
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amorphous porphyrin glasses exhibit near-infrared excimer luminescence†
Mitsuhiko Morisue *a,
Ikuya Uenoa,
Takayuki Nakanishib,
Takafumi Matsuib,
Sono Sasakic,
Masaki Shimizua,
Jun Matsuid and
Yasuchika Hasegawab
*a,
Ikuya Uenoa,
Takayuki Nakanishib,
Takafumi Matsuib,
Sono Sasakic,
Masaki Shimizua,
Jun Matsuid and
Yasuchika Hasegawab
aFaculty of Molecular Chemistry and Engineering, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan. E-mail: morisue@kit.ac.jp; Fax: +81-75-724-7806
bGraduate School of Engineering, Hokkaido University, North 13 West 8, Kita-ku, Sapporo 060-8628, Japan
cFaculty of Fiber Science and Engineering, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan
dDepartment of Material and Biological Chemistry, Faculty of Science, Yamagata University, Kojirakawa-cho, Yamagata, 990-8560, Japan
First published on 25th April 2017
Abstract
The amorphous nature of a series of zinc–porphyrins bearing two 3,4,5-tri((S)-3,7-dimethyloctyloxy)phenyl groups at the meso-positions, named “porphyrin glass”, were tolerant of π-conjugation engineering in ethynylene-linked dimers. The butadiyne-linked dimeric porphyrin glass formed an intermolecular excimer, which exhibited bright and exceptionally long-lived, near-infrared (NIR) luminescence at approximately 970 nm in the solid state. Therefore, porphyrin glasses overcame a general bottleneck for NIR-luminescence, such as an undesired π-stacked aggregation of a large porphyrin plane in addition to the energy gap law. The formation of amorphous molecular glasses from a series of meso-ethynylene-conjugated zinc–porphyrins, named “porphyrin glass”, is described. The butadiyne-linked dimeric porphyrin glass formed an intermolecular excimer, which exhibited solid-state, near-infrared (NIR) luminescence at approximately 970 nm.
Introduction
Metalloporphyrins have been one of the key photofunctional compounds at the forefront of materials science.1 Near-infrared (NIR) luminescent materials have versatile state-of-the-art applications, in fields such as in vivo imaging through the “biological optical window” (800–1350 nm),2 light-emitting diodes,3 and broadband optical amplifiers.4 However, the wavelength of NIR-luminescence of an organic chromophore barely exceeds 900 nm, with only a few exceptions, such as benzothiadiazole derivatives2d,e,3 and polymethine dyes,5 as well as porphyrins.6 An intrinsic obstacle for NIR luminescence comes from the “energy gap law”. As an optical band-gap narrows, excitons easily dissipate through thermal perturbation from vibrational oscillations.7 The meso-ethynylene-conjugated porphyrin oligomers exhibit outstanding NIR light-harvesting capacity,8,9 based not only on intense linear absorption but also on exceptional two-photon absorption cross-section values.10 The excellent photoelectronic properties of the porphyrins stem from extended meso-ethynylene-conjugation, but encounter undesired π-stacked aggregation. Such a drawback prevents the ethynylene-conjugated porphyrins from being used in NIR-luminescence applications under matrix-free conditions.11 Amorphous glassy molecules have introduced effective and challenging morphological strategies for attaining photoelectronic functionality that is relevant to molecular formulae.12,13 Hence, meso-ethynylene-conjugated porphyrin glasses may be potential NIR-luminescent materials.The present target is based on our serendipitous discovery that a zinc porphyrin bearing two 3,4,5-tri((S)-3,7-dimethyloctyloxy)phenyl groups at the meso-positions, such as 1, adopts the form of a solvent-free viscous fluid at room temperature (Fig. 1).14 Metallocomplexes, including metalloporphyrins, are usually crystalline and barely form amorphous solids with a few exceptions.15,16 A deliberate alkylation indicates a potential strategy to provide amorphous molecular glasses. For instance, the introduction of alkyl substituents is crucial for the morphology of π-conjugated polymers17 and nonvolatile fluids.18,19 Porphyrin fluid 1 prompted us to explore porphyrin glasses with meso-ethynylene-conjugations.
