
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel [NF2O]+ and [N3NFO]+-based energetic oxidizers for solid propellants with super high specific impulse
Yi Yu a,
Jifeng Chena,
Rubo Zhangb,
Yuchuan Li*a and
Siping Pang*a
a,
Jifeng Chena,
Rubo Zhangb,
Yuchuan Li*a and
Siping Pang*a
aSchool of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, PR China. E-mail: liyuchuan@bit.edu.cn; pangsp@bit.edu.cn
bSchool of Chemistry, Beijing Institute of Technology, Beijing 100081, PR China
First published on 3rd May 2017
Abstract
Novel [NF2O]+ and [N3NFO]+-based energetic oxidizers were designed, and their structures, thermal stabilities, and energetic properties were investigated via density functional theory (DFT). The analysis of the bond dissociation energies (from 93.4 to 120.8 kcal mol−1) for the screened salts suggests that they possess better thermal stabilities than the reported [NF2O]+SbF6− (89.8 kcal mol−1), and compound 5 was the most stable energetic salt. All the screened salts possess a positive oxygen balance ranging from 13% to 50%. Due to a positive oxygen balance, the specific impulses of the compounds 5, 11–14 (>300 s) were superior to those of ammonium perchlorate (AP) and ammonium dinitramide (ADN) when the optimized ratio of oxidizer/aluminium/PBAN (%) was 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:14. Considering their thermal stability and chemical reactivity, compounds 5 and 11 with super high specific impulses can be regarded as excellent candidates for novel potential solid propellants.
:10:14. Considering their thermal stability and chemical reactivity, compounds 5 and 11 with super high specific impulses can be regarded as excellent candidates for novel potential solid propellants.
Introduction
Energetic oxidizers, such as ammonium perchlorate (AP) and ammonium dinitramide (ADN), are compounds that release excess of oxygen, which react with fuels and produce large amounts of hot gases for propulsion.1,2 With high densities and heats of formation, high specific impulse, and high thermal stabilities, energetic salts have been a research hotspot of explosive compounds, which are useful as propellants, explosives, and pyrotechnics.3–7 As two parts of energetic salts, the cations and anions take the role of fuels and oxidizers, respectively. The introduction of non-oxygen containing cations into the energetic salts will decrease the oxygen balance of the salts, restricting the promotion of the energetic performance of the energetic salts; thus, it is often sufficient to oxidize the carbon content only to CO and not to CO2 to achieve near maximum performance.8 However, oxygen-containing cations remain elusive because once nitrated or oxidized, the ability of neutral Lewis bases to form cationic species is greatly reduced.9,10 Therefore, novel oxidizers with oxygen-containing cations will be helpful to improve the oxygen balance and energetic performance of the energetic salts; for example, [NF2O]+ and [N3NFO]+, which have been reported decades ago, but the research on these cations moved slowly due to their strong oxidizability and reactivity.11,12The study history of [NF2O]+ originated from the chemistry of trifluoramine oxide (NF3O) in the 1960s,13–16 and it was found in 1969 that it exists in an ionic form in its Lewis acid adducts.17 In 1999, [NF2O]+ was used as an intermediate to synthesize the amazing N5+ cation.18 To date, only several [NF2O]+-based salts with paired anions such as SbF6−, Sb2F11−, AsF6−, BF4−, and B2F7− have been synthesized, among which [NF2O]+SbF6− was the most stable salt.19 As another fluoride oxide cation, [N3NFO]+ was reported in the form of [N3NFO]+SbF6− in 2007,20 which exists in the form of two different configurations: trans-[N3NFO]+ and cis-[N3NFO]+. Since then, no novel [N3NFO]+-based salts have been experimentally or theoretically reported. These two cations compose of nitrogen, oxygen, and fluorine in their structures. On the one hand, contrary to the commonly used fuel cations, oxygen in their composition will help to improve the oxygen balance. On the other hand, fluorine can also act as a strong oxidizer to further improve the oxygen balance of the energetic salts. Moreover, hydrogen is not present in the cations, which helps to improve the entire density of the energetic salts. These characteristics make them excellent candidates as strong energetic oxidizers to be used as potential propellants when combined with appropriate anions. Fig. 1 demonstrates the superior specific impulse performance of oxygen-rich [NF2O]+ and [N3NFO]+-based oxidizers in contrast to that of traditional fuel cation-based oxidizers, based on theoretical calculations.
