in duplicate in serum-containing media using the PMO sequence 14 at a sequence concentration of 0.125-, 0.250- and 1.00 μM and 2′-OMeUtaPS (2.0 μM). Myotubes were also transfected in duplicate in serum-containing media using the PNA sequence 15 at a sequence concentration of 0.125-, 0.250- and 1.00 μM and dTtaPS (2.0 μM) for comparison purposes. The positive control 2′-OMe RNA phosphorothioate sequence 16 (0.25 μM) was delivered employing Lipofectamine® 2000, as the transfection reagent, in serum-free medium at a concentration recommended by the manufacturer.
RT-qPCR protocol
The transfected myotubes were washed with PBS and pooled in duplicates for RNA extraction using TriZol. The protocol was performed in accordance with the manufacturer's recommendation with the following modification: isopropanol precipitation of total RNA was performed at −20 °C overnight. The precipitated RNA (500 ng) was reverse-transcribed using qScript cDNA SuperMix as instructed by the manufacturer. cDNA (25 ng) was loaded on and analyzed using the 7900HT Fast Real-Time PCR system; each amplification reaction was performed in triplicate using mouse-specific TaqMan probes. The TaqMan gene expression probe (ID# AIOIXIL) for the skipped-exon product was designed to amplify the splice junction at exon 22–24, whereas the TaqMan gene expression probe (Mm0126935_m1) was used to amplify the region spanning exons 23–24 of the non-skipped exon product. Absolute quantitation of muscle-specific gene expression was calculated using a standard curve derived from a known quantity of DNA plasmids. The skipped-exon product fragment (i.e., exon 22–24) and the non-skipped exon product fragment (i.e., exon 23–24) were cloned separately into pMAT vectors, purified and concentrated. The copy number of each plasmid was calculated and a standard curve was loaded with a starting concentration of 300000 copies, which was then subjected to successive 10-fold dilution until no plasmid remained. The percentage of exon skipping was calculated using the following equation:
| % of exon skipping = [(average of triplicate reactions of skipped DMD exon)/(average of triplicate reactions of skipped DMD exon + average of triplicate reactions of non-skipped DMD exon)] × 100 |
Nested RT-PCR protocol
Total RNA (350 ng) of transfected mdx mouse myotubes was reverse-transcribed using a High Capacity cDNA Reverse Transcription Kit according to the manufacturer's protocol with the following modification: the primer mix was replaced with forward (5′-CAGAATTCTGCCAATTCGTGAG) and reverse (5′-TTCTTCAGCTTGTGTCATCC) DNA primers for amplification of exons 20 to 26. cDNA (350 ng) was PCR-amplified using the platinum TaqPCRx DNA polymerase with a hot start at 94 °C for 2 min, followed by 30 cycles of 95 °C (30 s), 55 °C (1 min) and 72 °C (2 min). Approximately 20 ng (3 μL) of the primary PCR products were re-amplified using the platinum TaqPCRx DNA polymerase with forward (5′-CCCAGTCTACCACCCTATCAGAGC) and reverse (5′-CAGCCATCCATTTCTGTAAGG) DNA primers amplifying exons 20 to 24. The secondary PCR was carried out with a hot start at 94 °C for 2 min, followed by 22 cycles of 95 °C (30 s), 55 °C (1 min) and 72 °C (2 min). Secondary PCR products were analyzed on a 1.5% agarose gel (see Fig. 11).