 | ||
| Fig. 1 The chemical structures of porphyrin 1 (A) and a photograph of neat 1 as a sticky fluid on a glass slide at room temperature (B). | ||
Results and discussion
Monomeric porphyrins 1 and 2 showed glass transition temperatures (Tg) at −6 and 19 °C, respectively, in differential scanning calorimetric (DSC) analyses (Scheme 1, Fig. 2), indicating the formation of glass. The results were in sharp contrast with porphyrins incorporating 3,4,5-tri(n-alkyloxy)phenyl groups at the meso-positions, which crystallize without a glass transition from both liquid and liquid crystalline (LC) states.19,20 Encouraged by these results, we synthesized 3 and 4 to expand the π-conjugation by connecting two glass-forming porphyrin subunits and aimed to achieve NIR-active porphyrin glasses. | ||
| Scheme 1 Chemical structures of porphyrin 2 and porphyrin dimers 3 and 4. | ||
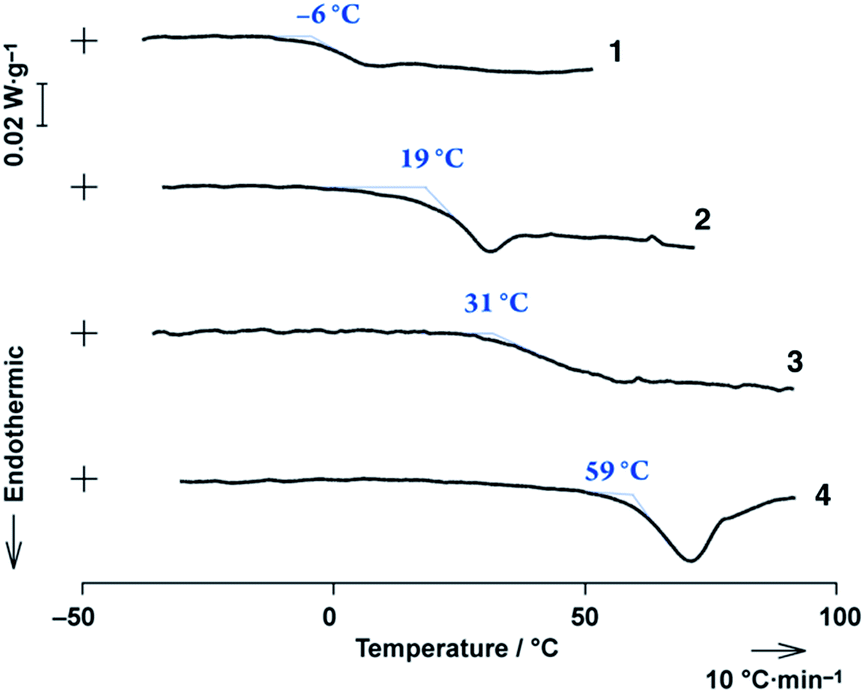 | ||
| Fig. 2 DSC profiles for 1–4 with a 10 °C min−1 heating rate. | ||
The DSC thermograms of 3 and 4 showed endothermic profiles at 31 °C and 59 °C, respectively, attributed to their glass transitions (Fig. 2), while the others involved enthalpic relaxation. The Tg point shifted to higher temperatures with extended π-conjugations. Porphyrins 1–4 provided neither crystals nor thermotropic LC-phases, even though 3,4,5-tri((S)-3,7-dimethyloctyloxy)phenyl groups are known as powerful LC-forming units,21,22 indicating that the 3,4,5-tri((S)-3,7-dimethyloctyloxy)phenyl group is an exquisite partner of porphyrin that encumbers the π-stacked interaction. Further investigations of 2–4 were examined in a uniform thin film prepared by a spin-cast method, as shown below.
Synchrotron microbeam glazing-incidence X-ray scattering (GIXS) found characteristic amorphous halo patterns at approximately a q (=2π/d, wherein d refers to spacing) of 13.5 nm−1 for 2–4 in spin-cast films on a silicon wafer. However, no periodic spacings were found other than primary intramolecular spacing (Fig. 3). Therefore, the blanched alkyl-chains did indeed govern the intermolecular interactions and disordered porphyrin arrangements in solvent-free bulk solid.