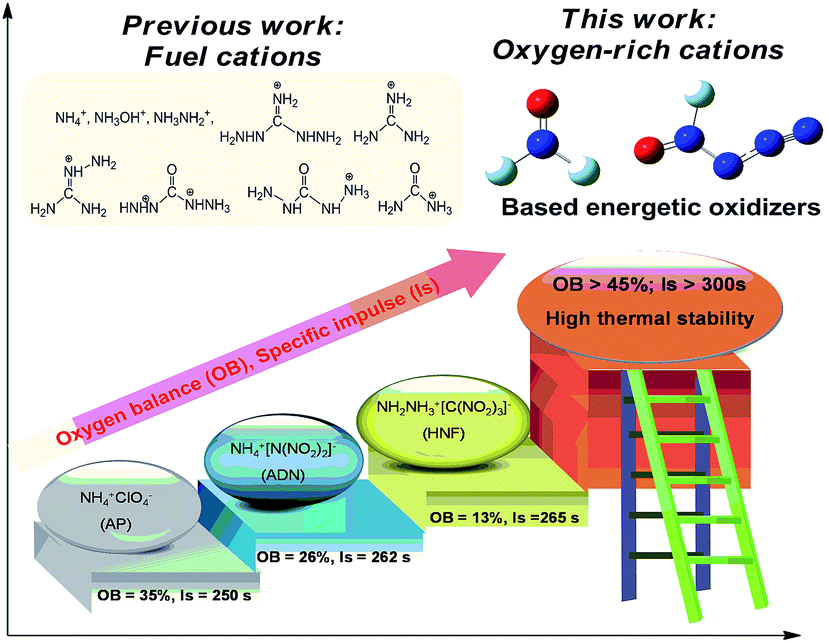 | ||
| Fig. 1 The superior performance of oxygen-rich [NF2O]+ and [N3NFO]+-based oxidizers in contrast to that of traditional fuel cation-based oxidizers. | ||
Our previous study indicated that the combination of AlF4− with a strong oxidizing N5+ cation may stabilize the salt,21 which prompted us to pair AlF4− with the two cations to evaluate their stabilities. Additionally, the possibility of synthesizing novel [NF2O]+ and [N3NFO]+ salts with good stability lead us to carry out further investigations. To evaluate the role of the central atom in SbF6− for the stability of [NF2O]+[SbF6]−, the Sb atom was substituted by Al to estimate its stability. Herein, a series of novel [NF2O]+ and [N3NFO]+ salts with paired anions (ClO4−, NO3−, N(NO2)2−, SO3CF3−, SO3NF2−, AlF4−, and AlF6−) were designed to study their structures, stabilities, and energetic performances. Via employing density functional theory, we screened out compounds with good stability and energetic performance, expanding the novel application of these cations.
Computational details
All the calculations were performed using the Gaussian 09 package. Geometry optimizations of the starting structures were carried out at the M06-2X level with the 6-311+G(d) basis set for the C, N, F, O, Cl, S, and Al atoms, and SDD for other heavy metal atoms.22 Note that the M06-2X functional is very suitable to multi-nitrogen or pure nitrogen molecules according to previous studies.23 Each optimized structure was confirmed as a local energy minima on the potential-energy surface without imaginary frequencies.Based on the Born–Haber energy cycle, as shown in Scheme 1, the heat of formation of a salt can be simplified by eqn (1), where ΔHL is the lattice energy of the ionic salts, which can be predicted by the formula suggested by Jenkins et al., as shown in eqn (2),24 where nM and nX depend on the nature of the ions Mp+ and Xq−, respectively, and are equal to 3 for monatomic ions, 5 for linear polyatomic ions, and 6 for nonlinear polyatomic ions. The equation for the lattice potential energy UPOT (kJ mol−1) has the form of (3), where ρ (g cm−3) is the density, M (g mol−1) is the chemical formula mass of the ionic compound, and the coefficients γ (kJ mol−1 cm) and δ (kJ mol−1) were obtained from the literature.24