Conclusions
The trans-acting phosphorothioate RNA element 2′-OMeUtaPS is a unique nucleic acid-based transfection reagent for the delivery of uncharged nucleic acid sequences to either HeLa pLuc 705 cells or mdx mouse myotube cells. 2′-OMeUtaPS is easily prepared from the 2′-O-methyluridine phosphoramidite monomers 3 and 4 according to standard solid-phase synthesis protocols. As expected, the properties of 2′-OMeUtaPS are similar to those of dTtaPS in terms of recognition of the polyA stretch of either PNA or PMO sequences that is necessary for internalization of polyA-tailed PNA or PMO sequences in mammalian cells. Although FACS studies indicated that the 2′-OMeUtaPS-assisted delivery of PNA sequence 8 was as efficient as that of PMO sequence 6 in HeLa pLuc 705 cells (Fig. 4B), the affinity of 2′-OMeUtaPS for the polyA-tail of PNA sequence 9 seemingly interfered with either its endosomal release or its nuclear membrane penetration; either outcome resulted in a ∼10-fold decrease in the production of luciferase in HeLa pLuc 705 cells when compared to that obtained with the polyA-tailed PMO sequence 10 (Fig. 3B). The FACS analyses are inspirational in terms of the requirements for endosomal trafficking of uncharged nucleic acid sequences, which are critical to the production of functional of nucleic acid-based drugs in the cytoplasm and/or nucleus of mammalian cells. These findings should encourage further investigations on the parameters associated with endosomal trafficking pathways. Similar to dTtaPS, the 2′-OMeUtaPS-mediated internalization of polyA-tailed uncharged sequences occurs through an energy-dependent mechanism and macropinocytosis appears to be the predominant endocytic pathway for cellular uptake. The recognition of polyA-tailed PNA or PMO sequences by 2′-OMeUtaPS is specific (Fig. 7) and likely to occur through weak base-pairing interactions; recognition of the polyA tail of these uncharged sequences is strongly inhibited in the presence of urea,12,13 which is a potent denaturant (Fig. 8). Furthermore, 2′-OMeUtaPS is nothing like commercial cationic lipid reagents, which preferably require serum-free media for optimal transfection performance; 2′-OMeUtaPS performed comparably well in both serum-free and serum-containing media (Fig. 9) at optimal transfection concentrations.
The functional activity of PMO sequence 10 when delivered by 2′-OMeUtaPS to HeLa pLuc 705 cells was confirmed by restoration of luciferase activity through alternate splicing of the incorrectly spliced pre-mRNA encoding luciferase. Likewise, the functionality of 2′-OMeUtaPS-mediated delivery of PMO sequence 14 to mdx mouse myotube cells was demonstrated by excision of the mutated exon 23 from the pre-mRNA encoding dystrophin through an alternate splicing event (Fig. 10 and 11). Efficient delivery of PMO sequences to muscle cells is particularly important for the treatment of DMD.
The findings reported herein strongly underscore the versatility of synthetic nucleic acid-based transfection reagents for the delivery of antisense nucleic acid-based drugs for the treatment of human diseases. Future work will focus on the development of modified amphipathic trans-acting phosphorothioate DNA and RNA elements. Specifically, these elements are intended to be decorated with a variable number of thermolytic positively charged and/or lipophilic functions for thiophosphate/phosphate protection; this should enable in vitro and in vivo delivery of uncharged and negatively charged nucleic acid-based drugs to mammalian cells/tissues while maintaining minimal cytoxicity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank Prof. Rudolph L. Juliano (University of North Carolina, Chapel Hill) and Dr Terry Partridge (Children's National Medical Center, Washington DC) for kindly providing the HeLa pLuc 705 and H-2Kb-tsA58 (H2K) mdx myoblast cell lines, respectively. FDA intramural funds supporting this research are gratefully acknowledged. K. N. is supported by the National Institutes of Health (5U54HD053177, K26OD011171, P50AR060836-01, 2R24HD050846-06), the Muscular Dystrophy Association and the US Department of Defense (W81XWH-05-1-0616, W81XWH-11-1-0782, W81XWH-11-1-0330).
Notes and references
- P. Jarver, L. O'Donovan and M. J. Gait, Nucleic Acid Ther., 2014, 24, 37 CrossRef PubMed.
- A. Aartsma-Rus and G. J. B. van Ommen, RNA, 2007, 13, 1609 CrossRef CAS PubMed.
- Y. Aoki, T. Nagata, T. Yokota, A. Nakamura, M. J. A. Wood, T. Partridge and S. Takeda, Hum. Mol. Genet., 2013, 22, 4914 CrossRef CAS PubMed.
- E. P. Hoffman, R. H. Brown and L. M. Kunkel, Cell, 1987, 51, 919 CrossRef CAS PubMed.
- T. Yokota, E. Pistilli, W. Duddy and K. Nagaraju, Expert Opin. Biol. Ther., 2007, 7, 831 CrossRef CAS PubMed.
