
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theory guided systematic molecular design of benzothiadiazole–phenazine based self-assembling electron-acceptors†
Dong-Chan Lee *a,
Youngjun Jeongb,
Lacie V. Brownella,
John E. Velascoa,
Kathleen A. Robinsa and
Youngu Lee*b
*a,
Youngjun Jeongb,
Lacie V. Brownella,
John E. Velascoa,
Kathleen A. Robinsa and
Youngu Lee*b
aDepartment of Chemistry and Biochemistry, University of Nevada Las Vegas, 4505 S. Maryland Parkway, Box 454003, Las Vegas, Nevada 89154-4003, USA. E-mail: Dong-Chan.Lee@unlv.edu
bDepartment of Energy Systems Engineering, Daegu Gyeongbuk Institute of Science & Technology (DGIST), 333, Techno Jungang-daero, Hyeonpung-Myeon, Dalseong-Gun, Daegu, 42988, Republic of Korea. E-mail: youngulee@dgist.ac.kr
First published on 3rd May 2017
Abstract
We report two new self-assembling n-type materials in which the design process was systematically analyzed using theoretical calculations prior to experimental synthesis. Benzothiadiazole (A′) and alkoxyphenazine (A) serve as our basic electron-deficient building blocks to construct two candidate molecules with an acceptor (A) – acceptor (A′) – acceptor (A) configuration. We conducted a computational characterization (B3LYP/6-31G*) of the electronic properties for structural subunits linked together and analyzed in a step-by-step fashion, thereby culminating in the target molecules of interest. We found that ELUMO was controlled primarily by benzothiadiazole as was evident by orbital localization on this subunit. Meanwhile, EHOMO was influenced by the dihedral angle between A and A′. The molecule with A and A′ coupled with a C–C triple bond (BTD-P-T) was found to be planar with a more stabilized ELUMO and a reduced Egap when compared to its C–C single bond counterpart (BTD-P-S). The two molecules were synthesized and characterized to verify the theoretical findings. Optical, electrochemical, and thermal properties were characterized with UV-vis absorption and fluorescence spectroscopy, cyclic voltammetry, and differential scanning calorimetry, respectively. In addition, the molecular packing was examined by X-ray powder diffraction. As predicted by theory, BTD-P-T exhibited a lower Egap based on the system's lower ELUMO in comparison to BTD-P-S. BTD-P-T exhibited a higher Tm and crystallinity due to its planarity. Both BTD-P-S and BTD-P-T exhibited excellent fibrillation ability upon solvent casting. Bulk heterojunction (BHJ) organic solar cell (OSC) devices were fabricated using BTD-P-S or BTD-P-T as an acceptor and P3HT as a donor at different weight ratios. For both P3HT:BTD-P-S and P3HT:BTD-P-T BHJs, the weight ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 produced the highest power conversion efficiency (PCE) of 0.17% and 0.44%, respectively. These results are consistent with fluorescence quenching experiments in which 90% of P3HT fluorescence was quenched at that ratio. Nearly two times higher PCE was observed for the BTD-P-T based device compared to that of the BTD-P-S based system, mainly due to the higher Jsc. Presumably, the flat geometry of BTD-P-T allows for more efficient intermolecular π-orbital overlap, enhancing charge transport.
:1 produced the highest power conversion efficiency (PCE) of 0.17% and 0.44%, respectively. These results are consistent with fluorescence quenching experiments in which 90% of P3HT fluorescence was quenched at that ratio. Nearly two times higher PCE was observed for the BTD-P-T based device compared to that of the BTD-P-S based system, mainly due to the higher Jsc. Presumably, the flat geometry of BTD-P-T allows for more efficient intermolecular π-orbital overlap, enhancing charge transport.
Introduction
Developing organic semiconductors that have tailored HOMO and LUMO energy levels (EHOMO and ELUMO) as well as a controllable solid-state molecular arrangement has been an important field of research.1–5 Understanding how structural constituents impact the electronic properties of whole molecules is an essential step for controlling and ultimately manipulating EHOMO and/or ELUMO. Current practices for designing organic semiconductors utilize structural entities with known electronic properties (either electron accepting or donating) as molecular building blocks. The typical design strategy is to combine building blocks with specific electronic properties in order to obtain a molecular combination that produces the desired electronic result. Numerous studies are available describing the role of certain structural units on electronic properties, making the design process more convenient and potentially predictable.6–13 However, consideration of individual subunit (building block) properties does not always produce the anticipated result in a larger system comprised of these individual units since overall molecular electronic properties do not always follow a simple additivity rule. This is more significant when the materials are composed of non-identical structural subunits, e.g., donor (D)–acceptor (A) type molecules.11–19 To predict and control the electronic properties for molecules before embarking on time-consuming synthesis, it is imperative to understand and assess how each smaller structural constituent contributes to these properties in a systematic manner. To that end, theoretical calculations can serve as a valuable tool.20–23 Although quantum mechanical theory has become a useful tool for understanding organic π-conjugated materials, the role of computation has mostly been employed as an “after-the-fact” analysis as opposed to predicting electronic properties before synthesizing a molecule. As one would expect, theoretically calculating molecular electronic properties will not produce numerical agreement with results obtained from experiment. This is particularly problematic when the sample set is small. In our work, we have obtained and correlated experimental and theoretical data for a wide range of related molecular systems using density functional theory and a modest basis set (B3LYP/6-31G*). Thus, we have established a large data set of related molecules that exhibit significantly high levels of correlation specific to electronic properties. Thus, our prediction of electronic properties for new, related systems is possible, and includes an ability to anticipate error. Specifically, our research group has achieved outstanding levels of correlation when treating substituted pyrazine–acene molecules. This previous work has given us a powerful tool for predicting the electronic properties of pyrazine–acene type molecules. In this study, our previous theoretical expertise handling pyrazine–acene derivatives has guided our current materials development and aided in identifying systems worthy of experimental preparation.In this paper, we developed two self-assembling electron-deficient organic semiconductors guided by theory. The title molecules have an A–A′–A configuration using benzothiadiazole (BTD) with two alkoxyphenazines. We chose BTD as a pendant (A′) due to its electron-deficiency4,24–26 and structural simplicity, as any bulky pendant moiety may disrupt molecular packing. Alkoxyphenazine (A) was chosen for its self-assembly property, with an ability to form fibrous structures through nonbonding interactions such as hydrogen bonding, π–π, and van der Waals interactions.27–29 Due to the presence of imine nitrogens, phenazine is considered an electron-deficient structural subunit.30 We also considered assembly properties since the molecular arrangement of organic semiconductors plays an important role in the solid-state properties. Both π-cores and alkyl side groups contribute to the assembly, and harmonious interplay between intermolecular forces is required to achieve well-defined molecular assembly.31–35 However, the assembly is very sensitive to many parameters including the type and location of alkyl side groups, therefore it is hard to predict the resulting morphology. To eliminate the uncertainty of structure-dependent molecular assembly, a reasonable strategy would be to start with structural components that have known assembly patterns.