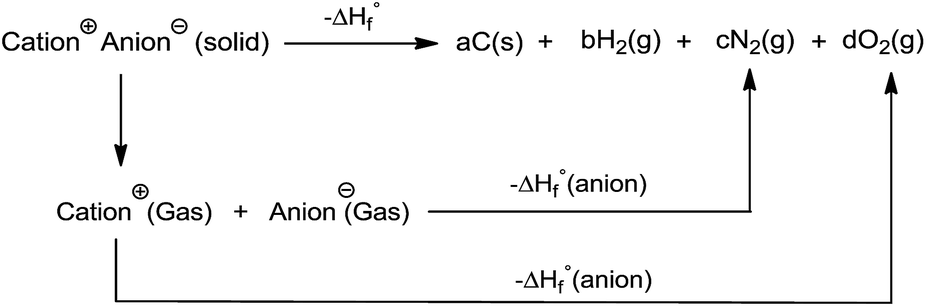 | ||
| Scheme 1 The Born–Haber energy cycle. | ||
For all the related compounds, the theoretical density was obtained from the molecular weight divided by the average molecular volume, which has been successfully applied to high-nitrogen compounds. For an ionic crystal with the formula unit MpXq, where M denotes the cation and X denotes the anion, its volume is simply the sum of the volumes of the ions contained in the formula unit. The volume of each ion is defined as inside a contour of 0.001 electrons bohr−3 density that was evaluated using a Monte Carlo integration. We performed 100 single-point calculations for the optimized structure of each ion to obtain an average volume. The detonation properties of these compounds were calculated using EXPLO 5 (v6.01).
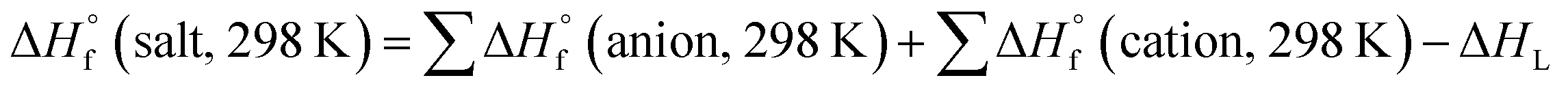 | (1) |
| ΔHL = UPOT + [p(nM/2 − 2) + q(nX/2 − 2)]RT | (2) |
| UPOT (kJ mol−1) = γ(ρm/Mm)1/3 + δ | (3) |
Results and discussion
Structures
Our previous study showed that the M06-2X method was reliable for the optimization of polynitrogen compounds,23 based on which we obtained the optimized structures of all the compounds, as depicted in Fig. 2. The structure of the separated [N3NFO]+ is planar, and it can exist in the form of two stereoisomers depending on whether the azido group and the fluorine ligand are cis (Z isomer) or trans (E isomer) with respect to each other. At our employed calculation level, the bond lengths of the N–F bonds in the two isomers significantly differ, with the Z isomer exhibiting a longer bond by 0.042 Å (reported 0.046 Å).8 The energy calculation also shows that the Z isomer is favoured by 0.78 kcal mol−1, which is slightly different from 0.6 kcal mol−1 reported in the literature.13 Thus, the stable Z isomer was selected for our further investigations.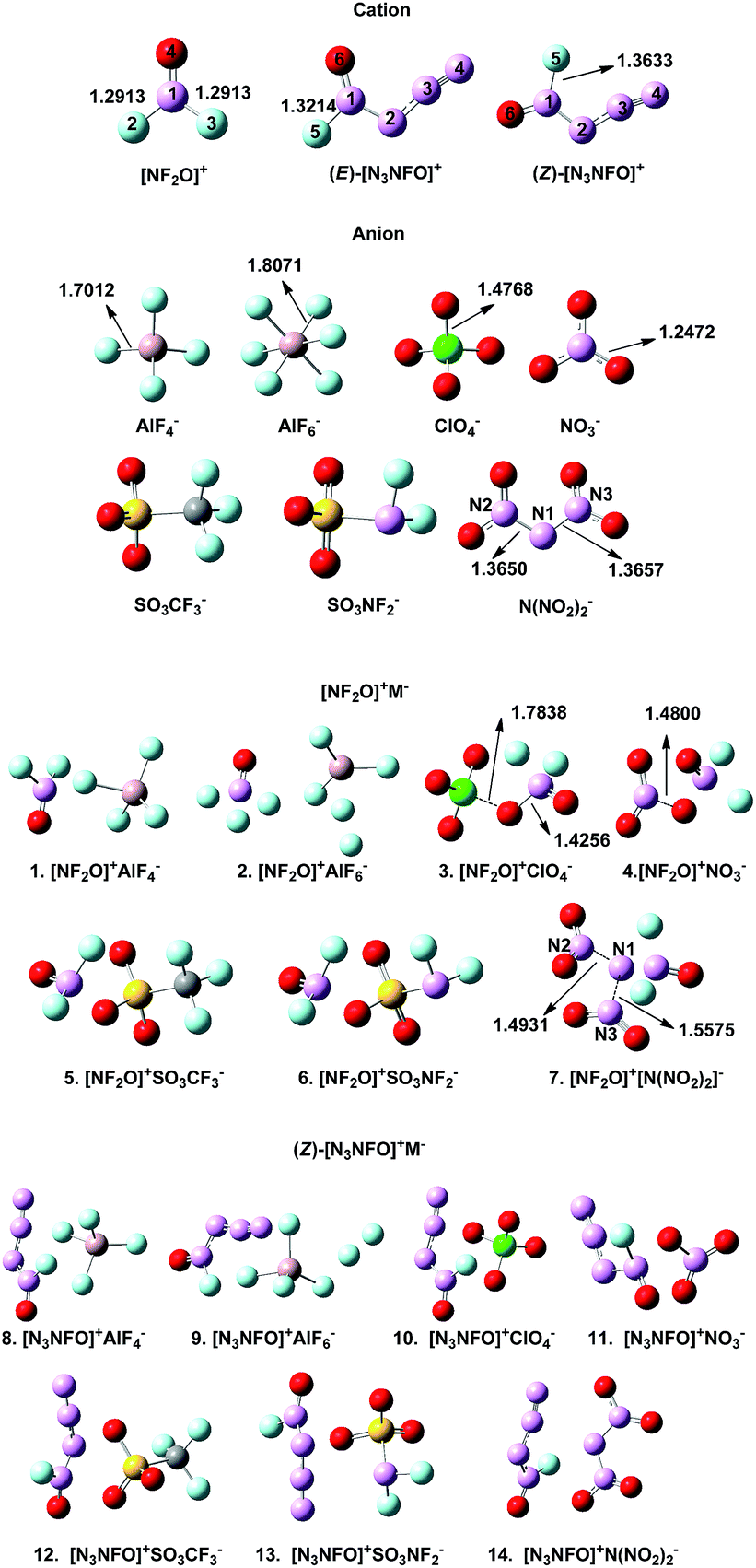 | ||