- M. C. Vila, M. B. Klimek and J. S. Novak, et al., Skeletal Muscle, 2015, 5, 44 CrossRef PubMed.
- F. Shabanpoor, G. McClorey, A. F. Saleh, P. Jarver, M. J. A. Wood and M. J. Gait, Nucleic Acids Res., 2015, 43, 29 CrossRef CAS PubMed.
- F. Muntoni and M. J. Wood, Nat. Rev. Drug Discovery, 2011, 10, 621 CrossRef CAS PubMed.
- S. Fletcher, K. Honeyman, A. M. Fall, P. L. Harding, R. D. Johnsen and S. D. Wilton, J. Gene Med., 2006, 8, 207 CrossRef CAS PubMed.
-
(a) L. O'Donovan, I. Okamoto, A. A. Arzumanov, D. L. Williams, P. Deuss and M. J. Gait, Nucleic Acid Ther., 2015, 25, 1 CrossRef PubMed;
(b) J. Bhadra, S. Pattanayak, P. P. Khan, J. Kundu and S. Sinha, Bioconjugate Chem., 2016, 37, 2254 CrossRef PubMed.
- B. Wu, P. Lu, C. Cloer, M. Shaban, S. Grewal, S. Milazi, S. N. Shah, H. M. Moulton and Q. L. Lu, Am. J. Pathol., 2012, 181, 392 CrossRef CAS PubMed.
- H. V. Jain, D. Verthelyi and S. L. Beaucage, RSC Adv., 2015, 5, 65245 RSC.
- H. V. Jain and S. L. Beaucage, in Current Protocols in Nucleic Acid Chemistry, ed. M. Egli, P. Herdewijn, A. Matsuda and Y. S Sanghvi, John Wiley & Sons, Inc., Hoboken, 2016, vol. I, p. 4.69.1 Search PubMed.
- J. Summerton, J. Drug Discov. Develop. Deliv., 2016, 3, 1019 Search PubMed.
- S. Karkare and D. Bhatnagar, Appl. Microbiol. Biotechnol., 2006, 71, 575 CrossRef CAS PubMed.
- A. Ellington and J. D. Pollard Jr, in Current Protocols in Nucleic Acid Chemistry, ed. S. L. Beaucage, D. E. Bergstrom, G. D. Glick and R. A. Jones, John Wiley & Sons, Inc., Hoboken, 2000, vol. I, p. A.3C.1 Search PubMed.
- I. A. Khalil, K. Kogure, H. Akita and H. Harashima, Pharmacol. Rev., 2006, 58, 32 CrossRef CAS PubMed.
- G. D. Ivanova, A. Arzumanov, R. Abes, H. Yin, M. J. A. Wood, B. Lebleu and M. J. Gait, Nucleic Acids Res., 2008, 36, 6418 CrossRef CAS PubMed.
- A. I. Ivanov, in Methods in Molecular Biology: Exocytosis and Endocytosis, ed. A. Ivanov, Humana Press, Inc., Totowa, 2008, vol. 440, p. 15 Search PubMed.
-
(a) C. J. Mann, K. Honeyman, A. J. Cheng, T. Ly, F. Lloyd, S. Fletcher, J. E. Morgan, T. A. Partridge and S. D. Wilton, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 42 CrossRef CAS PubMed;
(b) B. T. Le, K. Muyarama, F. Shabanpoor, H. Asanuma and R. N. Veedu, RSC Adv., 2017, 7, 34049 RSC.
- R. P. Iyer, L. R. Phillips, W. Egan, J. B. Regan and S. L. Beaucage, J. Org. Chem., 1990, 55, 4693 CrossRef CAS.
- J. E. Morgan, J. R. Beauchamp, C. N. Pagel, M. Peckham, P. Ataliotis, P. S. Jat, M. D. Noble, K. Farmer and T. A. Partridge, Dev. Biol., 1994, 162, 486 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and mass spectrometry spectra of compounds 1–4. See DOI: 10.1039/c7ra04247g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
J. F. Boehlerbc,
D. Verthelyid,
K. Nagarajube and
S. L. Beaucage
a,
J. F. Boehlerbc,
D. Verthelyid,
K. Nagarajube and
S. L. Beaucage