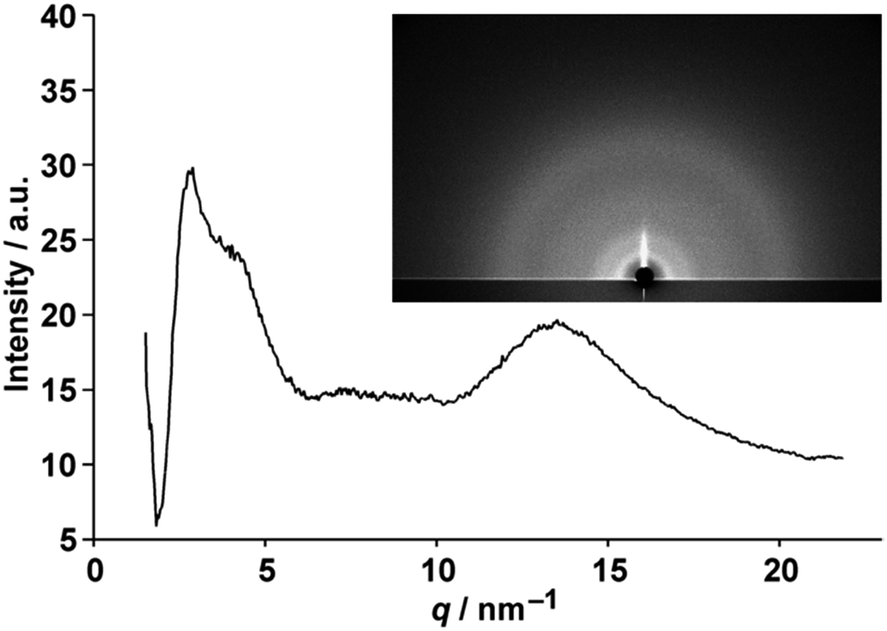 | ||
| Fig. 3 The GIXS pattern (inset) and one-dimensional profile of 4 on a silicon wafer observed with a synchrotron X-ray with 0.11° of the incident angle obtained from integrated radial azimuthal angles. | ||
Amorphous molecular glass is expected to be persistent to aggregation-caused fluorescence quenching. Remarkably, the porphyrin glass 4 exhibited broad NIR-luminescence at approximately 970 nm that extended to over 1100 nm (Fig. 4), and the monomeric fluorescence was weakened or extinguished instead. Since none of the electronic absorption bands of 4 exceeded 750 nm, the NIR-luminescence was assigned to the intermolecular excited dimer, i.e., the excimer comprised an excited-state intermolecular chromophore associated with a ground-state counterpart, as elucidated below.
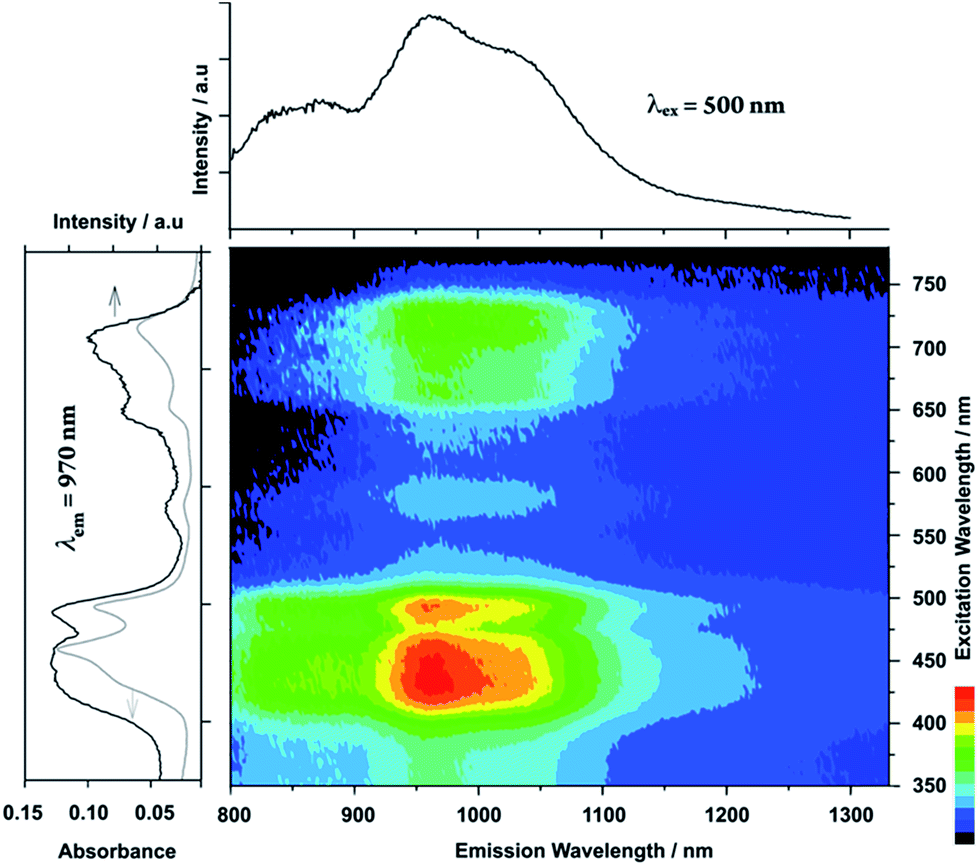 | ||
| Fig. 4 Emission–excitation correlations of 4 in a spin-cast film on a quartz substrate under air together with an emission spectrum (excitation wavelength = 500 nm) in the upper panel, and excitation (emission wavelength = 970 nm, black line) and absorption (grey line) spectra in the left panel. | ||
Intermolecular excimer formation was evidenced by the dispersion experiments in an amorphous polymer matrix, where 1 and 10 wt% of 4 were doped into an inert poly(methyl methacrylate) (PMMA) film. In the electronic absorption results, the spectral shape of 4 showed no substantial changes regardless of the fraction of 4 used in the PMMA films (1–100 wt%) (Fig. 5 and S2–S4†), suggesting the occurrence of marginal intermolecular interactions at the ground state in the neat spin-cast film.23 However, 4-doped PMMA films showed monomeric emission at 744 nm, and their NIR-luminescence dramatically weakened. The comparison clearly indicates that an intermolecular excimer formation is indispensable for the NIR-luminescence of porphyrin glass.