| Fig. 2 The optimized structures of the separated [NF2O]+, (Z)-[N3NFO]+, (E)-[N3NFO]+, and (Z)-[NF2O]+M− (M− = ClO4−, NO3−, N(NO2)2−, SO3CF3−, SO3NF2−, AlF4−, and AlF6−). | ||
There are two aspects worth noting in compound 2 (Fig. 2). One interesting case is that the three fluorine ions in AlF6− are obviously separated from the central Al ion. The partially dissociated Al–F bond suggests that the three F ions are not strongly bound to the central Al ion such that F ion tends to shift to [NF2O]+, promoting the decomposition of [NF2O]+. However, the shorter Al–F bond (1.7012 Å) in AlF4− indicates a stronger interaction between Al and F than that of AlF6− (1.8071 Å), which is relatively more difficult to be broken in the decomposition reaction. The bond strength of the central atom (Al) with the ligand (F−) in the counter-ion may be significant for the stability of [NF2O]+. A similar case exists in 9 where the two F ions are separated from the central Al ion. Another noteworthy case is the structural destruction of the anion after its combination with [NF2O]+, which may be caused by their intermolecular electrostatic interactions. For compounds 3, 4, and 7, when ClO4−, NO3−, and N(NO2)2− are combined with [NF2O]+, the stable structures of the anions are interrupted. The Cl–O bond noted in 3 is lengthened (1.7838 Å) when compared with the Cl–O bond (1.4768 Å) in separated ClO4−, resulting in Cl–O bond breakage and a newly formed N–O bond. A similar bond breakage occurs in 4 in which the N–O bond of NO3− is broken with much larger distance of N–O (1.4800 Å) than that of the separated NO3− (1.2472 Å). The structure of 7 obviously demonstrates its structural instability, where the distance of N1–N2 and N2–N3 in N(NO2)2− is 1.4931 Å and 1.5575 Å, respectively, completely decomposing to two separated NO2 molecules in the gas phase.