Two molecules were designed and prepared with different connectivity between A and A′, namely, C–C single (BTD-P-S) and C–C triple bonds (BTD-P-T). During the design process, theoretical analysis was conducted for each intermediate, from the dialkoxyphenazine to the title molecules. Our goal was to assess the impact of these subunits on both electronic properties as well as structural features. In addition, experimental characterization for the two title compounds included photophysical, electronic, thermal, and assembly properties. Furthermore, we investigated the efficacy of their function as an acceptor in BHJ OSCs to reveal the effect of small structural variations (connectivity). Detailed device characterizations are also presented.
Results and discussion
Theory assisted molecular design: understanding the role of each structural unit
Starting from the dialkoxyphenazine (1) as a base structure, the whole molecular build-up process to reach A–A′–A was calculated to assess the impact of each structural unit on the electronic properties (Fig. 1). First, when A′ (BTD) was attached to A (dialkoxyphenazine) directly to create 2, the major effect was a lowering of ELUMO by 0.43 eV.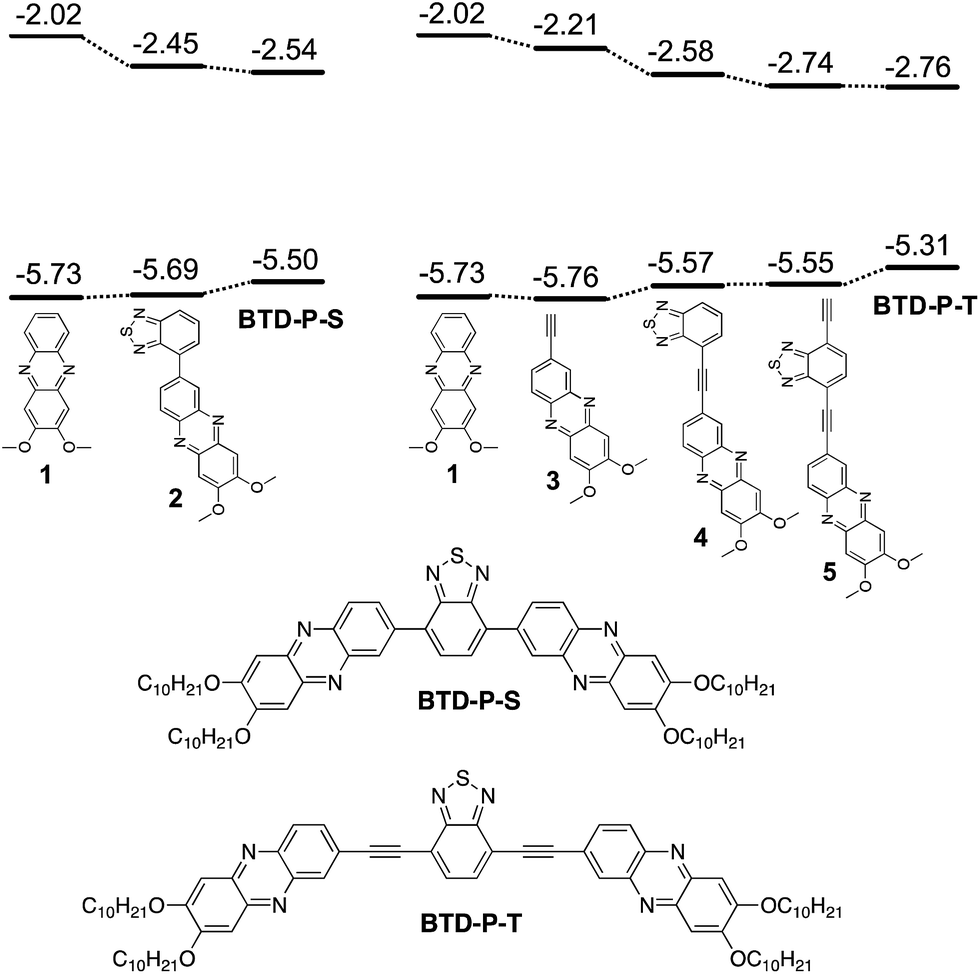 | ||
| Fig. 1 The structures of BTD-P-S and BTD-P-T and their respective build-up intermediates with theoretical HOMO and LUMO energy levels. Theoretical EHOMO and ELUMO for BTD are −6.61 and −2.35 eV, respectively. | ||
This is an expected result when one considers the electron affinity of BTD. In 1 and 2, a negligible change was found for EHOMO. Further addition of phenazine to produce the first A–A′–A molecule, BTD-P-S, influenced EHOMO more than ELUMO. EHOMO was raised by 0.19 eV while ELUMO was lowered by 0.09 eV. The same operation was carried out for BTD-P-T which connects phenazine and BTD through a C–C triple bond. Although structurally small, the influence of the triple bond was noticeable in terms of the electronic properties. The addition of a triple bond to 1 reduced ELUMO by 0.19 eV. Interestingly, EHOMO was negligibly affected. When BTD was added to 3 to form 4, EHOMO was increased by 0.09 eV and ELUMO was decreased far more significantly (−0.37 eV). Further addition of C–C triple bond to 4 produced the similar result as the process from 1 to 3, in which only ELUMO was affected. Finally, dialkoxyphenazine was added to produce BTD-P-T. Surprisingly, ELUMO remained almost the same, while EHOMO was raised by 0.24 eV. Considering the electron deficiency of phenazine, ELUMO was expected to be reduced, however, the computational result indicated otherwise. The changes in the electronic properties upon the structure build-up are presented in Table 1.