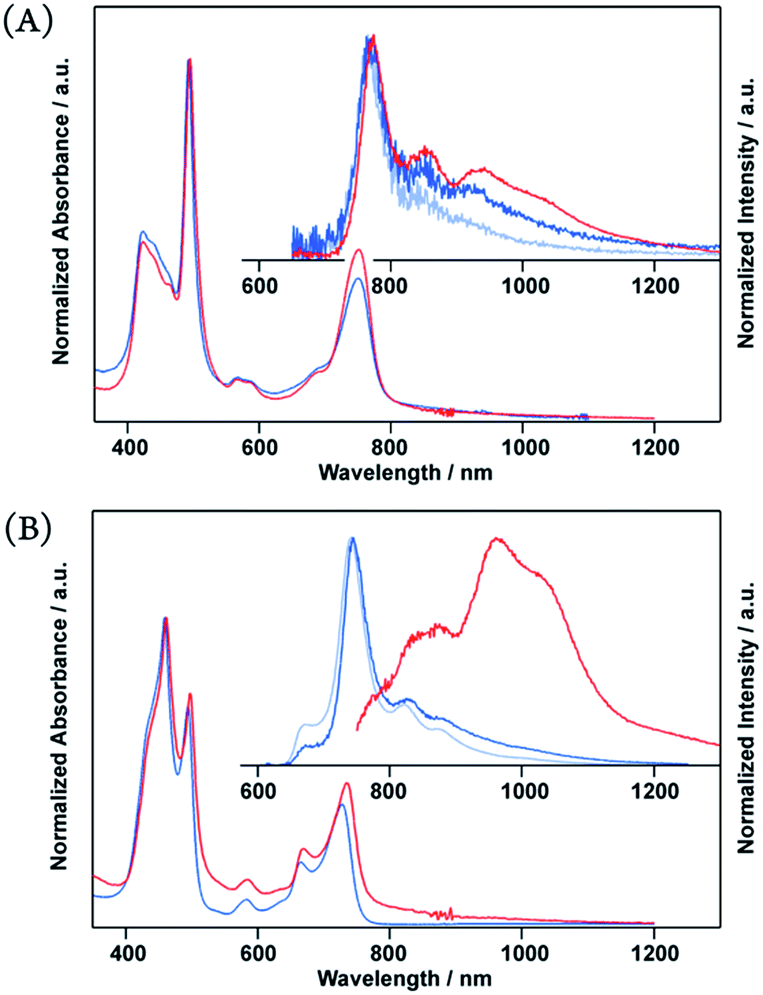 | ||
| Fig. 5 Normalized absorption and emission spectra of neat porphyrin (red) and porphyrin-doped PMMA (10 wt%, blue, and 1 wt% pale blue) (A: 3, B: 4) in spin-cast film on a quartz substrate. Emission obtained by excitation at 500 nm. | ||
Excimer formation was predominantly observed only in 4 among the present porphyrin glasses. For example, 2 and 3 mainly showed monomeric fluorescence, which was accompanied by a weak, broad emission at longer wavelengths. It was deduced that an appropriate internal porphyrin–porphyrin separation of 4 opens a space acceptable for intermolecular excimer formation.
It is remarkable that excimers in the porphyrin glasses surpass general obstacles due to the energy gap law.7 Moreover, large π-systems are prone to undesirable π-stacked aggregations. Thus, NIR-luminescent chromophore aggregates have rarely been reported,24 and porphyrins have never been the exception. Unlike structureless emissions from excimers in the solution, the solid-state NIR excimer luminescence bears vibronic features (Fig. 6). The vibronic spacing of approximately 1100 cm−1 for 3 and 1600 cm−1 for 4 were reminiscent of the resonance Raman vibrational modes of the porphyrin skeleton and ethynylene-linkage (Fig. 6),25 which suggests the presence of exciton–phonon coupling, such as Herzberg–Teller-type dynamic intensity borrowing.9,26 We propose that the exciton self-traps to resonantly facilitate radiative decay at the excimer site. Therefore, luminescence remains even at NIR wavelengths.
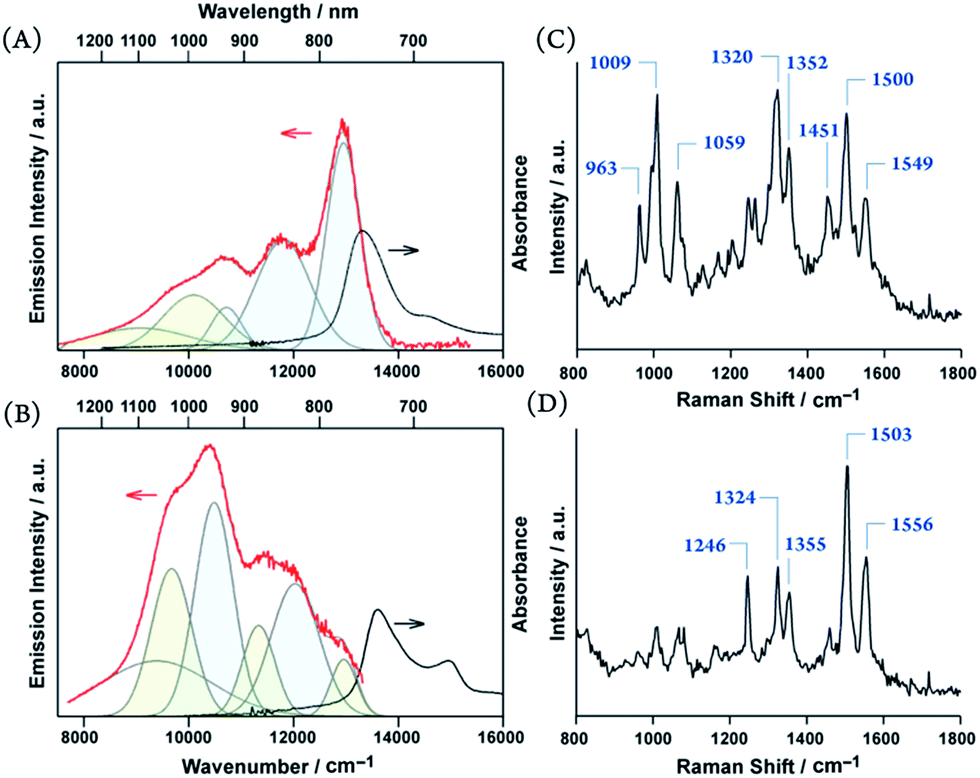 | ||
| Fig. 6 Emission spectra (red) and electronic absorption (black) (A and B), resonance Raman spectra excited at 532 nm (C and D) of 3 (A and C) and 4 (B and D). Deconvoluted emission peaks with periodic spacings of approximately 1100 cm−1 (A) and 1600 cm−1 (B) are colored in pale blue and green. | ||
All the experiments were performed under aerobic conditions, indicating that the NIR-luminescence was relatively persistent to oxygen. The emission lifetimes of the excimer components were considerably elongated (τ > 500 ns) from the monomeric fluorescence (τ ≈ 100 ns) (Table 1), in contrast to the very short lifetimes reported for the porphyrin films.27 It was difficult to assign such unusually long-lived luminescence to a singlet exciton, although the photophysical process is the subject under active investigation. The molar extinction coefficient (ε) of 4 displays an exceptional order of 105 M−1 cm−1 both at the Soret and Q bands. The brightness of the emission is defined as the product of the ε value and the absolute quantum yield (Φ).28 Although the luminescence efficiency (Φ = 0.11% as a tentative magnitude) was still comparable to those of the NIR-luminescent polymethine dyes,5 the excellent ε magnitude of porphyrin glass 4 ensured sufficient brightness of the NIR-luminescence.
| 770 nm | 970 nm | Φ/% | |||
|---|---|---|---|---|---|
| τ1(α1)/ns | τ2(α2)/ns | τ1(α1)/ns | τ2(α2)/ns | ||
| a Emission lifetime (τ) and normalized pre-exponential factor (α); excitation at 450 nm for 2 and at 500 nm for 3 and 4 (10 wt% 4-doped PMMA was employed to observe a monomeric component at 770 nm). Tentative emission quantum yield (Φ) was measured for opaque neat drop-cast films by excitation at 450 nm. | |||||
| 2 | 240(0.66) | 1311(0.34) | 76(0.59) | 606(0.41) | 0.056 |
| 3 | 231(0.66) | 1263(0.34) | 132(0.66) | 953(0.34) | 0.039 |
| 4 | 128(0.54) | 756(0.46) | 103(0.57) | 711(0.43) | 0.114 |
Conclusion
In conclusion, we have developed π-conjugated porphyrin glasses. The amorphous nature of the porphyrin glass was highly tolerant of the meso-ethynylene π-conjugation engineering. The excimer luminescence of 4 displayed a remarkable Stokes shift toward the NIR-wavelength region presumably through the aid of exciton–phonon coupling. Ethynylene-conjugated porphyrin glasses have highlighted a new fascinating approach towards developing solid-state NIR-luminescent materials. For instance, their amorphous nature was tolerant of further supramolecular π-electron engineering by employing our supramolecular approach14 toward NIR-luminescence beyond 1000 nm.29 A further study of NIR-luminescent porphyrin glasses and detailed mechanism of the solid-state NIR-luminescence are currently under active investigation.Acknowledgements
We are grateful to Prof. H. L. Anderson (University of Oxford) for fruitful discussions, Mr Mitsuru Ohta (Shimadzu Co., Kyoto) for DSC measurements, Ms Kiminori Etou, Dr Yoshihiro Osawa (Otsuka Electronics Co., Shiga) for quantum yield measurements of the NIR luminescence, and Kyoto Integrated Science & Technology Bio-Analysis Center (KIST-BIC) for Raman spectroscopic analysis. The synchrotron radiation experiments were performed at BL45XU in SPring-8 with the approval of RIKEN (Proposal No. 20160041). This study was partly supported by a Grant-in-Aid for Scientific Research on Innovative Areas “New Polymeric Materials Based on Element-Blocks (No. 2401)” (M. M., JP15H00741; J. M., JP15H00720; H. Y., No. 24102012) and KAKENHI (M. M., JP16K05749) from JSPS.Notes and references
- (a) A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed; (b) T. Higashino and H. Imahori, Dalton Trans., 2015, 44, 448–463 RSC; (c) J. P. Hill, Angew. Chem., Int. Ed., 2016, 55, 2976–2978 CrossRef CAS PubMed , and references cited therein.
- (a) R. Weissleder, Nat. Biotechnol., 2001, 19, 318–317 CrossRef PubMed; (b) A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef CAS PubMed; (c) G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed; (d) A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang, X. Zhang, O. K. Yaghi, Z. R. Alamparambli, X. Hong, Z. Cheng and H. Dai, Nat. Mater., 2016, 15, 235–242 CrossRef CAS PubMed; (e) Q. Yang, Z. Ma, H. Wang, B. Zhou, S. Zhu, Y. Zhong, J. Wang, H. Wan, A. Antaris, R. Ma, S. Zhang, J. Yang, X. Zhang, H. Sun, W. Liu, Y. Liang and H. Dai, Adv. Mater., 2017, 1605497 CrossRef PubMed; (f) G. Hong, A. L. Antaris and H. Dai, Nature Biomedical Engineering, 2017, 1, 0010 CrossRef.
- (a) G. Qian, B. Dai, M. Luo, D. Yu, J. Zhan, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2008, 20, 6208–6216 CrossRef CAS; (b) G. Qian, J. P. Gao and Z. Y. Wang, Chem. Commun., 2012, 48, 6426–6428 RSC.
- (a) S. Zhou, N. Jiang, K. Miura, S. Tanabe, M. Shimizu, M. Sakakura, Y. Shimotsuma, M. Nishi, J. Qiu and K. Hirao, J. Am. Chem. Soc., 2010, 132, 17945–17952 CrossRef CAS PubMed; (b) S. Zhou, Q. Guo, H. Inoue, Q. Ye, A. Masuno, B. Zheng, Y. Yu and J. Qiu, Adv. Mater., 2014, 26, 7966–7972 CrossRef CAS PubMed; (c) Y. Yu, Z. Fang, C. Ma, H. Inoue, G. Yang, S. Zheng, D. Chen, Z. Yang, A. Masuno, J. Orava, S. Zhou and J. Qiu, NPG Asia Mater., 2016, 8, e318 CrossRef CAS.
- (a) M. Casalboni, F. De Matteis, P. Prosposito, A. Quatela and F. Sarcinelli, Chem. Phys. Lett., 2003, 373, 372–378 CrossRef CAS; (b) S. Hatami, C. Würth, M. Kaiser, S. Leubner, S. Gabriel, L. Bahrig, V. Lesnyak, J. Pauli, N. Gaponik, A. Ecychmüller and U. Resch-Genger, Nanoscale, 2015, 7, 133–143 RSC.
- (a) T. V. Duncan, K. Susumu, L. E. Sinks and M. J. Therien, J. Am. Chem. Soc., 2006, 128, 9000–9001 CrossRef CAS PubMed; (b) M. Pawlicki, M. Morisue, N. K. S. Davis, D. G. McLean, J. H. Haley, E. Beuerman, M. Drobizhev, A. Rebane, A. L. Thompson, S. I. Pascu, G. Accorsi, N. Armaroli and H. L. Anderson, Chem. Sci., 2012, 3, 1541–1547 RSC.
- (a) W. Siebrand and D. F. Williams, J. Chem. Phys., 1968, 49, 1860–1871 CrossRef CAS; (b) R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- (a) V. S.-Y. Lin, S. G. DiMagno and M. J. Therien, Science, 1994, 264, 1105–1111 CAS; (b) V. S.-Y. Lin and M. J. Therien, Chem.–Eur. J., 1995, 1, 645–651 CrossRef CAS; (c) R. Kumble, S. Palese, V. S.-Y. Lin, S. G. DiMagno, M. J. Therien and R. M. Hochstrasser, J. Am. Chem. Soc., 1998, 120, 11489–11498 CrossRef CAS.
- (a) H. L. Anderson, Chem. Commun., 1999, 2323–2330 RSC; (b) M. U. Winters, J. Kärnbratt, M. Eng, C. J. Wilson, H. L. Anderson and B. Albinsson, J. Phys. Chem. C, 2007, 111, 7192–7199 CrossRef CAS; (c) M.-H. Chang, M. Hoffman, H. L. Anderson and L. M. Herz, J. Am. Chem. Soc., 2008, 130, 10171–10178 CrossRef CAS PubMed.
- (a) M. Pawlicki, H. A. Collins, R. G. Dennig and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed; (b) M. Drobizhev, Y. Stepanenko, Y. Dzenis, A. Karotki, A. Rebane, P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 2004, 126, 15352–15353 CrossRef CAS PubMed; (c) H. A. Collins, M. Khurana, E. H. Moriyama, A. Mariampillai, E. Dahlstedt, M. Balaz, M. K. Kuimova, M. Drobizhev, V. X. D. Yang, D. Phillips, A. Rebane, B. C. Wilson and H. L. Anderson, Nat. Photonics, 2008, 2, 420–424 CrossRef CAS.
- (a) J. C. Ostrowski, K. Susumu, M. R. Robinson, M. J. Therien and G. C. Bazan, Adv. Mater., 2003, 15, 1296–1300 CrossRef CAS; (b) O. Fenwick, J. K. Sprafke, J. Binas, D. V. Kondratuk, F. Di Stasio, H. L. Anderson and F. Cacialli, Nano Lett., 2011, 11, 2451–2456 CrossRef CAS PubMed.
- (a) Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC; (b) Y. Shirota, J. Mater. Chem., 2005, 15, 75–93 RSC.
- (a) A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed; (b) M. Grucela-Zajac, K. Bijak, S. Kula, M. Filapek, M. Wiacek, H. Janeczek, L. Skorka, J. Gasiorowski, K. Higerl, N. S. Sariciftci, N. Nosidlak, G. Lewinska, J. Sanetra and E. Schab-Balcerzak, J. Phys. Chem. C, 2014, 118, 13070–13086 CrossRef CAS PubMed.
- (a) M. Morisue, Y. Hoshino, K. Shimizu, M. Shimizu and Y. Kuroda, Chem. Sci., 2015, 6, 6199–6206 RSC; (b) M. Morisue, Y. Hoshino, M. Shimizu, S. Uemura and S. Sakurai, Chem.–Eur. J., 2016, 22, 13019–13022 CrossRef CAS PubMed.
- (a) M. R. Robinson, M. B. O'Regan and G. C. Bazan, Chem. Commun., 2000, 1645–1655 RSC; (b) Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito, H. Fueno, K. Tanaka, T. Satoh and Y. Hasegawa, Inorg. Chem., 2015, 54, 4364–4370 CrossRef CAS PubMed.