Based on the inspection of the optimized structures, we preliminarily screened out the possible stable compounds 1, 5, 6, 8, 10–14 to carry out further investigations.
Stabilities
The energies released upon the formation of salts by the isolated ions are reflected in the binding energy (ΔE) values, which can be expressed by the following equation.| ΔE = E0(cation) + E0(anion) − E0(salt) |
A larger binding energy means greater stability after the formation of the salts. For comparison, the calculated binding energy of [NF2O]+SbF6− and the screened compounds are listed in Table 1. Obviously, each compound has a larger binding energy ranging from 93.4 to 120.8 kcal mol−1 than that of [NF2O]+SbF6− (ΔE = 89.8 kcal mol−1), indicating they have relatively better stabilities than [NF2O]+SbF6−. For compounds 1 and 8 with the same paired anion AlF4−, 8 gives a larger ΔE, indicating that (Z)-[N3NFO]+ can better stabilize AlF4−-based salts when compared with [NF2O]+. In addition, our calculations show that for the same paired anions, (Z)-[N3NFO]+ and (E)-[N3NFO]+-based salts have close ΔE values with a difference of 2 kcal mol−1, which is due to the close free energies of separated (Z)-[N3NFO]+ and (E)-[N3NFO]+.25 The data in Table 1 shows compound 5 has the largest binding energy (ΔE = 120.8 kcal mol−1), indicating that it is the most stable salt, whereas 1 is the least stable (ΔE = 93.4 kcal mol−1). To sum up, the thermal stability order is 5 > 6 > 11 > 12 > 8 ≈ 14 > 10 > 13 ≈ 1 > [NF2O]+SbF6−, based on which it can be inferred that the screened compounds have great possibilities to be synthesized.
| Compd. | ΔE [kcal mol−1] | ΔELUMO–HOMO [eV] |
|---|---|---|
| [NF2O]+SbF6− | 89.8 | 7.85 |
| 1 | 93.4 | 8.17 |
| 5 | 120.8 | 11.02 |
| 6 | 114.1 | 9.92 |
| 8 | 96.4 | 7.49 |
| 10 | 95.6 | 5.12 |
| 11 | 99.0 | 8.72 |
| 12 | 97.6 | 5.14 |
| 13 | 93.8 | 5.11 |
| 14 | 96.3 | 4.77 |
The energy gap (ΔELUMO–HOMO) of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is essential for kinetic stability and chemical reactivity during the chemical processes with electron transfer or leap.26,27 The HOMOs and LUMOs of the selected salts are depicted in Fig. 3. For the series of (E)-[N3NFO]+ salts, the distributions of orbitals and ΔELUMO–HOMO values are similar to that of the Z isomers, which are not shown. An inspection of Fig. 3 demonstrates that the LUMO orbitals mainly locate on the part of the cations, and the HOMO orbitals mainly locate on the ions. It's well known that a higher energy gap implies lower chemical reactivity and a lower gap implies higher chemical reactivity. Therefore, the ΔELUMO–HOMO values in Table 1 indicate that compounds 1, 5, 6, and 11 have lower chemical reactivity than [NF2O]+SbF6−, with ΔELUMO–HOMO values ranging from 8.17 eV to 11.02 eV, and the compound with the lowest chemical reactivity should be 5 (11.02 eV). In summary, the screened salts have a chemical reactivity order of 5 < 6 < 11 < 1 < [NF2O]+SbF6− < 8 < 12 ≈ 13 ≈ 10 < 14.
