
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of g-C3N4 nanosheets loaded with WO3 nanoparticles with enhanced photocatalytic performance under visible light irradiation†
Jie Meng ,
Jingyuan Pei,
Zefang He,
Shiyan Wu,
Qingyun Lin,
Xiao Wei*,
Jixue Li and
Ze Zhang
,
Jingyuan Pei,
Zefang He,
Shiyan Wu,
Qingyun Lin,
Xiao Wei*,
Jixue Li and
Ze Zhang
Center of Electron Microscopy, State Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, 310027, P. R. China. E-mail: mseweixiao@zju.edu.cn; Fax: +86 571 87952797; Tel: +86 571 87952797
First published on 3rd May 2017
Abstract
Graphitic carbon nitride (g-C3N4) nanosheets loaded with WO3 nanoparticles were prepared via heat treatment of g-C3N4 together with WOx-EDA nanobelts. The thermal treatment temperature and WOx-EDA precursor play the key role in enhancing the interaction between WO3 nanoparticles and g-C3N4 nanosheets, because the temperature of thermal decomposition of WOx-EDA (∼400 °C) is close to the thermal exfoliation temperature of g-C3N4. The structure evolution and promotion effect of the nanocomposites in photocatalytic performance were well studied. It is found that WO3 nanoparticles uniformly dispersed on the surface of the g-C3N4 nanosheets, and the close integration of WO3 and g-C3N4 lead to the high photocatalytic activity in the degradation of RhB under visible light irradiation. Meanwhile, a larger specific area and increase of visible light absorption are also of benefit to speed up the degradation of organic dye. Also, the mechanism of the photocatalytic reaction and its reutilization properties were investigated.
Introduction
Photocatalysts have been receiving much attention as a promising material in recent years due to their applications in alleviating global environmental pollution and the energy crisis, such as waste pollutant degradation,1,2 water splitting3 and CO2 reduction.4 However, there are some major challenges that hamper the requirements of their practical applications, for instance, a high recombination rate of photo-excited charge carriers, low solar energy conversion efficiency resulting from a wide band gap, and other restrictions.5,6 Accordingly, it is essential to develop a high performance photocatalyst that could not only enhance the separation and transportation efficiency of electron–hole pairs but also improve the light absorption properties under solar radiation.Graphitic carbon nitride (g-C3N4), a great potential material for photocatalytic reaction first reported by Wang et al. in early 2009,7 has received intensive attention on account of its unique electronic and optical properties, such as visible light response, good reduction ability, easy synthesis, and high stabilities. What's more, the band gap of g-C3N4 is around 2.7 eV, which could absorb visible light up to 460 nm correspondingly.8,9 The conduction band edge potential of g-C3N4 is more negative (about −1.12 eV vs. NHE), therefore, photo-induced electrons would have high reduction capacity.10,11 Nevertheless, it has been concluded that the low photocatalytic activities of g-C3N4 are mainly attributed to high recombination of photo-generated charge carriers and low specific surface area.12–14 Many efforts have been made to overcome these bottlenecks. It was reported that ultrasonic vibration,15 thermal exfoliation16 and hydrothermal treatment17 could be used to change the BET surface area. Meanwhile, doping,18 noble metal loading19 and coupling with other semiconductors8 are also effective methods to achieve the separation of carriers and then further increase the photocatalytic performance under visible light illumination. Among all the strategies, coupling with other semiconductors is an efficient attempt to solve above two questions due to the suitable band position and enhanced hole–electron pairs transfer between the two semiconductors by appropriate combination style. Especially, g-C3N4 nanosheets which show distinct physical and chemical features with larger specific area and outstanding electronic mobility,20,21 are considered to be an appropriate candidate to couple with other semiconductors to form high-performance photocatalysts. As a matter of fact, many progresses have been made on g-C3N4 nanosheets based composites,22 such as TiO2/g-C3N4,23 ZnO/g-C3N4,24 Bi2WO6/g-C3N4,25 MoS2/g-C3N4,26 etc. These composite systems have deservedly achieved enhanced photocatalytic performance which is generally ascribed to the improved reaction sites, charge separation and migration processes. Nevertheless, it is necessary for a practical photocatalyst to be provided with stable crystal structure, narrow band gap, low material cost, and easy preparation. So there is still a long way to seek a proper material coupling with g-C3N4 nanosheets to form highly active composite photocatalysts.