- (a) M. Ottmar, T. Ishisaka, L. R. Subramanian, M. Hanack and Y. Shirota, Chem. Lett., 2001, 788–789 CrossRef CAS; (b) A. Meunier and O. Lebel, Org. Lett., 2010, 12, 1896–1899 CrossRef CAS PubMed.
- T. Marszalek, M. Li and W. Pisula, Chem. Commun., 2015, 52, 10938 RSC.
- (a) S. S. Babu, M. J. Hollamby, J. Aimi, H. Ozawa, A. Saeki, S. Seki, K. Kobayashi, K. Hagiwara, M. Yoshizawa, H. Möhwald and T. Nakanishi, Nat. Commun., 2013, 4, 1969 Search PubMed; (b) H. Li, J. Choi and T. Nakanishi, Langmuir, 2013, 29, 5394–5406 CrossRef CAS PubMed.
- (a) A. Nowak-Król, D. Gryko and D. T. Gryko, Chem.–Asian J., 2010, 5, 904–909 CrossRef PubMed; (b) S. Maruyama, K. Sato and H. Iwahashi, Chem. Lett., 2010, 39, 714–716 CrossRef CAS.
- (a) X. Zhou, S.-W. Kang, S. Kumar, R. R. Kulkarni, S. Z. D. Cheng and Q. Li, Chem. Mater., 2008, 20, 3551–3553 CrossRef CAS; (b) T. Sakurai, K. Shi, H. Sato, K. Tashiro, A. Osuka, A. Saeki, S. Seki, S. Tagawa, S. Sasaki, H. Masunaga, K. Osaka, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 13812–13813 CrossRef CAS PubMed.
- (a) K. Kishikawa, S. Furusawa, T. Tamaki, S. Kohmoto, M. Yamamoto and K. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 1597–1605 CrossRef CAS PubMed; (b) S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem.–Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed; (c) F. Helmich, M. M. J. Smulders, C. C. Lee, P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 12238–12246 CrossRef CAS PubMed.
- (a) A. P. H. J. Schenning, M. Fransen and E. W. Meijer, Macromol. Rapid Commun., 2002, 23, 265–270 CrossRef CAS; (b) V. Percec, E. Aquad, M. Peterca, J. G. Rudick, L. Lemon, J. C. Ronda, B. B. De, P. A. Heiney and E. W. Meijer, J. Am. Chem. Soc., 2006, 128, 16365–163672 CrossRef CAS PubMed.
- The spectral shape was reminiscent of the planar conformer similarly to the recent report: H. Doan, S. L. Raut, D. Yale, M. Balaz, S. V. Dzyuba and Z. Gryczynski, Chem. Commun., 2016, 52, 9510–9513 RSC.
- (a) K. Cai, J. Xie and D. Zhao, J. Am. Chem. Soc., 2013, 136, 28–31 CrossRef PubMed; (b) M. Tanioka, S. Kamino, A. Muranaka, Y. Ooyama, H. Ota, Y. Shirasaki, J. Horigome, M. Ueda, M. Uchiyama, D. Sawada and S. Enomoto, J. Am. Chem. Soc., 2015, 137, 6436–6439 CrossRef CAS PubMed.
- D. Beljonne, G. E. O'Keefe, P. J. Hamer, R. H. Friend, H. L. Anderson and J. L. Brédas, J. Chem. Phys., 1997, 106, 9439–9460 CrossRef CAS.
- (a) M. H. Perrin, M. Gouterman and C. L. Perrin, J. Chem. Phys., 1969, 50, 4137–4150 CrossRef CAS; (b) H. Kano, T. Saito and T. Kobayashi, J. Phys. Chem. A, 2002, 106, 3445–3453 CrossRef CAS.
- (a) A. Huijser, B. M. J. M. Suijkerbuijk, R. J. M. K. Gebbink, T. J. Savenije and L. D. A. Siebbeles, J. Am. Chem. Soc., 2008, 130, 2485–2492 CrossRef CAS PubMed; (b) X. Wang, G. Brisard, D. Fortin, P.-L. Karsenti and P. D. Harvey, Macromolecules, 2015, 48, 7024–7038 CrossRef CAS.
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232–1242 CrossRef CAS PubMed.
- M. Morisue, Y. Hoshino, M. Shimizu, T. Nakanishi, Y. Hasegawa, M. A. Hossain, S. Sakurai, S. Sasaki, S. Uemura and J. Matsui, Macromolecules DOI:10.1021/acs.macromol.7b00316.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, GIXS profiles, and emission decay profiles. See DOI: 10.1039/c7ra02752d |
| This journal is © The Royal Society of Chemistry 2017 |