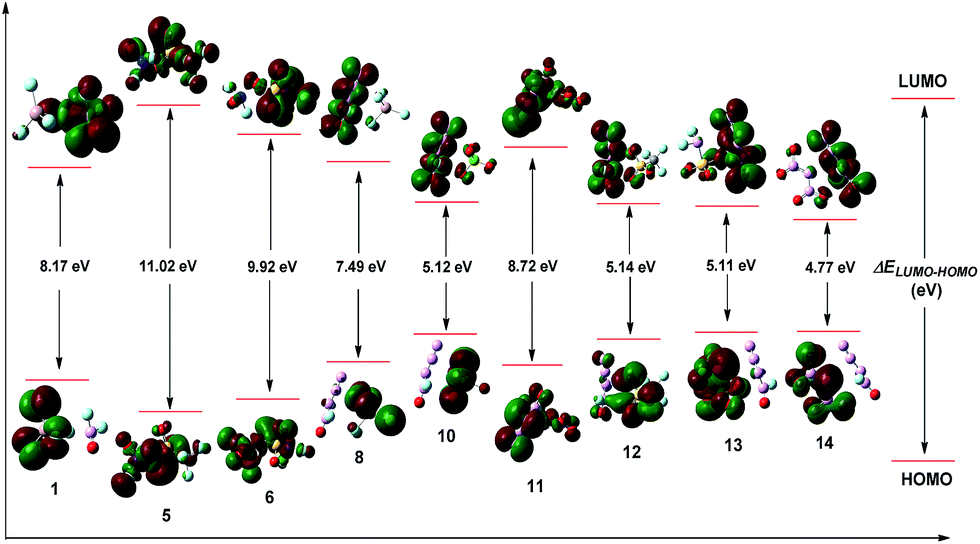 | ||
| Fig. 3 The depicted HOMOs, LUMOs, and energy gaps (ΔELUMO–HOMO) of the selected compounds. | ||
Energetic properties
The heat of formation (HOF) is an important parameter to obtain the detonation properties of a compound, and the accurate heats of formation of the cations and anions of the screened compounds were calculated at the G2 level of theory. The lattice energy (ΔHL) was mainly determined by the volume and type of the salt. For all the salts, the ratio of the number of cations to anions was' 1:1 and the calculated ΔHL values in Table 2 range from 507 to 569 kJ mol−1. Due to the low HOF of AlF4−, compounds 1 and 8 with AlF4− as the anion show lower HOFs when compared with other compounds, whose values are −1517 and −1160 kJ mol−1, respectively, among which 14 has the highest positive heat of formation up to 637 kJ mol−1. Moreover, all the related salts exhibit certain detonation properties such as detonation velocities (D) and detonation pressures (P), among which 14, composed of N(NO2)2− and [N3NFO]+, shows a comparable energetic performance with AP and ADN, better than that of other compounds with NO3−, ClO4−, AlF4−, SO3CF3−, and SO3NF2− as paired anions.
| Compd. | OBa [%] | ρb [g cm−3] | ΔHLc [kJ mol−1] | ΔHfd [kJ mol−1] | VDe [km s−1] | Pf [GPa] | Isg [s] | Ish [s] | Isi [s] | Isj [s] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Oxygen balance.b Density.c Lattice energy.d Calculated enthalpy of formation (kJ mol−1).e Detonation velocity.f Detonation pressure.g Specific impulse(s) of neat compounds.h Specific impulse(s) of mixtures with aluminium and/or PBAN binder as 76:10:14. Binder composition 6% polybutadiene acrylic acid, 6% polybutadiene acrylonitrile and 2% bisphenol A ether.i Original NASA composition (70:16:14) for the space shuttle solid rocket boosters.j Specific impulse(s) of mixtures with aluminium and/or PBAN binder as 80:20:0. |
||||||||||
| 1 | 23 | 2.18 | 569 | −1517 | 2.69 | 2.3 | 138 | 216 | 227 | 239 |
| 5 | 18 | 1.82 | 507 | −947 | 4.51 | 12.0 | — | 324 | 263 | 244 |
| 6 | 32 | 1.76 | 532 | −344 | 2.72 | 2.1 | — | 279 | 224 | 257 |
| 8 | 17 | 2.03 | 539 | −1160 | 5.14 | 11.1 | 187 | 220 | 217 | 205 |
| 10 | 50 | 1.97 | 538 | 389 | 6.61 | 19.0 | 236 | 297 | 289 | 271 |
| 11 | 47 | 1.80 | 557 | 413 | 7.03 | 20.0 | 264 | 309 | 296 | 284 |
| 12 | 13 | 2.06 | 512 | −624 | 5.34 | 13.8 | 191 | 464 | 124 | 247 |
| 13 | 25 | 2.01 | 519 | −3 | 6.17 | 15.4 | 206 | 361 | 80 | 263 |
| 14 | 45 | 1.85 | 524 | 637 | 7.56 | 23.9 | 260 | 309 | 294 | 286 |
| ADN28 | 26 | 1.81 | — | −150 | 7.86 | 23.6 | 202 | 264 | 269 | 202 |
| AP29 | 26 | 1.95 | — | −296 | 6.37 | 15.8 | 157 | 256 | 261 | 244 |
The oxygen balance (OB) indicates the degree to which an explosive can be oxidized. As an important index for identifying the potential of oxidants, compounds with positive OBs indicate that there is enough oxygen in their composition to convert all the carbon and hydrogen atoms to carbon monoxide and water, respectively, whereas negative OBs indicate an insufficient oxygen content for complete oxidation. All the screened salts possess positive OBs ranging from 13% to 50%. Compounds 1 and 13 have positive OBs of 23% and 25%, respectively, which are comparable to those of AP and ADN. Particularly, the OBs of 10 (50%), 11 (47%), and 14 (45%) are much higher than those of AP and ADN (26%).