Alternatively, tungsten(VI) oxide (WO3) which is deemed as a promising material of photocatalysts27 with a narrow band gap of about 2.7 eV, is a potential candidate to form composites with g-C3N4 nanosheets to get excellent photocatalytic performance. Meanwhile, its stable crystal structure, resistance to photo-corrosion, and inexpensiveness are all important issues for the practical application.28 The VB edge potential of WO3 is more positive, implying that it has a better oxidation capability in photocatalytic activities. However, the CB position of WO3 limits its photocatalytic reaction under visible light illumination since this CB level is not beneficial to the single-electron reaction of O2.29,30 It could decrease the recombination of hole–electron pairs and make up the defects of the conduction band by synergistic effect of two semiconductors when WO3 coupling with g-C3N4.30 Therefore, WO3 is a proper candidate to form composites with g-C3N4 to achieve excellent photocatalytic performance under visible light irradiation. However, for photocatalytic composites, the separation efficiency of photo-induced carriers is not only related to appropriate electronic structures, but most importantly determined by effective combination with better contact, that is still a challenge for most semiconductor composites. Many attempts have been applied to form WO3/g-C3N4 composites to achieve the above goals, but there are no reports about using the WOx-EDA nanobelts, as the precursor of WO3 to synthesize WO3/g-C3N4 composites with a suitable combination method which could not only enlarge the specific surface area but also restrain the recombination of photo-excited carriers.29–33 The WOx-EDA nanobelts was synthesized by solvothermally treating WO3 in ethanediamine (EDA) following a so-called solvent-coordination molecular template (SCMT) mechanism.34
In this paper, we prepared the WO3/g-C3N4 composites through heat treatment of WOx-EDA and g-C3N4. Since the decomposition temperature (around 400 °C) of WOx-EDA is close to thermal exfoliation temperature of g-C3N4, both of two reaction processes conduct simultaneous, which was found to facilitate WO3 nanoparticles to uniformly disperse on g-C3N4 nanosheets. The photocatalytic performance of the WO3/g-C3N4 composites was evaluated by the degradation of Rhodamine B under visible light illumination. The decrease of recombination of photo-generated hole–electron pairs evaluated by photoluminescence (PL) spectra, larger specific surface area calculated by BET method and enhanced visible light absorption characterized by diffuse reflectance spectrum (DRS) are main reasons to increase photocatalytic activity. Based on the systematically characterizations of the photocatalysts, a possible mechanism for the enhanced photocatalytic performance of WO3/g-C3N4 composites is proposed.
Experimental section
Materials and synthesis
All of the chemical reagents used in this experiment were of analytical stage without further purification. | ||
| Scheme 1 Schematic presentation of the synthesis process of WO3/g-C3N4 composites. | ||
Characterization
The crystalline phases of the as-synthesized samples were characterized by X-ray diffraction (XRD, PANalytical X'Pert PRO) with Cu Kα radiation with 2θ from 10–80°. The morphology and composition of the as-obtained samples were investigated by field emission scanning electron microscopy (SEM, Hitachi SU-70) and transmission electron microscopy (TEM, FEI Tecnai G2 F20 S-TWIN) equipped with an energy dispersive X-ray spectrometer (EDX). Specific surface area (SBET) was determined with a surface area analyser (Quantachrome AUTOSORB-IQ2-MP) by the BET method. Diffuse reflectance spectra (DRS) was measured by a UV-vis spectrophotometer (Persee TU-1900) equipped with an integrating sphere unit using BaSO4 as the reflectance standard. Photoluminescence (PL) spectra was obtained by fluorescence spectrometer (Edinburgh instruments FLS920). Fourier transform infrared (FTIR) spectra of samples was recorded using a Nicolet Avatar-370 spectrometer at room temperature. The surface electronic states of the samples were measured by X-ray photoelectron spectroscopy (XPS, Escalab 250Xi). The concentration of phenol was characterized by liquid chromatograph (C18-ODS column).Photocatalytic measurement
The photocatalytic performances of the WO3/CN samples were characterization by the degradation of Rhodamine B (RhB) under visible light. A 300 W xenon lamp acts as a visible light source equipped with a UV cutoff filter (λ > 420 nm) during the experiment. For photocatalytic test, 0.05 g of the photocatalyst was dissolved in 100 mL of 10 mg L−1 RhB solution in a glass beaker. Before irradiation, the solution was magnetically stirred in the dark for 30 minutes to achieve adsorption–desorption equilibrium between photocatalyst and dye. After the illumination, 5 mL of solution was taken from the reaction suspension and centrifuged to remove the particles every time before measuring the absorption spectra. The UV-vis spectrophotometer (Persee TU-1900) was used to study the concentration of RhB in solution. The characteristic absorption peak of RhB at 554 nm was used to determine the extent of its degradation.Electrochemical analysis
Electrochemical measurements were conducted on a CHI 760 electrochemical workstation in a standard three-electrode cell, using a Pt plate as counter electrode and an Ag/AgCl electrode as reference electrode and photocatalyst-coated FTO conductive glass slides (ca. 2 cm2) as working electrode, respectively. Na2SO4 aqueous solution was selected as the electrolyte. The working electrode was prepared by follow deposition method: 20 mg sample powder was mixed in 40 mL PEG10000 solution, sonicated for 30 min and as-prepared solution was drop onto FTO. Then, parallel FTO glasses were placed in the drying oven for at 100 °C for 6 h. Finally, as-prepared FTO glasses with sample film were annealed at 300 °C for 3 h in muffle furnace. The bias voltage was set at −0.5 V vs. Ag/AgCl. 300 W Xe lamp with a UV filter (λ > 420 nm) served as light source in the photocurrent tests obtained at 0.5 V vs. Ag/AgCl.Results and discussion
Characterization of WO3/g-C3N4 composites
Fig. 1 shows the XRD patterns of g-C3N4, WO3 and serials WO3/g-C3N4 composite photocatalysts. It is observed that two diffraction peaks areas around 27.7° and 12.9° for g-C3N4 (JCPDS 87-1526), are corresponding to the (002) and (100) diffraction planes, respectively. According to the reported results,36 the stronger peak at 27.7° corresponds to the interlayer stacking of the aromatic systems with the interlayer distance of 0.322 nm. The weaker one at 12.9° which attributes to a distance around d = 0.686 nm is related to in-plane tris-s-triazine structural packing. For WO3, it can be index to the monoclinic structure (JCPDS card no. 83-0950 with lattice parameters of a = 7.301 Å, b = 7.539 Å, c = 7.690 Å and α = γ = 90°, β = 90.892°) and the main diffraction peaks at 23.3°, 23.7°, 24.5°, 26.7°, 28.8°, 34.3°, 41.7°, 50.0°, and 55.94° are corresponding to (002), (020), (200), (120), (112), (202), (222), (400), and (420) planes, respectively. For WO3/g-C3N4 composites, the XRD patterns reveal a coexistence of WO3 and g-C3N4. The intensity of characteristic (002) peak assigned to g-C3N4 becomes weaker with the increasing WO3 content in the composites gradually. The (100) peak ascribed to g-C3N4 is not observed in the composites which may be due to the relative low intensity compared to WO3. The main peak changes slightly compared to original 27.7° for g-C3N4 resulted from the introduction of WO3 indicate that a small shift of interlayers for the interaction between WO3 and g-C3N4. | ||
| Fig. 1 XRD patterns of g-C3N4, WO3 and WO3/g-C3N4 composite photocatalysts. | ||
The morphologies and microstructures of the as-obtained samples are shown in Fig. 2 and S3.† As seen in Fig. 2(a), g-C3N4 displays a sheet-like structure with different thickness through high or low contrast in the picture. This microstructure of g-C3N4 is consistent with our assumption after the process of ultrasonic and thermal exfoliation. As can be seen in Fig. 2(b), WO3 particles are deposited on the surface of g-C3N4 in the WO3/g-C3N4 composite (WECN40) morphology. The crystal phase of particles is attributed to monoclinic WO3 which could be demonstrated by high resolution transmission electron microscopy (HRTEM) in Fig. 2(c). The fringes of particles match the crystallographic plane of WO3 described in Fig. 2(c) and the interfaces in WO3/g-C3N4 composites are thought to be beneficial to suppress the recombination of hole and election under photo-excited environment. The Fig. 2(d) presents the morphology of pure WO3 nanobelts prepared without the g-C3N4. From the SEM images, it is clearly found that WO3 nanobelts are mainly composed of nanoparticles due to the thermal decomposition of EDA.37 These results are according with the XRD results. As shown in Fig. S4,† the peaks of EDS spectrum for WO3/g-C3N4 composites are ascribed to C, N, O and W, the presence of Cu and partly high intensity of C are due to the influence of the micro grid, implying that the composites are composed of WO3 and g-C3N4 without any kind of impurity. The FT-IR spectra of g-C3N4, WO3 and WO3/g-C3N4 composite (WECN40) are presented in Fig. 3. For pure g-C3N4, several strong bands in the 1000–1650 cm−1 region and the peak at 810 cm−1 were observed, which belong to the s-triazine ring modes.38,39 The absorption peaks at 1638 cm−1 and 1238 cm−1 are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N stretching modes respectively.40–43 The sharp peak at 1638 cm−1 is an indication of good crystallinity of g-C3N4 crystals. Also, broad bands of stretching and deformation modes of the NH2 (as well as an overlapping band of O–H stretching possibly) groups at 3414 cm−1.44 The broad absorption peak at 817 cm−1 is assigned to O–W–O stretching vibrations for WO3. Obviously, the characteristic peaks of g-C3N4 between 800 and 1600 cm−1 weaken to some extent when combined with WO3. The peak at 3414 cm−1 for all samples is attributed to O–H stretching vibration and the bending vibration of adsorbed water molecules is located at 1620 cm−1 for samples.45
N and C–N stretching modes respectively.40–43 The sharp peak at 1638 cm−1 is an indication of good crystallinity of g-C3N4 crystals. Also, broad bands of stretching and deformation modes of the NH2 (as well as an overlapping band of O–H stretching possibly) groups at 3414 cm−1.44 The broad absorption peak at 817 cm−1 is assigned to O–W–O stretching vibrations for WO3. Obviously, the characteristic peaks of g-C3N4 between 800 and 1600 cm−1 weaken to some extent when combined with WO3. The peak at 3414 cm−1 for all samples is attributed to O–H stretching vibration and the bending vibration of adsorbed water molecules is located at 1620 cm−1 for samples.45
 | ||
| Fig. 2 TEM images of (a) CN; (b) WECN40; (c) high resolution TEM of WECN40; (d) WO3 (the SEM images could be observed by the inset). | ||
 | ||
| Fig. 3 FT-IR spectra of g-C3N4, WO3 and WO3/g-C3N4 composite (WECN40). | ||
XPS spectra are shown in Fig. 4 to analyse the surface chemical composition and chemical state. It is observed that as-synthesized WO3/g-C3N3 composites are primarily composed of the elements like W, O, C and N from the XPS survey spectrum in Fig. 4(a). It is clearly seen that g-C3N4 and WO3 are coexist in composite and slight shift of peaks indicates the interaction between g-C3N4 and WO3 is exist indeed. The W4f spectrum (Fig. 4(b)) centred at 35.0 eV is ascribed to 4f7/2 and the other peak at 37.1 eV is ascribed to 4f5/2, which indicate that they are the typical valence state peaks of W6+.46–48 Two peaks (Fig. 4(c)) are showed at about 529.4 eV and 531.8 eV for O1s XPS spectra, which could be associated with W–O–W and W–O–H, respectively.49 The C1s peaks (Fig. 4(d)) could be separated into three characterization parts: the peak located at 287.3 eV is typically related to the sp2-bonded NC–N, the peak centred at 285.9 eV is attributed to the C–O species on the g-C3N4 surface and the peak at 284.6 eV can be assigned to C–C may due to the surface adventitious carbon or defect in the g-C3N4.50,51 Additionally, the N1s peaks (Fig. 4(e)) can be deconvoluted into three peaks at 398.4 eV, 399.7 eV, and 400.8 eV which refer to triazine rings (CN–C), tertiary nitrogen (N–(C)3), and amino functions (C–N–H).52 Fig. 4(f) shows valence band spectra of samples and valence band potentials of g-C3N4 and WO3 are 1.62 and 2.70 eV respectively. The changes of electronic structure for WO3/g-C3N4 (WECN40) indicate that the existence of interaction between g-C3N4 and WO3 compared to pure WO3 and g-C3N4 (Fig. S5 and S6†) and that would be beneficial to photocatalytic performance.
 | ||
| Fig. 4 (a) XPS survey spectra of g-C3N4, WO3 and WO3/g-C3N4 composite (WECN40); high revolution XPS spectra of (b) W4f, (c) O1s, (d) C1s, (e) N1s; (f) valence band spectra of g-C3N4 and WO3. | ||
As can be seen in Fig. 5(a), the UV-vis diffuse reflectance spectra of g-C3N4, WO3, WO3/g-C3N4 composites are exhibited. For WO3/g-C3N4 composites, they are red-shifted in the region of 300–750 nm and the absorption ability increases with the increasing amount of WO3, which is consistent with gradually darken colour of samples (inset of Fig. 5(a)). It is observed that the composite sample WECN40 shows stronger absorption in the visible region at wavelengths more than around 400 nm compared with g-C3N4 and WO3. The band gap of samples can be estimated by the following equation:
| (ahν)n/2 = A(hν − Eg) |
| EVB = X − Ec + 0.5Eg |
| ECB = EVB − Eg |
 | ||
| Fig. 5 (a) UV-vis diffuse reflectance spectra of g-C3N4, WO3 and WO3/g-C3N4 composites (the different number colors marked in samples are consistent with the nomination of samples); (b) the band gap determination of g-C3N4, WO3 and WECN40 by Tanc's approach; (c) the optical transition type determination of g-C3N4 and WO3; (d) PL spectra of g-C3N4, WO3 and WO3/g-C3N4 composites. | ||
In this equation, EVB, X, Ec, ECB, andEg are defined as the VB edge potential, the electronegativity of the semiconductor, the energy of free electrons on the hydrogen scale (around 4.5 eV), the CB edge potential and the band gap energy of the semiconductor respectively.55 The valence band edge potential of g-C3N4 and WO3 are 1.62 and 2.70 eV respectively from above XPS analysis (Fig. 5(f)). And the conduction band potentials of g-C3N4 and WO3 are calculated to be −1.09 eV and −0.17 eV, respectively.
Fig. 5(d) shows the PL spectra of g-C3N4, WO3 and WO3/g-C3N4 composites under the excitation wavelength of 325 nm. For g-C3N4, a strong emission band centred at about 465 nm could be attributed to the band–band PL phenomenon of photo-excited electron–hole.56 While a weak emission peak of WO3 could be related to localized state in the band gap with the existence of defects or oxygen vacancies.57 It is clear that the peak intensities for WO3/g-C3N4 composites decrease with the increase of WO3. This means that an increasing number of charge carriers take part in photocatalytic reactions since the lower PL intensities implied a slower recombination rate of the photo-generated electron–hole pairs. Meanwhile, this result also indicates that the interfaces between g-C3N4 and WO3 do promote the separation and migration of photo-induced charge carriers.
As shown in Fig. 6(a), the BET surface area of samples is determined by nitrogen adsorption and desorption isotherms. The SBET of WECN40 composites (Table S1†) increased by 1.81 and 1.96 times compared to pure g-C3N4 and WO3 photocatalysts, indicating that the interaction between g-C3N4 nanosheets and WO3 nanoparticles (about 19.8 nm calculated by Scherrer equation) enlarges the BET surface area and corresponding composite materials provide more reaction sites for photodegradation. From Fig. 6(b) and Table S1,† average pore diameters of g-C3N4, WO3 and WO3/g-C3N4 composite (WECN40) are distributed at about 51.796, 2.105, 19.844 nm, which corresponds to the pore volume of 0.166, 0.167, 0.046 cm g−1. It means that WECN40 is a photocatalyst with smaller pore diameter and larger pore volume. The pore volume of WO3/g-C3N4 composite is almost the same as pure g-C3N4 but it has smaller pore diameter, which can be relate to the presence of WO3 particles dispersing on g-C3N4 nanosheets.
 | ||
| Fig. 6 (a) N2 adsorption–desorption isotherm and (b) pore size distribution curves of g-C3N4, WO3 and WO3/g-C3N4 composite (WECN40). | ||
Photocatalytic performance and stability of the WO3/g-C3N4 composites
The photocatalytic performance of samples was investigated by the degradation of RhB under visible light illumination. As depicted in Fig. 7(a), for WO3/g-C3N4 composite (WECN40), the absorption peak of the RhB solution decreased with the increase of time which indicates the photodegradation of RhB under visible light. And the blue shift from 554 to 503 nm for the wavelength of maximum characteristic peak could be attributed to N-deethylation mechanism in photocatalytic reaction.58 The (Ci/C0) of all the samples are shown in Fig. 7(b), it is obvious that WO3/g-C3N4 composites display much higher photocatalytic capability compared with pure g-C3N4 and WO3. The photocatalyst of WECN40 exhibits best degradation rate of contamination. In order to further estimate the photocatalytic reactions of pollutants in aqueous solutions, the first order equation of the Langmuir–Hinshelwood model is used to as follows:59| ln(Ci/C0) = kt |
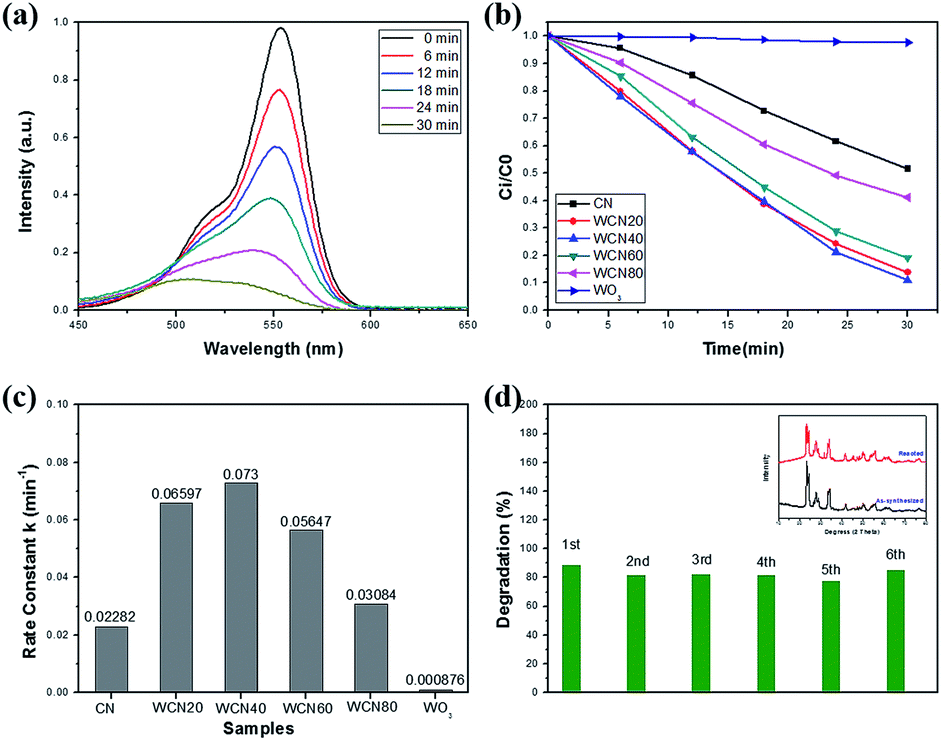 | ||
| Fig. 7 (a) Photodegradation of RhB with WECN40; (b) Ci/C0 of all the samples; (c) rate constants k values of all the photocatalysts; (d) recyclability of WECN40. | ||
To demonstrate the advantages of WOx-EDA precursor, commercial WO3 (bought from Aladdin) was selected instead of WOx-EDA as precursor to prepare WO3/g-C3N4 composites. According to different mass ratio between commercial WO3 and CN, the samples were named CN, WCN20, WCN40, WCN60, WCN80, and WO3(C). The above samples were prepared by the same process according to the g-C3N4 nanosheets loading with WO3 nanoparticles using WOx-EDA as precursor and its photocatalytic performance was tested with the same conditions under visible light illumination (Fig. S8†). Among the above six samples, WCN60 shows the highest photocatalytic activity with the 68% degradation of RhB after 30 min visible light irradiation, but it is still lower than WECN20 (89%) which was prepared using WO3-EDA nanobelts as precursor. The poor photocatalytic performance could be attributed to its large particles size of commercial WO3, which can be seen in Fig. S7.†
Recycling tests of the WO3/g-C3N4 composites are carried out to assess photostability and recyclability as shown in Fig. 7(d). The degradation of RhB for WECN40 sample, from 89% to 85% in the first and sixth cycle respectively, remains relative stable without distinct fluctuation throughout six runs under visible light irradiation. Furthermore, the main diffraction peaks of XRD of the catalyst, before and after reaction, retain almost the same which implies the structural stability. Only the 2θ at around 31° may ascribe to catalyst contamination or reaction between sample and aqueous solution. The stability of photocatalytic performance and crystal phase implies the interaction between g-C3N4 and WO3 is relative strong.
Electrochemical characterization
To further support the restrained recombination of photo-induced electron–hole pairs for the WO3/g-C3N4 composites, the transient photocurrent response tests were carried out for several on–off cycles under visible light irradiation (Fig. S9†). The WECN40 exhibits higher photocurrent intensity than that of CN, which is in accordance with its enhanced photocatalytic degradation rate under visible light irradiation. The fact indicates that the WO3/g-C3N4 composites show better separation or transfer of photo-induced electrons and holes, which is also in accordance with the PL spectra results. For WO3 sample, it holds high photocurrent intensity, nevertheless, the inappropriate CB position limits its photocatalytic activity under visible light illumination.24,25Possible photocatalytic mechanism
To investigate the photocatalytic mechanism of the WO3/g-C3N4 composites, several scavengers were applied to check the active species during the photodegradation process under visible light irradiation (Fig. 8). Ethylenediaminetetraacetic acid disodium salt (EDTA-2Na), tert butyl alcohol (TBA) and 1,4-benzoquinone (BQ) were employed as the hole (h+) scavenger, hydroxyl radical (˙OH) scavenger and superoxide radical (˙O2−) scavenger. It is clear that the degradation of organic pollutants decreases by the addition of scavengers compared with catalyst without any scavengers, implying that reactive species are significant for the RhB degradation in aqueous solution under visible light. What's more, the effect of the scavengers of TBA and BQ based on suppression of RhB degradation is much bigger than that of EDTA-2Na, indicating that the species of hydroxyl radical (˙OH) and superoxide radical (˙O2−) are the major active species in this photocatalytic system. | ||
| Fig. 8 The effects of various scavengers on the photodegradation of WO3/g-C3N4 composite (WECN40) under visible light. | ||
Based on the above analysis, a proposed mechanism for the improved photocatalytic activity is briefly described in Fig. 9. For g-C3N4, the CB and VB potentials are 1.62 and −1.09 eV respectively, whereas these values of WO3 are 2.70 and −0.17 eV respectively. The g-C3N4 and WO3 would be excited and the transition of electrons from VB to CB will happen in both semiconductor under visible light irradiation. Because the VB level of g-C3N4 is more negative than that of WO3 and the CB potential of WO3 is more positive than that of g-C3N4, the photo-generated holes can transfer from the VB of WO3 to that of g-C3N4. Meanwhile, the photo-excited electrons in the CB of g-C3N4 could move to the CB of WO3. These electrons can react with W6+ species in the WO3 and then W6+ be reduced into W5+. The W5+ ions in the surface of WO3 can be re-oxidized into W6+ through the reaction with oxygen to generate superoxide radical (˙O2−). Superoxide radical (˙O2−) can also react with H2O molecules to form ˙OH.30 Hydroxyl radical (˙OH) and superoxide radical (˙O2−) are the main species to degrade RhB into CO2 and H2O under visible light irradiation. Hence, it is concluded that g-C3N4 coupling with WO3 increases the separation efficiency of the photo-induced holes and electrons through reduction or oxidation reaction in the surface of composites under visible light illumination.
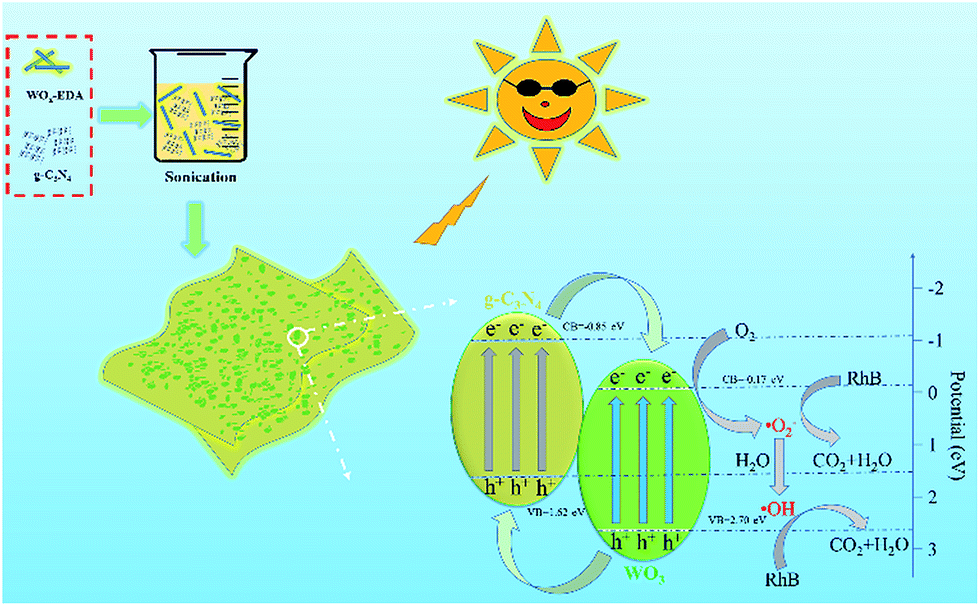 | ||
| Fig. 9 Possible degradation mechanism of RhB for photocatalysts under visible light illumination. | ||
Conclusions
The WO3/g-C3N4 composites were facile prepared through heat treatment of WOx-EDA nanobelts and g-C3N4 powder. The heat treatment temperature of 400 °C is the key factor to obtain g-C3N4 nanosheets with uniformly dispersed WO3 nanoparticles. The enhanced photocatalytic performance is considered to arise from the increased reaction sites, enhanced visible light absorption and the restrained recombination of photo-induced electron–hole pairs under visible light irradiation. The WO3/g-C3N4 composite exhibits best activity for the degradation of RhB when the amount of WOx-EDA reached 40 wt% in the mixture with g-C3N4. Meanwhile, it is concluded that the species of hydroxyl radical (˙OH) and superoxide radical (˙O2−) are the major active species in the photocatalytic system. Therefore, this work demonstrates a method using WOx-EDA as precursor, which could improve the photocatalytic degradation under visible light illumination. And it also provides the reference for other composites synthesis.Acknowledgements
This work was supported by National Nature Science Foundation of China (grant number: 11234011, 11327901, 51102208), the Fundamental Research Funds for the Central Universities (2014QNA4008).Notes and references
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J. M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- W. Ren, Z. Ai, F. Jia, L. Zhang, X. Fan and Z. Zou, Appl. Catal., B, 2007, 69, 138–144 CrossRef CAS.
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K. Q. Zhang, S. S. Al Deyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801 CAS.
- O. Ola and M. M. Maroto-Valer, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- S. W. Cao, X. F. Liu, Y. P. Yuan, Z. Y. Zhang, J. Fang, S. C. Loo, J. Barber, T. C. Sum and C. Xue, Phys. Chem. Chem. Phys., 2013, 15, 18363–18366 RSC.
- F. Raziq, Y. Qu, X. Zhang, M. Humayun, J. Wu, A. Zada, H. Yu, X. Sun and L. Jing, J. Phys. Chem. C, 2016, 120, 98–107 CAS.
- J. Fu, Y. Tian, B. Chang, F. Xi and X. Dong, J. Mater. Chem., 2012, 22, 21159–21166 RSC.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC.
- K. Dai, L. Lu, C. Liang, Q. Liu and G. Zhu, Appl. Catal., B, 2014, 156–157, 331–340 CrossRef CAS.
- K. Dai, L. Lu, Q. Liu, G. Zhu, X. Wei, J. Bai, L. Xuan and H. Wang, Dalton Trans., 2014, 43, 6295–6299 RSC.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- T. Sano, S. Tsutsui, K. Koike, T. Hirakawa, Y. Teramoto, N. Negishi and K. Takeuchi, J. Mater. Chem. A, 2013, 1, 479–485 Search PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
- L. Ge, C. Han, J. Liu and Y. Li, Appl. Catal., A, 2011, 409–410, 215–222 CrossRef CAS.
- W. Jiang, W. Luo, J. Wang, M. Zhang and Y. Zhu, J. Photochem. Photobiol., C, 2016, 28, 87–115 CrossRef CAS.
- X. Dong and F. Cheng, J. Mater. Chem. A, 2015, 3, 23642–23652 CAS.
- G. Mamba and A. K. Mishra, Appl. Catal., B, 2016, 198, 347–377 CrossRef CAS.
- L. Shen, Z. Xing, J. Zou, Z. Li, X. Wu, Y. Zhang, Q. Zhu, S. Yang and W. Zhou, Sci. Rep., 2017, 7, 41978–41989 CrossRef CAS PubMed.
- X. Yuan, C. Zhou, Q. Jing, Q. Tang, Y. Mu and A. K. Du, Nanomaterials, 2016, 6, 173–185 CrossRef PubMed.
- F. Chen, D. Li, B. Luo, M. Chen and W. Shi, J. Alloys Compd., 2017, 694, 193–200 CrossRef CAS.
- Y. Cao, Q. Li and W. Wang, RSC Adv., 2017, 7, 6131–6139 RSC.
- H. Reiche, W. W. Dunn and A. J. Bard, J. Phys. Chem., 1979, 83, 2248–2251 CrossRef CAS.
- S. G. Kumar and K. S. R. K. Rao, Appl. Surf. Sci., 2015, 355, 939–958 CrossRef.
- S. Chen, Y. Hu, S. Meng and X. Fu, Appl. Catal., B, 2014, 150–151, 564–573 CrossRef CAS.
- I. Aslam, C. Cao, M. Tanveer, W. S. Khan, M. Tahir, M. Abid, F. Idrees, F. K. Butt, Z. Ali and N. Mahmood, New J. Chem., 2014, 38, 5462–5469 RSC.
- M. A. Gondal, A. A. Adesida, S. G. Rashid, S. Shi, R. Khan, Z. H. Yamani, K. Shen, Q. Xu, Z. S. Seddigi and X. Chang, React. Kinet., Mech. Catal., 2014, 114, 357–367 CrossRef.
- H. Katsumata, Y. Tachi, T. Suzuki and S. Kaneco, RSC Adv., 2014, 4, 21405–21409 RSC.
- J. Ding, Q. Liu, Z. Zhang, X. Liu, J. Zhao, S. Cheng, B. Zong and W. L. Dai, Appl. Catal., B, 2015, 165, 511–518 CrossRef CAS.
- X. Hu, Q. Ji, J. P. Hill and K. Ariga, CrystEngComm, 2011, 13, 2237–2241 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- L. Ye, J. Liu, Z. Jiang, T. Peng and L. Zan, Appl. Catal., B, 2013, 142–143, 1–7 CAS.
- R. Wu, J. Zhang, Y. Shi, D. Liu and B. Zhang, J. Am. Chem. Soc., 2015, 137, 6983–6986 CrossRef CAS PubMed.
- V. N. Khabashesku, J. L. Zimmerman and J. L. Margrave, Chem. Mater., 2000, 12, 3264–3270 CrossRef CAS.
- J. L. Zimmerman, R. Williams, V. N. Khabashesku and J. L. Margrave, Nano Lett., 2001, 1, 731–734 CrossRef CAS.
- Y. J. Bai, B. Lü, Z. G. Liu, L. Li, D.-L. Cui, X. G. Xu and Q. L. Wang, J. Cryst. Growth, 2003, 247, 505–508 CrossRef CAS.
- J. H. Kaufman, S. Metin and D. D. Saperstein, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 13053–13060 CrossRef CAS.
- A. Bousetta, M. Lu, A. Bensaoula and A. Schultz, Appl. Phys. Lett., 1994, 65, 696–698 CrossRef CAS.
- Q. Fu, C. B. Cao and H. S. Zhu, Chem. Phys. Lett., 1999, 314, 223–226 CrossRef CAS.
- H.-X. Han and B. J. Feldman, Solid State Commun., 1988, 65, 921–923 CrossRef CAS.
- Z. A. Huang, Q. Sun, K. Lv, Z. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420–427 CrossRef CAS.
- R. J. Colton and J. W. Rabalais, Inorg. Chem., 1976, 15, 236–238 CrossRef CAS.
- M. Sun, N. Xu, Y. Cao, J. Yao and E. Wang, J. Mater. Res., 2000, 15, 927–933 CrossRef CAS.
- M. Feng, A. L. Pan, H. R. Zhang, Z. A. Li, F. Liu, H. W. Liu, D. X. Shi, B. S. Zou and H. J. Gao, Appl. Phys. Lett., 2005, 86, 141901–141903 CrossRef.
- Y. Liu, S. Xie, C. Liu, J. Li, X. Lu and Y. Tong, J. Power Sources, 2014, 269, 98–103 CrossRef CAS.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8525–8531 RSC.
- A. Vinu, K. Ariga, T. Mori, T. Nakanishi, S. Hishita, D. Golberg and Y. Bando, Adv. Mater., 2005, 17, 1648–1652 CrossRef CAS.
- H. Katsumata, T. Sakai, T. Suzuki and S. Kaneco, Ind. Eng. Chem. Res., 2014, 53, 8018–8025 CrossRef CAS.
- H. Zhang, J. Yang, D. Li, W. Guo, Q. Qin, L. Zhu and W. Zheng, Appl. Surf. Sci., 2014, 305, 274–280 CrossRef CAS.
- H. Song, Y. Li, Z. Lou, M. Xiao, L. Hu, Z. Ye and L. Zhu, Appl. Catal., B, 2015, 166–167, 112–120 CrossRef CAS.
- C. Bi, J. Cao, H. Lina, Y. Wang and S. Chen, Appl. Catal., B, 2016, 195, 132–140 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CAS.
- J.-H. Ha, P. Muralidharan and D. K. Kim, J. Alloys Compd., 2009, 475, 446–451 CrossRef CAS.
- H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432–22439 CrossRef CAS PubMed.
- K. Hayat, M. A. Gondal, M. M. Khaled, S. Ahmed and A. M. Shemsi, Appl. Catal., A, 2011, 393, 122–129 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02297b |
| This journal is © The Royal Society of Chemistry 2017 |