| EHOMO (eV) | ELUMO (eV) | |
|---|---|---|
| 1 to 2 | +0.04 | −0.43 |
| 2 to BTD-P-S | +0.19 | −0.09 |
| 1 to 3 | +0.03 | −0.19 |
| 3 to 4 | +0.19 | −0.37 |
| 4 to 5 | +0.02 | −0.16 |
| 5 to BTD-P-T | +0.24 | −0.02 |
The comparative impacts of structural entities during the construction of two BTD-P molecules can be summarized as follows: (1) the addition of BTD lowered ELUMO at a similar level, but the impact on EHOMO was not consistent, (2) C–C triple bond reduced ELUMO, and (3) the addition of phenazine at the final step raised EHOMO while negligibly impacting ELUMO.
To elucidate the origin of these results, two parameters were examined: frontier molecular orbital distributions and the dihedral angles between A and A′. A visual inspection of the HOMO and LUMO orbital distribution can give valuable insight into the subunits that impact the electronic properties. The orbital pictures of intermediates are shown in Fig. 2 for BTD-P-S and Fig. 3 for BTD-P-T. The most profound feature common to both compounds is the LUMO diagram for those systems where BTD is present. For example, the orbital coefficients are more localized on BTD for 2 and 4 when compared to 1 and 3. This accounts for the large ELUMO lowering when BTD is added. The addition of phenazine to 2 and 5 that lead to BTD-P-S and -T, respectively, did not alter the LUMO pictures significantly in spite of the electron-deficiency of phenazine, which resulted in only small change in ELUMO. Lower ELUMO for BTD-P-T compared to BTD-P-S is due to the inclusion of the C–C triple bond which has sp-hybridized carbons. Increased EHOMO when BTD is present is due to the delocalization of the HOMO orbital on the benzene in BTD. However, the degree of EHOMO raising is more significant for the BTD-P-T case. To investigate this phenomenon, we examined the dihedral angle between A and A′.
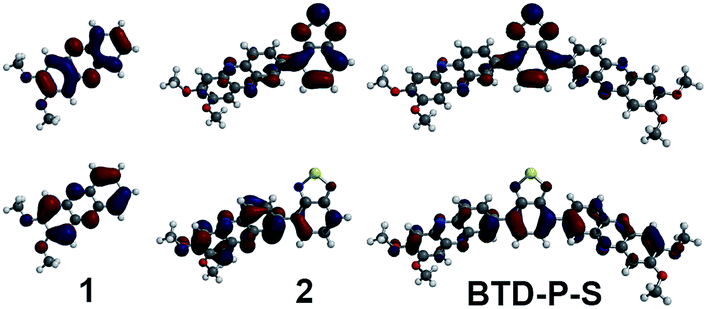 | ||
| Fig. 2 HOMO (bottom row) and LUMO (top row) diagrams of build-up intermediates for BTD-P-S. | ||
 | ||
| Fig. 3 HOMO (bottom row) and LUMO (top row) diagrams of build-up intermediates for BTD-P-T. | ||
The B3LYP/6-31G* optimized geometries resulted in a 36.40° dihedral angle between A (phenazine) and A′ (BTD) for BTD-P-S while BTD-P-T was found to be completely planar (Fig. 4). The relatively large dihedral angle of BTD-P-S is due to the steric repulsion between two hydrogen atoms on BTD and phenazine. On the contrary, separation of the two hydrogen atoms by a C–C triple bond allows BTD-P-T to adapt a flat geometry. Such planarity enables more efficient orbital delocalization, which accounts for the higher EHOMO.
 | ||
| Fig. 4 Optimized geometry of BTD-P-S (a) and BTD-P-T (b) calculated by B3LYP/6-31G*. | ||
In summary, BTD-P-T is superior to BTD-P-S with lower ELUMO, more reduced HOMO–LUMO gap (Egap), and potentially better self-assembly driven by molecular planarity that supports more efficient π–π interactions.
The unique finding from our bottom-up theoretical molecular build-up process is the discovery of individual structural component's impact on the electronic properties that defy prediction using a simple additivity rule. In fact, it would be difficult to elucidate the electronic contributions from the individual subunits by a single computation of the whole molecule only.
Synthesis, optical, electrochemical, thermal, and self-assembly properties
To verify theoretical findings experimentally, the two molecules (BTD-P-S and -T) were synthesized. The key reaction to prepare BTD-P-S was a Suzuki coupling between 2,3-bis(decyloxy)-7-iodophenazine and 2,1,3-benzothiadiazole-4,7-bis(boronic acid pinacol ester). Meanwhile, a Sonogashira coupling of 2,3-bis(decyloxy)-7-iodophenazine with 4,7-diethynyl-2,1,3-benzothiadizole produced BTD-P-T. 2,3-Bis(decyloxy)-7-iodophenazine was prepared from a cyclization reaction between 4-iodobenzen-1,2-diamine and 2,5-dihydroxybenzoquinone followed by a Williamson ether synthesis with bromodecane. The yields for both of the final C–C coupling reactions were ca. 40%. More detailed synthetic procedures with full structural characterizations are provided in ESI.†The optical properties of these title compounds were characterized by UV-vis and FL spectroscopy. As shown in Fig. 5, BTD-P-S showed a broad absorption caused by a π–π* transition with λmax at 426 nm in dichloromethane solution (5 × 10−6 M). In the case of BTD-P-T, a more structured absorption pattern appeared with a red-shifted λmax at 446 nm which is accompanied by shoulders at ca. 470 nm and 420 nm. From the absorption edge, the optical Egap was calculated. Egap of BTD-P-T was found to be 2.51 eV, which is more compressed than that of BTD-P-S by ca. 0.1 eV. In the solid-state, the two compounds showed distinctively different absorption behaviors. The λmax of the BTD-P-S cast film was found to be the same as in solution, however the absorption became broader with tailing at longer wavelengths. On the contrary, the BTD-P-T cast film showed a blue-shifted λmax at 421 nm relative to the solution λmax and red-shifted shoulder at ca. 485 nm. A similar behaviour has been assigned to pseudo H-aggregates.36 Egap's in the solid-state showed the same trend as those in solution; though more reduced for BTD-P-T. Meanwhile, the fluorescence maxima (λem) for BTD-P-S and BTD-P-T were nearly the same: 505 nm and 508 nm in dichloromethane (5 × 10−6 M), when excited at 430 nm and 446 nm, respectively (Fig. 6). The fluorescence quantum efficiencies for both compounds were quite high, reaching 58% for BTD-P-S and 66% for BTD-P-T which were measured using 9,10-diphenylanthracene as a standard reference (ΦF = 90% in cyclohexane).
 | ||
| Fig. 5 UV-vis absorption spectra of BTD-P-S and -T in dichloromethane (5 × 10−6 M) (broken line) and as a film (solid line). | ||
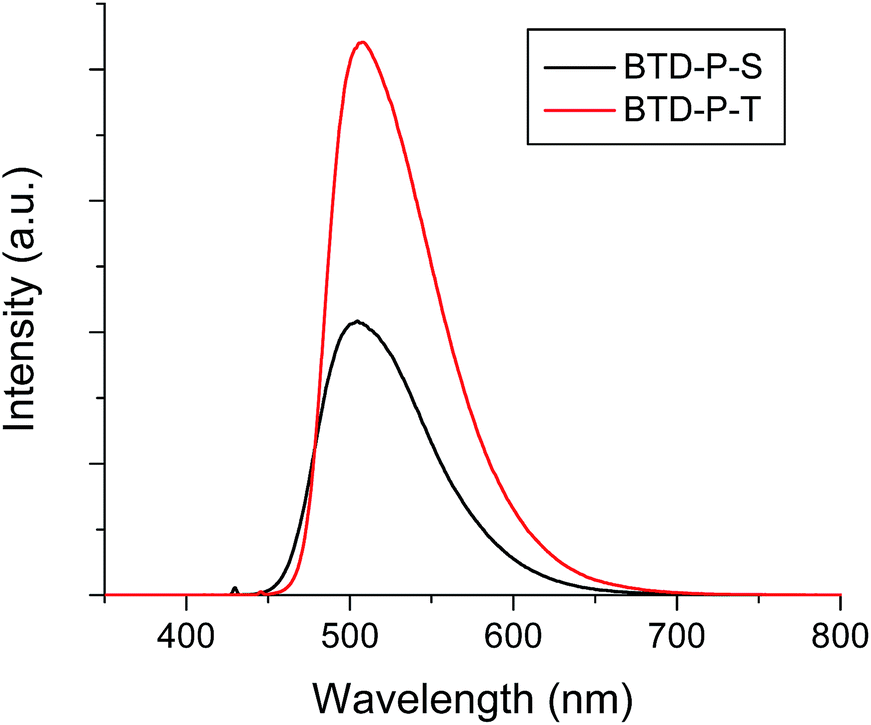 | ||
| Fig. 6 Fluorescence spectra of BTD-P-S and BTD-P-T in dichloromethane (5 × 10−6 M). λex = 430 nm for BTD-P-S and 446 nm for BTD-P-T. | ||
The electrochemical properties of the title compounds were investigated with cyclic voltammetry (CV) (Fig. 7). Our primary focus was on the reduction potential, which is directly related to the electron affinity. Both compounds showed one reversible reduction potential within the characterized potential window. From the onset of the reduction potential, we deduced the lowest unoccupied molecular orbital energy level (ELUMO).
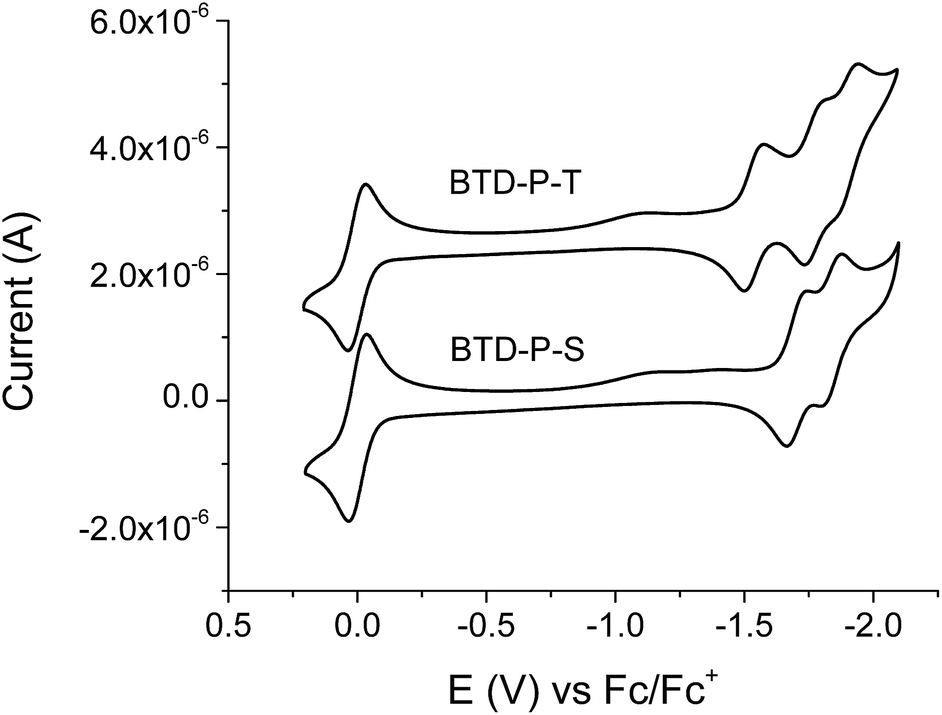 | ||
| Fig. 7 Cyclic voltammograms of BTD-P-S and BTD-P-T in dichloromethane with tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte. Scan rate = 100 mV s−1. | ||
The CV experiments were conducted using ferrocene as an internal reference, therefore ELUMO was calculated relative to the oxidation potential of ferrocene to ferrocenium, which is −4.8 eV from vacuum. Due to the electron-deficient nature of the title compounds, it was difficult to observe the oxidation potential, and therefore, EHOMO was calculated from ELUMO (from CV) and optical Egap (from UV-vis). These values are listed in Table 2. In solution, ELUMO of BTD-P-T was found to be −3.33 eV, which is more stabilized than that of BTD-P-S by 0.16 eV. In the case of EHOMO, the difference was marginal.
A differential scanning calorimetry (DSC) was employed to characterize thermal properties (Fig. 8).
 | ||
| Fig. 8 DSC thermograms of title compounds. 2nd heating/cooling scans at 5 °C min−1. | ||
Both compounds showed reversible melting and crystallization behaviors. A sharp melting transition appeared at 142 °C for BTD-P-S and 164 °C for BTD-P-T. The heat of fusion of BTD-P-T (33.3 J g−1) was clearly higher than that of BTD-P-S (26.4 J g−1), indicating higher crystallinity of BTD-P-T. While BTD-P-T revealed only one crystallization transition at 153 °C, BTD-P-S had a small exothermic peak at 138 °C in addition to the major crystallization peak at 130 °C. This appeared repeatedly only at the cooling scan, which suggests monotropic liquid crystalline behavior.
To examine the molecular packing, XRD experiments were conducted (Fig. 9). The XRD samples were prepared by drop-casting chlorobenzene solutions (1 mg mL−1) on a Si wafer in a glove box. These samples were annealed at 160 °C for 10 min. The annealing process was included to mimic the OSC device preparation which will be discussed later. The most distinctive difference in the diffraction pattern was found at 2θ below 5°. BTD-P-T showed sharp primary diffractions at 2θ = 3.58° and 4.16°, which corresponds to d-spacings of 24.84 Å and 21.39 Å, respectively, in addition to broad peaks at higher angles. It should be noted that the theoretically calculated π-core length excluding alkyl side groups is ca. 25 Å (Fig. S7, ESI†) which corresponds to d-spacing from primary diffraction. Absence of strong, higher order peaks of the primary d-spacing indicates that long-range order is not present in the film. In the case of BTD-P-S, the diffraction was quite featureless, indicating less crystalline character.
 | ||
| Fig. 9 X-ray powder diffraction patterns of BTD–phenazine compounds. | ||
More significant π–π interactions from the solid-state absorption, higher melting transition with larger heat of fusion, and more crystalline character suggest that BTD-P-T has tighter molecular arrangement than BTD-P-S. Solving the crystal structure of both systems would provide direct verification of such a hypothesis, however single crystals of the title compounds were not available since they both have a tendency to fibrillate. Presumably, the planarity of BTD-P-T as ascertained by computation allows for more compact molecular packing, which explains the observed difference in optical, thermal, and diffraction properties.
Both BTD-P-S and BTD-P-T showed excellent fibrillation capability. As presented in Fig. 10, cast films of the two molecules from dichloromethane solutions (3 mg mL−1) exhibit morphologies which are composed of long fibers. In particular, BTD-P-T formed longer, less branched fibers compared to BTD-P-S. The initial assembly results support our strategy of incorporating dialkoxyphenazine to stimulate 1D assembly. Further detailed study on the self-assembly including organogelation is in progress.
 | ||
| Fig. 10 Optical micrographs of cast films: (a) BTD-P-S and (b) BTD-P-T. Scale bar: 20 μm. | ||
We note that optical, electronic, thermal, and assembly properties of BTD-P compounds are better understood when coupled with our theoretical findings.
Application to organic solar cell as electron acceptors
We investigated the efficacy of the title compounds as electron acceptors in BHJ OSCs. In recent years, there has been significant advancement in small-molecule-based non-fullerene acceptors for OSCs.37–45 We note that our objective in this work is to investigate the impact of small structural differences on device performance, rather than to achieve the state-of-art PCE. Before an OSC device was fabricated, fluorescence quenching experiments were conducted to determine the optimum ratio between P3HT and BTD-P compounds. An optimum ratio of these two compounds facilitates maximum electron transfer. The blended films were fabricated via spin-coating from a chlorobenzene solution (20 mg mL−1) at 1200 rpm for 40 s. The chlorobenzene solution contained DIO at 3 v/v% to mimic the actual layer of the OSC device that will be fabricated. The films were further annealed at 160 °C for 10 min. Interestingly, the fluorescence quenching was efficient even with a small content of BTD-P acceptors. For both P3HT:BTD-P-S (Fig. S8, ESI†) and P3HT:BTD-P-T (Fig. S9, ESI†) blend films, 90% of P3HT FL was quenched at a weight ratio of 6:1. Further increase in the BTD-P content did not facilitate more FL quenching. This result indicates that the electron transfer from P3HT to the acceptor is most efficient when the wt% of acceptor is only ca. 14.
To evaluate the performance of the BTD-P molecules as acceptors in organic solar cell (OSC) devices, inverted bulk heterojunction (BHJ) OSC devices were fabricated with a structure of ITO/ZnO (40 nm)/active layer (P3HT:BTD-P blend) (100 nm)/MoO3 (10 nm)/Ag (100 nm). Detailed descriptions on the device fabrication process are provided in ESI. For both P3HT:BTD-P-S and P3HT:BTD-P-T, four different donor:acceptor ratios were tested. Representative characteristics of inverted BHJ OSCs such as open circuit voltage (Voc), short circuit current (Jsc), fill factor (FF), and power conversion efficiency (PCE) are summarized in Table 3, and current density–voltage (J–V) characteristics of the BHJ OSC devices under AM 1.5G illumination at 100 mW cm−2 are shown in Fig. 11. In both cases, the donor:acceptor ratio of 6:1 gave the highest PCE. The device based on P3HT:BTD-P-S BHJ showed a Voc of 0.49 V, a Jsc of 0.57 mA cm−2, a FF of 60.38% and a PCE of 0.17%. On the other hand, the device based on P3HT:BTD-P-T BHJ exhibited highly improved photovoltaic performance with a Voc of 0.63 V, a Jsc of 1.18 mA cm−2, a FF of 58.53% and a PCE of 0.44%. Compared to the devices with P3HT:BTD-P-S BHJ, the improvement in the photovoltaic performance of the devices with P3HT:BTD-P-T BHJ mainly resulted from an improved Jsc value.
| Acceptor | Ratioa | Voc (V) | Jsc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|---|
| a P3HT:acceptor (weight ratio). |
|||||
| BTD-P-S | 1:1 |
0.33 | 0.17 | 47.67 | 0.03 |
| 2:1 |
0.41 | 0.35 | 55.06 | 0.08 | |
| 4:1 |
0.48 | 0.50 | 57.47 | 0.14 | |
| 6:1 |
0.49 | 0.57 | 60.38 | 0.17 | |
| BTD-P-T | 1:1 |
0.56 | 0.41 | 42.92 | 0.10 |
| 2:1 |
0.61 | 0.83 | 50.04 | 0.26 | |
| 4:1 |
0.62 | 0.73 | 57.81 | 0.25 | |
| 6:1 |
0.63 | 1.18 | 58.53 | 0.44 | |
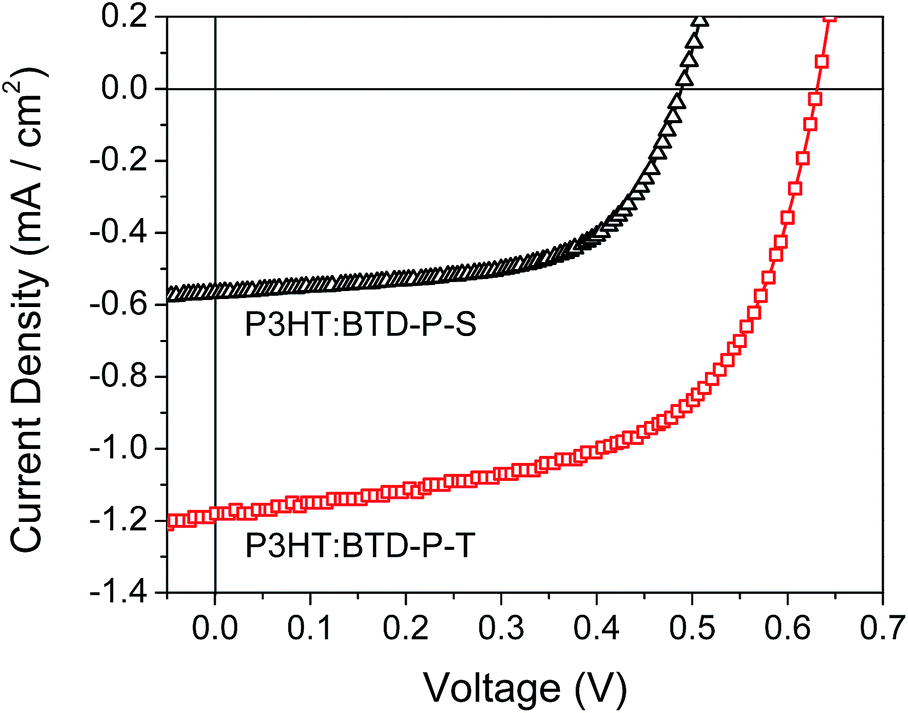 | ||
| Fig. 11 J–V curves of P3HT:BTD-P BHJs (6:1 wt ratio for both devices) under AM 1.5G condition. | ||
Surprisingly, increased acceptor content from 14% (6:1) to 50% (1:1) decreased Jsc by ca. 70% for both BHJs. Meanwhile, Voc decreased by ca. 30% for P3HT:BTD-P-S and ca. 10% for P3HT:BTD-P-T when the ratio changed from 6:1 to 1:1. Lower PCE at higher acceptor content may be due to the increased phase separation and poor light harvesting as P3HT is the major contributor for the light absorption.
To further confirm the accuracy of the measurements, the external quantum efficiency (EQE) of P3HT:BTD-P-T BHJ OSC devices at different ratios was measured as shown in Fig. 12. All the tested donor:acceptor ratios exhibited a maximum EQE at ca. 500 nm, which is consistent with the absorption maximum of P3HT (Fig. S10, ESI†). The highest EQE was found at the P3HT:BTD-P-T ratio of 6:1, which gave the highest PCE. The morphology of the active layers (P3HT:BTD-P-T) were investigated by atomic force microscopy (AFM). The AFM images with donor:acceptor ratios of 1:1 and 6:1 did not exhibit any drastic morphological differences (Fig. S11, ESI†). Both blends exhibited smooth surfaces with root mean square roughness of 2.8 nm for 1:1 blend and 3.5 nm for 6:1 blend. Due to the low content of the acceptor, it would be difficult to observe fibrous textures seen in the cast films.
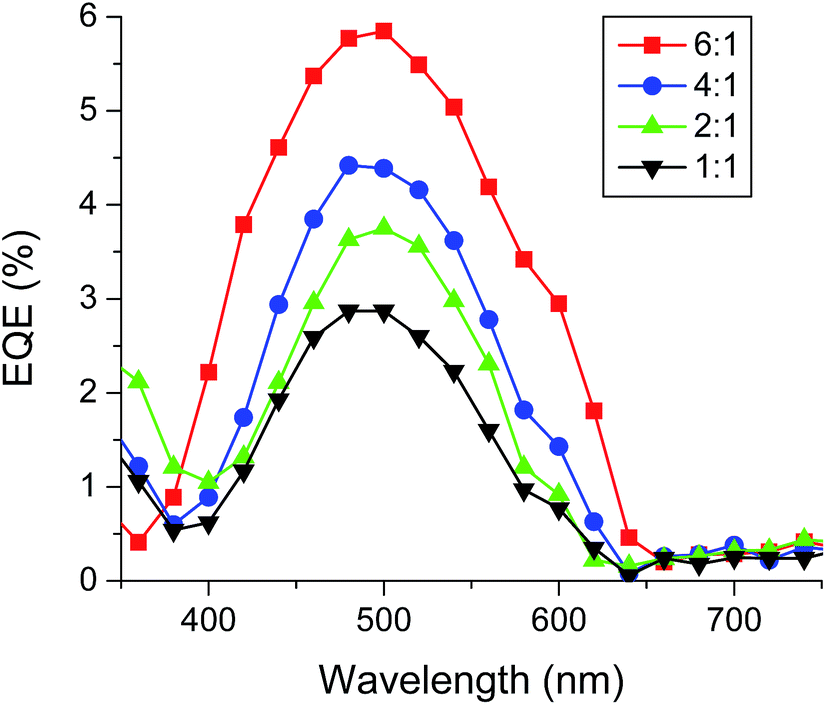 | ||
| Fig. 12 EQE spectra of P3HT:BTD-P-T based BHJ. | ||
Conclusions
In this work, we demonstrated how structural entities impact the electronic properties of two A–A′–A systems by analyzing each structural intermediate theoretically during the design process. The most profound effect on ELUMO was found when more electron-deficient BTD was added to the less electron-deficient phenazine. The connectivity between A and A′ affected the dihedral angle significantly which influenced EHOMO since the HOMO orbital was distributed along the whole molecule while the LUMO orbital was localized more on the electron-deficient component. Both BTD-P-T and BTD-P-S were successfully synthesized and characterized. From optical, electrochemical, and thermal property characterizations, BTD-P-T showed lower Egap and ELUMO, and higher Tm with larger heat of fusion. X-ray powder diffraction also revealed higher crystallinity for BTD-P-T. Both BTD-P-S and BTD-P-T showed excellent 1D assembling properties by producing fibers upon solvent casting. Theoretical calculations revealed a planar conformation for BTD-P-T, which accounts for more enhanced intermolecular π–π interactions. Even with moderate electron deficiency compared to fullerene, the title compounds functioned successfully as an electron-acceptor for bulk heterojunction OSCs with P3HT as a donor. For both P3HT:BTD-P-S and P3HT:BTD-P-T BHJs, the weight ratio of 6:1 produced the highest PCE of 0.17% and 0.44%, respectively. The higher Jsc of the BTD-P-T device was a major contribution for the PCE, and nearly double that of BTD-P-S. We hypothesize that the flat geometry of BTD-P-T allows for more efficient intermolecular π-orbital overlap, thus enhancing charge transport. The theory guided molecular building process we present in this work should make the experimental design process more effective and efficient by screening and optimizing molecular architectures before embarking on lengthy synthetic procedures.
Acknowledgements
DCL gratefully acknowledges financial support from NSF Career Award (DMR-0846479). YL acknowledges financial support from the Technology Development Program to Solve Climate Changes of the National Research Foundation (NRF-2015M1A2A2056216) and Basic Science Research Program through the National Research Foundation of Korea (NRF-2015R1A2A2A01003622) funded by the Ministry of Education, Science, and Technology. This work was also supported by the DGIST R&D Program of the Ministry of Science, ICT and Future Planning (17-BD-0404).Notes and references
- J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS PubMed.
- C. R. Newman, D. D. Frisbie, D. A. da Silva Filho, J.-L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436 CrossRef CAS.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Mater., 2011, 23, 583 CrossRef CAS.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef CAS PubMed.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876 CrossRef CAS PubMed.
- P. Sonar, J. P. F. Lim and K. L. Chan, Energy Environ. Sci., 2011, 4, 1558 CAS.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- A. Facchetti, Mater. Today, 2013, 16, 123 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- T. Xu and L. Yu, Mater. Today, 2014, 17, 11 CrossRef CAS.
- L. Lu and L. Yu, Adv. Mater., 2014, 26, 4413 CrossRef CAS PubMed.
- H. A. M. van Mullekom, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1 CrossRef.
- A. Ajayaghosh, Acc. Chem. Res., 2005, 38, 449 CrossRef CAS PubMed.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- J. Peet, A. J. Heeger and G. C. Bazan, Acc. Chem. Res., 2009, 42, 1700 CrossRef CAS PubMed.
- O. Inganäs, F. Zhang and M. R. Andersson, Acc. Chem. Res., 2009, 42, 1731 CrossRef PubMed.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394 RSC.
- L. Pandey, C. Risko, J. E. Norton and J.-L. Brédas, Macromolecules, 2012, 45, 6405 CrossRef CAS.
- D.-C. Lee, K. Jang, K. K. McGrath, R. Uy, K. A. Robins and D. W. Hatchett, Chem. Mater., 2008, 20, 3688 CrossRef CAS.
- L. V. Brownell, K. Jang, K. A. Robins, I. C. Tran, C. Heske and D.-C. Lee, Phys. Chem. Chem. Phys., 2013, 15, 5967 RSC.
- L. V. Brownell, K. A. Robins, Y. Jeong, Y. Lee and D.-C. Lee, J. Phys. Chem. C, 2013, 117, 25236 CAS.
- Y. Cao, T. Lei, J. Yuan, J.-Y. Wang and J. Pei, Polym. Chem., 2013, 4, 5228 RSC.
- D. Mori, H. Benten, H. Ohkita, S. Ito and K. Miyake, ACS Appl. Mater. Interfaces, 2012, 4, 3325 CAS.
- C. Guo, Y.-H. Lin, M. D. Witman, K. A. Smith, C. Wang, A. Hexemer, J. Strzalka, E. D. Gomez and R. Verduzco, Nano Lett., 2013, 13, 2957–2963 CrossRef CAS PubMed.
- J.-L. Pozzo, G. M. Clavier and J.-P. Desvergne, J. Mater. Chem., 1998, 8, 2575 RSC.
- D.-C. Lee, B. Cao, K. Jang and P. M. Forster, J. Mater. Chem., 2010, 20, 867 RSC.
- K. Jang, L. V. Brownell, P. M. Forster and D.-C. Lee, Langmuir, 2011, 27, 14615 CrossRef CAS PubMed.
- T. Yamamoto, T. Maruyama, Z. Zhou, T. Ito, T. Fukuda, Y. Yoneda, F. Begamn, T. Ikeda, S. Sasaki, H. Takezoe, A. Fukuda and K. Kubota, J. Am. Chem. Soc., 1994, 116, 4832 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS PubMed.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 CrossRef CAS PubMed.
- L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed.
- A. Ajayaghosh, C. Vijayakumar, R. Varghese and S. J. George, Angew. Chem., Int. Ed., 2006, 45, 456 CrossRef CAS PubMed.
- H. Li, T. Earmme, G. Ren, A. Saeki, S. Yoshikawa, N. M. Murari, S. Subramaniyan, M. J. Crane, S. Seki and S. A. Jenekhe, J. Am. Chem. Soc., 2014, 136, 14589 CrossRef CAS PubMed.
- Z. Mao, W. Senevirathna, J.-Y. Liao, J. Gu, S. V. Kesava, C. Guo, E. D. Gomez and G. Sauvé, Adv. Mater., 2014, 26, 6290 CrossRef CAS PubMed.
- H. Li, Y.-J. Hwang, B. A. E. Courtright, F. N. Eberle, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2015, 27, 3266 CrossRef CAS PubMed.
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610 CAS.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156 CrossRef CAS PubMed.
- Y.-J. Hwang, H. Li, B. A. E. Courtright, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2016, 28, 124 CrossRef CAS PubMed.
- Y. Lin, Q. He, F. Zhao, L. Huo, J. Mai, X. Lu, C.-J. Su, T. Li, J. Wang, J. Zhu, Y. Sun, C. Wang and X. Zhan, J. Am. Chem. Soc., 2016, 138, 2973 CrossRef CAS PubMed.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955 CrossRef CAS PubMed.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterizations, molecular length of BTD-P-T, fluorescence spectra of P3HT:BTD-P blends, UV-vis absorption spectra of P3HT:BTD-P-T, AFM images of P3HT:BTD-P-T blends. See DOI: 10.1039/c7ra03612d |
| This journal is © The Royal Society of Chemistry 2017 |