The value of excellent oxidizers can be reflected in the application as propellants. As an important parameter, the specific impulse (Is) is used to determine the performance of solid propellants or binders. The Is values of the relevant compounds were calculated under isobaric conditions at 7 MPa with an initial temperature of 3300 K using EXPLO5 (v6.01). Compounds 10, 11, and 14 show impulse values of 236 s, 264 s, and 260 s, which are much higher than those of AP (Is = 157 s) and ADN (Is = 202 s) as neat compounds. The impulse calculations for neat compounds 5 and 6 were not performed due to the problem of temperature limits during the calculations. The addition of aluminium and PBAN binder as a fuel drastically increases the Is for all the compounds. We carried out Is calculations with different combustion parameters: oxidizer/aluminium/PBAN [%], and the calculations indicate that the optimized ratio of the mixtures with aluminium and PBAN binder was composed of 76% oxidizer, 10% aluminium, and 14% binder. Surprisingly the calculations show compound 12 achieves the highest Is value at 464 s, far exceeding that of any other already known solid propellant. This unexpected effect may be caused by the introduction of carbon in 12 when compared with the other salts. Moreover, compounds 5, 11, 13, and 14 are quite promising oxidizers, with superior Is values (>300 s) relative to those of AP (Is = 256 s) and ADN (Is = 264 s) with similar formulations. Our calculations show that the Is values of all the screened oxidizers were superior to those of AP and ADN, except for 1 and 8. Considering the relative thermal stability and chemical reactivity discussed above, the calculation results suggest that compounds 5 and 11 may be used as a new generation of superenergetic oxidizers with potential applications as solid rocket propellants.
Conclusions
A series of [NF2O]+ and [N3NFO]+-based energetic oxidizers were designed employing density functional theory. The calculated binding energies indicate that compounds 1, 5, 6, 8, 10–14 (ΔE > 93 kcal mol−1) have better thermal stabilities than previously reported NF2OSbF6 (ΔE = 89.8 kcal mol−1). The energy gap (ΔELUMO–HOMO) analysis predicted that 1, 5, 6, and 11 exhibit a lower chemical reactivity than NF2OSbF6, in which 5 has the lowest chemical reactivity. Amazingly with good thermal stabilities, compounds 5, 11–14 show superior Is values (>300 s) to those of ammonium perchlorate (AP) and ammonium dinitramide (ADN) with an optimized ratio of oxidizer/aluminium/PBAN (%) of 76:10:14. While considering the kinetic stability, compounds 5 and 11 may be used as potential solid propellants. Moreover, our exploration using experiments is currently underway. We hope our work may supply new insight into the expansion of novel energetic oxidizers and candidates for future solid propellants.
Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the financial support received from the National Natural Science Foundation of China (21576026 and U1530262).Notes and references
- X. X. Zhao, S. H. Li, Y. Wang, Y. C. Li, F. Q. Zhao and S. P. Pang, J. Mater. Chem. A, 2016, 4, 5495–5504 CAS.
- Q. J. Axthammer, B. Krumm and T. M. Klapötke, J. Org. Chem., 2015, 80, 6329–6335 CrossRef CAS PubMed.
- R. P. Singh, R. D. Verma, D. T. Meshri and J. n. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584–3601 CrossRef CAS PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 CrossRef CAS PubMed.
- C. Darwich, T. M. Klapötke and C. M. Sabaté, Chem.–Eur. J., 2008, 14, 5756–5771 CrossRef CAS PubMed.
- R. Wang, H. Xu, Y. Guo, R. Sa and J. n. M. Shreeve, J. Am. Chem. Soc., 2010, 132, 11904–11905 CrossRef CAS PubMed.
- T. Fendt, N. Fischer, T. M. Klapötke and J. Stierstorfer, Inorg. Chem., 2011, 50, 1447–1458 CrossRef CAS PubMed.
- C. B. Jones, R. Haiges, T. Schroer and K. O. Christe, Angew. Chem., Int. Ed., 2006, 45, 4981–4984 CrossRef CAS PubMed.
- W. Liu, Q.-H. Lin, Y.-Z. Yang, X.-J. Zhang, Y.-C. Li, Z.-H. Lin and S.-P. Pang, Chem.–Asian J., 2014, 9, 479–486 CrossRef CAS PubMed.
- G. W. Drake, S. Bolden, J. Dailey, M. J. McQuaid and D. Parrish, Propellants, Explos., Pyrotech., 2012, 37, 40–51 CrossRef CAS.
- C. A. Wamser, W. B. Fox, B. Sukornick, J. R. Holmes, B. B. Stewart, R. Juurik, N. Vanderkooi and D. Gould, Inorg. Chem., 1969, 8, 1249–1253 CrossRef CAS.
- K. O. Christe and D. A. Dixon, J. Am. Chem. Soc., 1992, 114, 2978–2985 CrossRef CAS.
- V. Plato, W. D. Hartford and K. Hedberg, J. Chem. Phys., 1970, 53, 3488–3494 CrossRef CAS.
- W. B. Fox, J. S. MacKenzie and R. Vitek, Inorg. Nucl. Chem. Lett., 1970, 6, 177–179 CrossRef CAS.
- W. H. Kirchhoff and D. R. Lide Jr, J. Chem. Phys., 1969, 51, 467–468 CrossRef CAS.
- R. R. Smardzewski, J. Chem. Phys., 1974, 60, 2193 CrossRef CAS.
- S. Abramowitz and I. W. Levin, J. Chem. Phys., 1969, 51, 463–464 CrossRef CAS.
- K. O. Christe, W. W. Wilson, J. A. Sheehy and J. A. Boatz, Angew. Chem., Int. Ed., 1999, 38, 2004–2009 CrossRef CAS.
- K. O. Christe and W. Maya, Inorg. Chem., 1969, 8, 1253–1257 CrossRef CAS.
- W. W. Wilson, R. Haiges, J. A. Boatz and K. O. Christe, Angew. Chem., Int. Ed., 2007, 46, 3023–3027 CrossRef CAS PubMed.
- Y. Yu, Y.-c. Li, J.-f. Chen, C.-h. Sun, J.-s. Li, G.-j. Fan, S.-p. Pang and R.-b. Zhang, RSC Adv., 2015, 5, 104841–104845 RSC.
- M. T. Nguyen and T.-K. Ha, Chem. Phys. Lett., 2000, 317, 135–141 CrossRef CAS.
- C. Qi, R.-B. Zhang and S.-P. Pang, RSC Adv., 2013, 3, 17741–17748 RSC.
- H. D. B. Jenkins, D. Tudela and L. Glasser, Inorg. Chem., 2002, 41, 2364–2367 CrossRef CAS PubMed.
- T. Yu, Y.-Z. Liu, R. Haiges, K. O. Christe, W.-P. Lai and B. Wu, RSC Adv., 2014, 4, 28377–28389 RSC.
- Z. Zhou and R. G. Parr, J. Am. Chem. Soc., 1989, 111, 7371–7379 CrossRef CAS.
- Y.-F. Li, X.-W. Fan, Z.-Y. Wang and X.-H. Ju, J. Mol. Struct.: THEOCHEM, 2009, 896, 96–102 CrossRef CAS.
- M. r. Y. Nagamachi, J. I. S. Oliveira, A. M. Kawamoto and R. d. C. s. L. Dutra, J. Aerosp. Technol. Manage., 2009, 1, 153–160 CrossRef CAS.
- E. Gökcinar and T. M. Klapötke, Turk. J. Chem., 2010, 34, 953–967 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |