
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The phosphorescence and excitation-wavelength dependent fluorescence kinetics of large-scale graphene oxide nanosheets†
Litao Zhaoa,
Xiantong Yua,
Sanjun Zhanga,
Xiaoxiao Hea,
Lei Lia,
Menghui Jiaab,
Mengfang Changa,
Haifeng Pana,
Jinquan Chen a,
Wenjun Wang*c and
Jianhua Xu*a
a,
Wenjun Wang*c and
Jianhua Xu*a
aState Key Laboratory of Precision Spectroscopy, East China Normal University, Shanghai, 200062, China. E-mail: jhxu@phy.ecnu.edu.cn
bShanghai Institute of Optics and Fine Mechanics, Chinese Academy of Science, 201800, Shanghai, China
cShandong Key Laboratory of Optical Communication Science and Technology, School of Physical Science & Information Technology of Liaocheng University, 252059, Shandong Province, China. E-mail: phywwang@163.com
First published on 25th April 2017
Abstract
In this study, phosphorescence emission and a strong excitation-wavelength dependent fluorescence has been found in large-scale graphene oxide (GO) nanosheets. GO was covalently functionalized with triphenylamine (GO-CONH-TPA) in order to enhance the GO fluorescence quantum yield to 18%. The intersystem crossing (ISC) dynamics were studied using a femtosecond transient absorption technique, which appeared in the same timescale as the fluorescence dynamics of GO-CONH-TPA in both a polar solvent and solid film. Therefore, both, the solvation (several hundreds of picoseconds) and the intersystem crossing (ISC) gave rise to the strong excitation-wavelength dependent fluorescence. Moreover, the phosphorescence emission of the GO-CONH-TPA film at room temperature has been described for the first time in this report, and the lifetime of phosphorescence was found to be 6.95 μs. The fluorescence kinetics of GO-CONH-TPA were attributed to the aromatic hydrocarbon-carboxylic domain structure of GO.
1. Introduction
Since two-dimensional (2D) graphene possesses a unique linear band structure, it has been reported for various applications during the past decade, such as optoelectronic devices,1–3 optical imaging, therapy4–6 and drug delivery.4 Graphene oxide (GO) has an opening band with a band gap range of around several electron volts (eV) compared with graphene (zero eV);7 however, its weak photoluminescence (PL) was unresolved, and thus its fluorescence mechanisms require more clarification. Conventional viewpoints state that GO comprises a quasi-2D network of small sp2 carbon domains in a sp3-bonded matrix,8 which can be regarded as graphene with oxygen functional groups decorating the basal plane and edges. An extreme perspective is that a GO nanosheet is a quasi-2D oxidized derivative of graphene, which contains isolated polyaromatic clusters dispersed in the sp3 C–O matrix.To date, only a few groups have reported the PL spectroscopy of GO, which is found in the following wavelength ranges: the blue-centered (<450 nm),7,9–12 green-centered (450–490 nm)4–6,10,13–17 and red-centered (≥590 nm).9,11,13,16–20 This is in accordance with Wu's theory, where the fingerprint photoluminescence of the functional groups in GO was certified. Three PL peaks originate from the σ*–n, π–π* and π*–n electronic transitions between the antibonding and bonding molecular orbitals. In other words, these peaks are associated with the C–OH, aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO groups in GO. As is known to all, GO's PL has signatures including a large full width at half maximum (FWHM), pH sensitive emission, extreme low quantum yield and indeterminate emission peaks. These signatures limit the applications of GO in the fields of luminescent materials, optoelectronics, biolabeling, and bioimaging, etc.
C and CO groups in GO. As is known to all, GO's PL has signatures including a large full width at half maximum (FWHM), pH sensitive emission, extreme low quantum yield and indeterminate emission peaks. These signatures limit the applications of GO in the fields of luminescent materials, optoelectronics, biolabeling, and bioimaging, etc.
There are several literature studies reporting the phenomena of phosphorescence from carbon dots and graphene quantum dots.21,22 In this study, we demonstrate the existence of phosphorescence from a large-scale graphene oxide sheet at room temperature for the first time. Time-resolved fluorescence spectroscopy and ultra-fast femtosecond transient absorption were used to reveal the intersystem crossing (ISC) dynamics as well as the mechanism of the ISC induced excitation-wavelength dependent fluorescence.
In this study, GO covalently functionalized with triphenylamine (GO-CONH-TPA) was synthesized and the excitation-wavelength dependent fluorescence found both in solvent and a solid matrix. The ISC between the singlet and triplet states of the aromatic carbonyl group is a reasonable interpretation for the ‘red-edge’ effect. The transient absorption (TA) measurements showed that the ISC lifetime was about 2 ns and the phosphorescent emission peak was located around 500 nm with an average lifetime of 6.95 μs.
2. Experimental
GO nanosheets were synthesized from natural graphite using the modified Hummer's method.23 Details of the syntheses of graphene oxide and GO-CONH-TPA are reported in the ESI.† Fig. S1 gives the typical TEM images of GO and the GO-CONH-TPA nanosheets in the ESI file.† The size of the GO and GO-CONH-TPA nanosheets were around several micrometers.The fluorescence lifetime was measured using a homebuilt time-correlated single photon counting (TCSPC) system (4 ps per channel and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 channels) with electronics (PicoHarp 300 from Picoquant, Germany) and a super-continuum pulsed fiber laser (SC400-pp-4 from Fianium, UK). The pulse duration of our fiber laser was about 190 ps. An acousto-optic tunable filter (AOTF) and an OD4 short-pass filter were used to select the excitation wavelength. A monochromater and an OD4 long-pass filter were employed to choose the detection wavelength.
500 channels) with electronics (PicoHarp 300 from Picoquant, Germany) and a super-continuum pulsed fiber laser (SC400-pp-4 from Fianium, UK). The pulse duration of our fiber laser was about 190 ps. An acousto-optic tunable filter (AOTF) and an OD4 short-pass filter were used to select the excitation wavelength. A monochromater and an OD4 long-pass filter were employed to choose the detection wavelength.
The phosphorescence lifetime was measured using a homebuilt system; a mode-locked Ti:sapphire laser (Tsunami, New-Port) generated a 400 mW seed pulse train with a typical pulse duration of 50 fs at a repetition rate of 80 MHz. The femtosecond laser pulses were used to seed a Ti:sapphire generative amplifier (Spitfire Pro, New-Port) to generate a 2 W pulse train centered at 800 nm with a pulse width of 50 fs and a repetition rate of 1 kHz. Furthermore, 60% of the fundamental pulse energy was then used to pump a parametric amplifier (Topas-C, New-Port) to generate the excitation pulses at 400 nm. An attenuator was inserted in the light path before the samples. A monochromater (Zolix, Omni-λ 500) was used to select the detection wavelength, an avalanche photodiode (APD) was used to collect the weak emission signal and a gate switch was used to control the APD; the gate switch was synchronous triggered using the excitation laser pulse with the APD starting to work after a 50 ns delay. An oscilloscope (LeCroy, Wave surfer 44MXS-B) was used to distinguish and integrate the output signal from the APD.
A homebuilt femtosecond transient absorption set-up was used to obtain the transient absorption kinetics. A Ti:Sapphire laser system (Spitfire Pro, center wavelength at 800 nm, pulse duration of 35 fs, pulse energy of 1.6 mJ and 1 kHz repetition rate) was used to generate both the pump and the probe light beams. A small portion (0.45 mW) of the 800 nm beam was used to generate a stable white-light continuum (WLC) probe beam by focusing onto a sapphire plate. The 400 nm wavelength pump beam was generated from the second harmonic of the fundamental output and was chopped at 500 Hz using a synchronized chopper (MC2000, Thorlabs). The kinetic traces were then obtained using a home-built set-up equipped with a PMT detector (CR317, Hamamatsu) for higher sensitivity. The probe wavelength in the kinetic measurements was selected using a set of band-pass filters (10 nm). The signals from the PMT were sent into a lock-in amplifier (Model SR830, Stanford Research Systems) and recorded using data collection software written by LabWindows. The instrument response function (IRF) of this system was determined to be ∼150 fs by measuring the solvent responses under the same experimental conditions.
3. Results and discussion
Fig. 1(a) shows the synthesis process used to prepare the GO-CONH-TPA. The surface covalent attachment of triphenylamine (TPA) was supported by the FT-IR data as shown in Fig. 1(b). The FT-IR data show a new strong stretching-vibration band around 1700 cm−1 due to the C–N stretching in the –CONH group. In addition, strong C–H stretching peaks were observed at both 2960 cm−1 and 2881 cm−1. The carboxylic group bands at both 1730 cm−1 and 1233 cm−1 from the original GO disappear after the chemical treatment step. These results clearly suggest that the TPA was covalently attached on the GO surface via the formation of an amide bond. The epoxide band at 1055 cm−1 completely disappears in GO-CONH-TPA. However, the bands at 1700 cm−1 and 1270 cm−1 were assigned to the C–N–H asymmetric stretching of the attached triphenylamine. Triphenylamine does not only successfully remove the epoxy groups but is also covalently attached on the surface of GO via a ring-opening amination of the epoxides. The band at 3250 cm−1 was assigned to the N–H vibration of the acylamino groups. As shown in Fig. 1(c), the absorption peak of GO (around 230 nm) is assigned to the π–π* transitions of CC. After the treatment with triphenylamine, the center of the absorption band shifts to 270 nm. The peak can be regarded as an overlap of the 230 nm peak of GO and the 300 nm absorption peak of triphenylamine.10,24 After the reaction with triphenylamine, these groups, such as carboxyl and epoxy groups, are removed. The above results indicate the formation of new luminescent centers on the surface of the GO-CONH-TPA nanosheets, resulting in approximately nine hundred fold enhancement in the fluorescence compared to that of the original GO nanosheets (0.02%).10
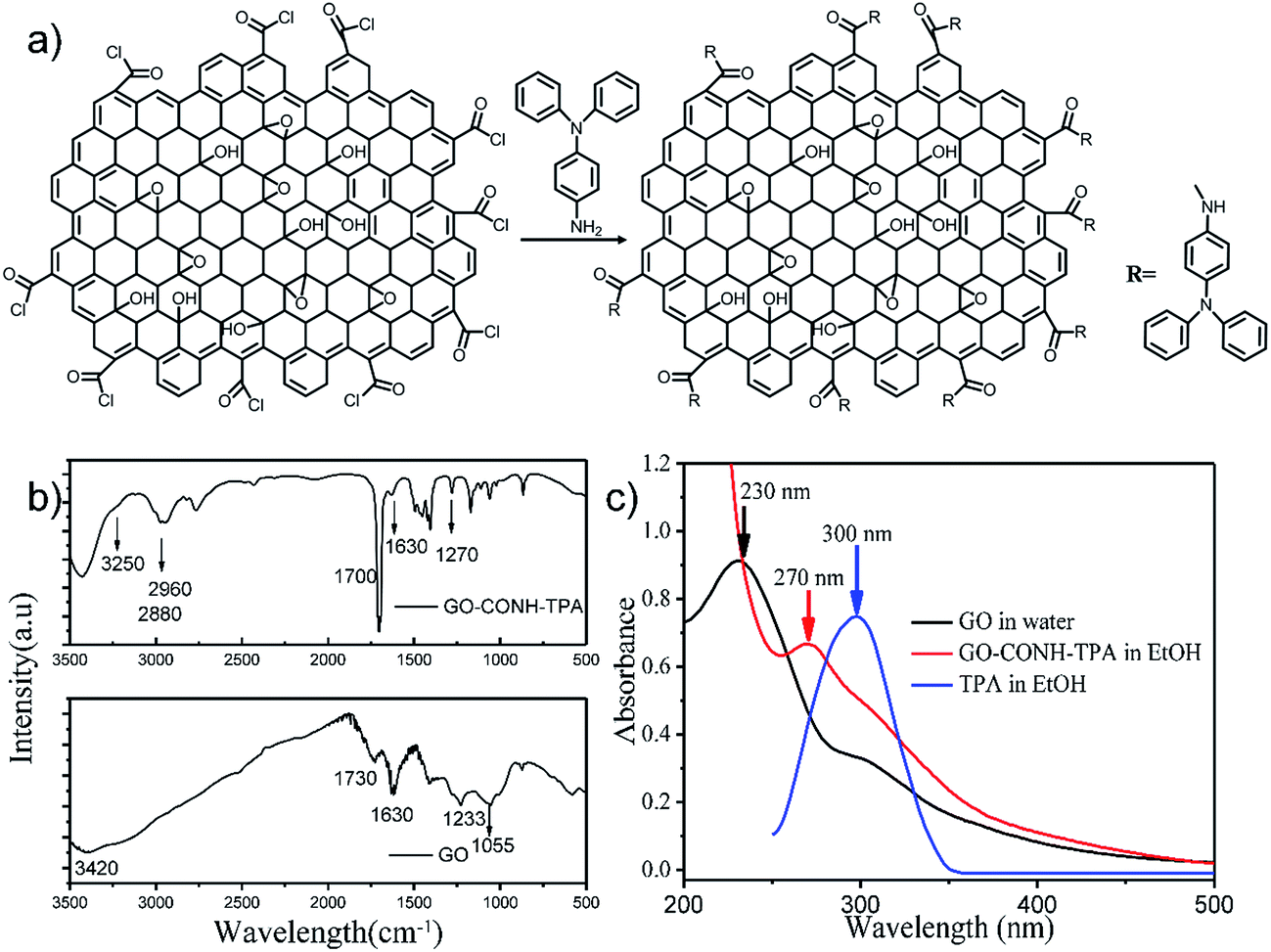 | ||
| Fig. 1 The synthetic process used to prepare GO-CONH-TPA and its basic characterization. (a) The reaction scheme of the preparation of the GO-CONH-TPA material. (b) The FT-IR spectra of GO-CONH-TPA and graphene oxide (GO). (c) The UV-vis absorption spectra of GO in water, GO-CONH-TPA and TPA in ethyl alcohol, respectively. | ||
The quantum yield (QY) of GO-CONH-TPA was measured to be 18% using a reference method,25 which was much higher than that of GO. The nucleophilic reaction between the amido-groups and epoxy/carboxylic groups can distinctly break off the intramolecular charge-transfer (CT) process, making the red-centered fluorescence disappear. At the same time, the removal of non-radiative recombination sites increase the quantum yield from the aromatic hydrocarbons-carboxylic domain in GO.7,10,19 The reaction removes the groups that often induce non-radiative recombination of localized e–h pairs, such as epoxide groups and carboxylic groups. Interestingly, the emission peaks of GO-CONH-TPA in ethyl alcohol are dependent on the excitation-wavelength as shown in Fig. 2(a). There are three primary reasons to explain the phenomenon of the excitation-wavelength dependent fluorescence from GO. The first explanation is that the excitation-wavelength dependent fluorescence originates from the sp2 domains and these sp2 domains open the heterogeneous electronic band gaps, which are intrinsically correlated to the sp2 domains sizes, shapes, and fractions.7,10 With an increase in the excitation wavelength (red-shift), only the large size sp2 domains with a small energy gap can be excited, which makes the PL peak red-shift. The second explanation is that the wavelength dependent fluorescence of GO is a result of the interaction between the graphene basal plane and different functional groups (i.e. –OH, –COOH, and epoxide) as well as the edge states.26 The third explanation is that the excitation-wavelength dependent fluorescence comes from the ‘giant red-edge effect’.17 In other words, the solvation dynamics are slowed down to the same time-scale as that of fluorescence in a polar solvent due to the local environment of the GO-like materials.17 In this study, both, the samples in solution (Fig. 2(a)) and as a solid film (Fig. 2(b)) show the phenomenon of a strong excitation wavelength dependent fluorescence from GO-CONH-TPA.
 | ||
| Fig. 2 The photoluminescence spectra of GO-CONH-TPA in either ethyl alcohol (a) or PMMA (b) at different excitation wavelengths. | ||
The ‘red-edge effect’ induced by intersystem crossing (ISC)
Previous reports have described the ‘red-edge effect’ induced by solvation dynamics.17 Wu's study proved that in water, the solvation dynamics with the same time-scale as the fluorescence τfluo ≅ τsolv can induce the ‘red-edge effect’ of GO in an aqueous solution. Consequently, the fluorescence will broaden and red-shift with an increase in the excitation wavelength. The steady-state fluorescence spectrum is unchanged for the red-shift at a certain excitation ω, which means the integral of the time-resolved emission spectrum have the same spectral form for all excitation ω. However, when the solvation dynamics were taken into account, the steady-state fluorescence intensity was reduced and shifted to a lower energy by Δω. The ratio of Δλexcitation/Δλemission was constant and a linear slope existed for the ‘giant red-edge effect’ in GO when ωexc < ω(∞).17In this study, we would like to take the ISC into account for the ‘red-edge effect’ of GO-CONH-TPA both in solution and a solid matrix. The excitation-wavelength dependent fluorescence was related to the weak and slow ISC process during the fluorescence emission. As the ISC and fluorescence lifetimes have the same order of magnitude, the fluorophore emits when the excited state's energy is reduced. This results in a time-dependent emission energy.25 This phenomenon is also referred to as the ‘red-edge effect’. Therefore, time-resolved fluorescence (TRF) measurements were used to obtain the origin of the ‘red-edge effect’ in the graphene oxide samples. A mathematical model was applied to describe the ISC relaxation process using the time-dependent emission energy, which can be written as25
| U′em(t) = Uexc(0) − Uisc(t) − Usolve(t) | (1) |
In our experimental data, there is a linear slope for the ‘giant red-edge effect’ in GO-CONH-TPA when ωexc < ω(∞). The change in the time-dependent emission energy due to ISC was independent of the excitation-wavelength. As long as the energy relaxation is not completed during the fluorescence emission, the same relaxation process could lead to the different excitation-wavelength with the same spectral emission form only offset by the initial difference in the excitation wavelength. The excited states, which associated with the different excitation-wavelengths, undergo the same relaxation process and the fluorescence emissions with the same spectral form. At the same time, the ratio between the change in the peak emission wavelength and that of the excitation wavelength Δλexcitation/Δλemission should be constant. The Δλexcitation/Δλemission of GO-CONH-TPA in solution and PMMA film matrix are 0.75 and 0.8, respectively (as shown in Fig. S2 in the ESI†). This value implies the existence of ISC relaxation in both a solution and the solid system. Moreover, the tiny difference (0.75 and 0.8) can be attributed to other types of energy relaxation channels occurring in solution, such as the slow solvent relaxation caused by the local environment of the GO nanosheets.17
The TCSPC count baselines elevate abnormally as shown in Fig. 3(a) with an increase in the detection wavelength. A linear relationship between dark-noise and the detection time was found in our exclusion test (Fig. S3 in the ESI†). Under normal circumstances, the TCSPC count baseline elevates slightly with the red-shift in the detection wavelength because of the dark-noise count (Fig. S3†). The dark-noise count signal is recorded randomly as the phosphorescence component photons. The dark-noise count directly leads to the rise of the baseline in the TRF curves. Fig. S3 in the ESI† file proves that the relationship between the dark-noise count and TCSPC acquisition time was linear. The function of the relationship was clear, the acquisition time was recorded and the dark-noise could be easily calculated. In this study, after the dark-noise count is ruled out, the region under the fluorescence-count baseline can be regarded as the photoluminescence-count for a particular emission wavelength; the ratio of the fluorescence and phosphorescence counts denotes their intensity ratio in our TCSPC experiments; the acquisition time was not fixed and was decided by the time a certain count number was reached. Therefore, different count times were observed at different emission wavelengths. If there is a much longer decay time component, such as the phosphorescence component, and its lifetime is much longer than the TCSPC detection time window (50 ns), the TRF curves baseline increase in this situation. The counts for the fluorescence and photoluminescence were both integrated in our experiments. Then, the counts are divided by the acquisition time, and their ratio should be the same as the ratio of the two amplitudes in the steady-state PL curve. For a certain detection wavelength, if the counts for the photons (from the fluorescence and phosphorescence components) per unit time are integrated, the ratio of the counts can represent the intensity ratio of these components. In addition, we changed the detection wavelength every 10 nm from 460 to 560 nm. After the intensity ratios of these components with the different detection wavelengths are clear, the steady-state PL of GO-CONH-TPA in the PMMA film (excited at 400 nm) can be traced out. Therefore, the rough curves obtained for the fluorescence and phosphorescence were traced out based on the PL curve obtained for GO-CONH-TPA in PMMA film, as shown in Fig. 3(b). Fig. 3(c) shows the phosphorescence spectra of GO-CONH-TPA in a PMMA film matrix and the phosphorescence peak is around 485 nm. Fig. 3(d) shows the photoluminescence decay curve recorded by a homebuilt experimental setup. The curve can be fitted using a multi-exponential function with two lifetimes of 1.2 μs (75%) and 9.2 μs (25%), and thus the average lifetime is found to be 6.95 μs. This value was close to that of the triplet states and electronic relaxation in photoexcited graphene quantum dots (4 μs).21 The results are in accordance with the literature,21 which reported that the triplet states and electronic relaxation in photoexcited graphene quantum dots have a lifetime of 4 μs. Furthermore, as far as we know, the fluorescence lifetime has a lifetime in the time-scale of nanoseconds; generally, fluorescence lifetimes are near 10 ns.25 Then, the component of 6.95 μs can be regarded as the phosphorescent emission. Similar room temperature phosphorescence (RTP) has been observed in carbon dots, and the reported RTP comes from the triplet state of the aromatic carbonyl group.22 What calls for special attention is that energy of the singlet and triplet were 2.76 (450 nm) and 2.48 eV (500 nm), respectively, in our experiment, suggesting that the energy gap between phosphorescence and fluorescence was narrow. Generally, it was previously assumed that the S1 level was 0.5–1.0 eV higher in energy than the T1 level due to the electron exchange energy between these levels. In H. Uoyama's report,27 they designed a molecular system with a small ΔEST = 100 meV and found that careful design of the organic molecules can lead to a small energy gap between the S1 and T1 levels. Other literature reports support the same phenomenon.28,29 They have demonstrated an organic molecule with an energy gap between its singlet and triplet excited states of almost zero (ΔEST ∼ 0 eV). Considering the situation of the energy level of GO and its derivatives is complicated and their energy structures remain controversial issues. These special attentions mentioned above can be regarded as the direct evidence to prove our assumption that the 500 nm emission comes from the phosphorescence. When its singlet and triplet state energies are close, the spin–orbit coupling will be efficient, and the intersystem crossing will also be efficient.21,22 Therefore, we suppose that the RTP, observed in this study, comes from aromatic hydrocarbons-carboxylic domain structure in the GO-CONH-TPA in a PMMA film, and the PMMA molecules effectively protect their energy from rotational or vibrational loss by rigidifying these groups with hydrogen bonds. RTP cannot be observed from the GO-CONH-TPA dispersed in other solvents.
 | ||
| Fig. 3 (a) The fluorescence decay curves obtained for GO-CONH-TPA in a PMMA film on the nanosecond time-scale. The samples were excited at 405 nm and detected at various wavelengths. (b) The fluorescence and phosphorescence of GO-CONH-TPA in a PMMA film. (c) The fitted line of the phosphorescence spectra of GO-CONH-TPA in a PMMA film. (d) Lifetime studies of the phosphorescence of GO-CONH-TPA in a PMMA film, monitored at 500 nm. | ||
Fig. 4(a) shows the fluorescence decay curves obtained for GO-CONH-TPA in an ethyl alcohol (EtOH) solution on the nanosecond time-scale. The sample was excited at 405 nm and the emission was detected at distinct wavelengths. Upon increasing the detection wavelength, the fluorescence decay curves in EtOH become narrower, which means the lifetime of the sample in the EtOH solution decreases. In the multi-exponential model, the intensity is assumed as the sum of individual single exponential decay times:
 | (2) |
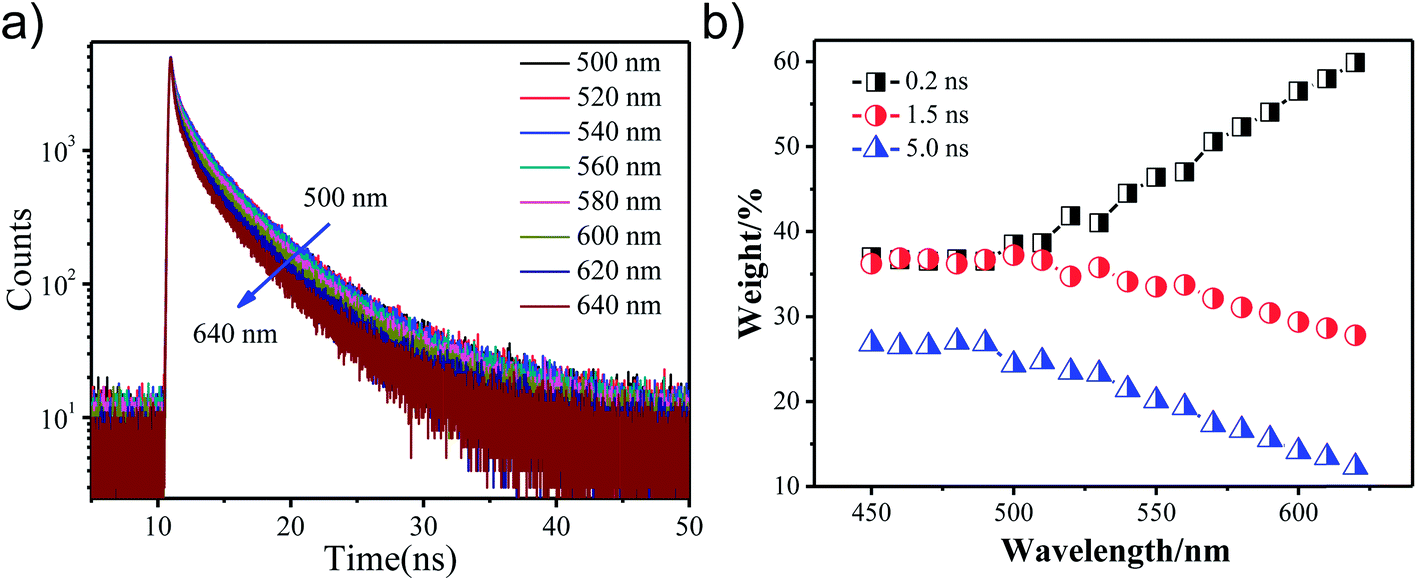 | ||
| Fig. 4 (a) The fluorescence decay curves obtained for GO-CONH-TPA in an EtOH solution on the nanosecond time scale. The samples were excited at 405 nm and detected at the different wavelengths. (b) The three lifetime components weight percentage of the GO-CONH-TPA sample. | ||
In this expression τi is the decay time, αi represents the amplitude at t = 0 and n is the number of decay times. Fig. 4(b) shows three lifetime components (0.2 ns, 1.5 ns and 5 ns) for the GO-CONH-TPA sample in EtOH. Upon increasing the detection wavelength, the amplitude of the smallest lifetime component (0.2 ns) gradually increases. As for the GO-CONH-TPA in EtOH or other usual organic solvents, there is no phosphorescent channel, but it does have an ISC channel. It was noticed that the average lifetime of the GO-CONH-TPA sample in EtOH decreases. The turning point of the wavelength (500 nm) is near the phosphorescence emission peak position of 485 nm. Therefore, it is evident that the ISC channel mainly influences the two longer lifetime components. Briefly, the excited singlet state in GO-CONH-TPA can be relaxed via both the fluorescence and ISC channels. When the energy gap between the excited singlet state and the excited triplet state decreases, there is a competition between the two types of energy relaxation channels. As for the fluorescence channel in GO-CONH-TPA, there are three lifetime components as mentioned above (Fig. 4(b)). After the ISC occurs in solution, more and more molecules in the excited singlet state are relaxed via ISC, and this can explain the phenomenon shown in Fig. 4(a). Both the long-lifetime (1.5 ns and 5 ns) components have a very near energy gap between their related excited singlet state and triplet state. The related excited singlet state can be easily relaxed via ISC instead of fluorescence emission, and the amplitude of the long-lifetime (1.5 ns and 5 ns) components is gradually reduced because of ISC. The direct consequence is that the fluorescence decay curves in EtOH become more and more narrower, and the weight percentage of the smallest lifetime component gradually increases upon increasing the detection wavelength. The two long-lifetime components are classified as molecular luminescence (several nanoseconds).16 The shortest lifetime component related to the excited singlet state has a large energy band gap compared to the excited triplet state, and thus the spin–orbit coupling is inefficient (the ISC is forbidden). On the contrary, the ISC of the long-lifetime components are efficient, and thus ISC mainly decreases the long-lifetime fluorescence emission. It is clear that the weak phosphorescence emission (or ISC) can affect both the steady-state fluorescence and the time-resolved fluorescence. The ISC induced steady-state fluorescence of GO-CONH-TPA shows the excitation-wavelength dependent fluorescence (‘red-edge effect’) both in solution and solid film. It also can affect the time-resolved fluorescence of GO-CONH-TPA in solid and solution. In the solid film, the phosphorescence elevates the TCSPC count baseline abnormally in the form of radiative transition and the time-resolved fluorescence decay curves become more and more slow. In the solution, the two long-lifetimes and radiative transition components of GO-CONH-TPA fluorescence quench in the form of ISC, which makes the time-resolved fluorescence decay become faster with the increase of the detection wavelength.
A home-built femtosecond transient absorption setup was used to study the photoluminescence kinetics. Fig. 5 shows a typical kinetic trace recorded with a pump wavelength of 400 nm and a probe wavelength of 500 nm. The kinetics were obtained by red-shifted probing. Half the energy difference of the pump and probe photons was 310 meV and the kinetics of GO were observed to be positive.8
 | ||
| Fig. 5 The transient decay kinetics of graphene oxide (GO) and GO-CONH-TPA at 500 nm after excitation using a 400 nm laser pulse. | ||
The kinetic signals were fitted using the sums of the multi-exponential functions, i.e. 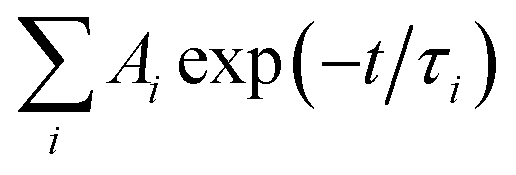 , using a global fitting procedure (Igor Pro. program Version 6.37, Wavemetrics. Inc.,Portland, OR). The τ1 = 4.1 ps component was associated with the carrier phonon scattering process for both GO in EtOH and GO-CONH-TPA.30,31 It was observed that this component was larger than that of the accumulation type samples.32 The accumulation type samples have a τ1 component smaller than 2.5 ps. The τ2 = 33.5 ps and τ3 = 282.6 ps components are associated with the different de-trapping of the trapped electrons in the different trap depths. Our experiments are in accordance with the literature.30–37 It is noted that GO-CONH-TPA has a much longer component τ4 = 2 ns, which has not been reported in the previous studies. Yu et al. were inclined to identify the longest time component associated with the lowest excited state (LUMO) rather than the trap states (>400 ps).35
, using a global fitting procedure (Igor Pro. program Version 6.37, Wavemetrics. Inc.,Portland, OR). The τ1 = 4.1 ps component was associated with the carrier phonon scattering process for both GO in EtOH and GO-CONH-TPA.30,31 It was observed that this component was larger than that of the accumulation type samples.32 The accumulation type samples have a τ1 component smaller than 2.5 ps. The τ2 = 33.5 ps and τ3 = 282.6 ps components are associated with the different de-trapping of the trapped electrons in the different trap depths. Our experiments are in accordance with the literature.30–37 It is noted that GO-CONH-TPA has a much longer component τ4 = 2 ns, which has not been reported in the previous studies. Yu et al. were inclined to identify the longest time component associated with the lowest excited state (LUMO) rather than the trap states (>400 ps).35
This 2 ns lifetime component is comparable to that of the fluorescence emission. The 2 ns component is a positive signal in the transient absorption spectra, and this positive signal was observed in the wavelength region of the excited-state absorption.38 This can be associated with the intersystem crossing (ISC), which has a time of duration from picoseconds to nanoseconds.25 There are several reasons to explain the disappearance of the 2 ns component in GO. First, GO has a lot of defect levels, and the excited singlet state energy can be easily reduced rather than via ISC. Second, for GO in most instances, such as in solutions, its excited triplet state energy can be easily reduced by oxygen. Finally, GO has a very low PL quantum yield, and hence the probability of ISC in GO is very small. In this study, since the broad and structureless charge-transfer fluorescence band of GO is reduced, it has a higher PL quantum yield than GO. The organic solution and organic solid matrix instead of water can isolate the triplet state energy from being reduced by oxygen.
It should be noted that the aromatic carbonyl group has been generally considered as the origin of RTP.21,22 Because the singlet and triplet states are close in energy, the spin–orbit coupling is efficient and the intersystem crossing is also efficient. It is reasonable to suppose that the RTP observed here comes from the aromatic carbonyls in GO-CONH-TPA.
4. Conclusions
In summary, we have obtained a pure organic RTP based on an enhanced fluorescence type GO-PMMA composite film. The phosphorescence peak was located around 485 nm with an average lifetime of 6.95 μs under 400 nm excitation. It is proven that the phosphorescence originates from the aromatic hydrocarbons-carboxylic domain structure in GO. The GO-CONH-TPA dispersed in a polymethyl methacrylate (PMMA) film matrix, the intramolecular motions are reduced as well as the oxygen quenched triplet states energy. The ISC can help us to explain the phenomenon of the ‘red-edge effect’ in GO-CONH-TPA. The 2 ns ISC is quite similar to the fluorescence lifetime in its time-scale, the fluorophore can emit at the same time of the excited state's energy being reduced, and this results in time-dependent emission energy. The transient absorption (TA) was detected at 500 nm after being excited at 400 nm, and the new 2 ns lifetime component can be associated with the intersystem crossing (ISC). This can be explained from three aspects: (1) the excited singlet state energy is inclined to reduce to the defect levels rather than the triplet state, (2) oxygen or other factors can easily result in reducing the triplet state energy and (3) the PL quantum yield of GO is very low (0.02%) and the ISC in GO is difficult to detect. Both the time-resolved fluorescence and ultra-fast time-resolved transient absorption reveal the existence of GO phosphorescence near the fluorescence with a wide emission peak. The original finding of the graphene oxide phosphorescence has potential applications in chemical imaging and organic semiconductor devices, such as carrier collection in photovoltaic devices.Acknowledgements
This study was partly supported by the National Science Foundation of China (61108077, 61178085, 61008003, 61275147), the Key Project of Science and Technology of Shandong Province of China (No. 2010GGX10127) and the Shandong Province Natural Science Foundation of China (No. ZR2012AL11) (No. ZR2013EML006).References
- M. Yi, L. Zhao, Q. Fan, X. Xia, W. Ai, L. Xie, X. Liu, N. Shi, W. Wang, Y. Wang and W. Huang, J. Appl. Phys., 2011, 110, 063709 CrossRef.
- S.-S. Li, K.-H. Tu, C.-C. Lin, C.-W. Chen and M. Chhowalla, ACS Nano, 2010, 4, 3169–3174 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- J.-L. Li, H.-C. Bao, X.-L. Hou, L. Sun, X.-G. Wang and M. Gu, Angew. Chem., Int. Ed., 2012, 51, 1830–1834 CrossRef CAS PubMed.
- J. Qian, D. Wang, F.-H. Cai, W. Xi, L. Peng, Z.-F. Zhu, H. He, M.-L. Hu and S. He, Angew. Chem., Int. Ed., 2012, 51, 10570–10575 CrossRef CAS PubMed.
- G. Eda, Y.-Y. Lin, C. Mattevi, H. Yamaguchi, H.-A. Chen, I. S. Chen, C.-W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505–509 CrossRef CAS PubMed.
- Q. Zhang, H. Zheng, Z. Geng, S. Jiang, J. Ge, K. Fan, S. Duan, Y. Chen, X. Wang and Y. Luo, J. Am. Chem. Soc., 2013, 135, 12468–12474 CrossRef CAS PubMed.
- X.-F. Zhang, X. Shao and S. Liu, J. Phys. Chem. A, 2012, 116, 7308–7313 CrossRef CAS PubMed.
- Q. Mei, K. Zhang, G. Guan, B. Liu, S. Wang and Z. Zhang, Chem. Commun., 2010, 46, 7319–7321 RSC.
- C.-T. Chien, S.-S. Li, W.-J. Lai, Y.-C. Yeh, H.-A. Chen, I. S. Chen, L.-C. Chen, K.-H. Chen, T. Nemoto, S. Isoda, M. Chen, T. Fujita, G. Eda, H. Yamaguchi, M. Chhowalla and C.-W. Chen, Angew. Chem., Int. Ed., 2012, 51, 6662–6666 CrossRef CAS PubMed.
- D. Kozawa, Y. Miyauchi, S. Mouri and K. Matsuda, J. Phys. Chem. Lett., 2013, 2035–2040, DOI:10.1021/jz400930f.
- J.-L. Chen, X.-P. Yan, K. Meng and S.-F. Wang, Anal. Chem., 2011, 83, 8787–8793 CrossRef CAS PubMed.
- T. V. Cuong, V. H. Pham, Q. T. Tran, S. H. Hahn, J. S. Chung, E. W. Shin and E. J. Kim, Mater. Lett., 2010, 64, 399–401 CrossRef CAS.
- K. S. Subrahmanyam, P. Kumar, A. Nag and C. N. R. Rao, Solid State Commun., 2010, 150, 1774–1777 CrossRef CAS.
- C. Galande, A. D. Mohite, A. V. Naumov, W. Gao, L. Ci, A. Ajayan, H. Gao, A. Srivastava, R. B. Weisman and P. M. Ajayan, Sci. Rep., 2011, 1, 85 CrossRef PubMed.
- S. K. Cushing, M. Li, F. Huang and N. Wu, ACS Nano, 2013, 8, 1002–1013 CrossRef PubMed.
- J.-L. Chen and X.-P. Yan, Chem. Commun., 2011, 47, 3135–3137 RSC.
- L. Zhengtang, P. M. Vora, E. J. Mele, A. T. C. Johnson and J. M. Kikkawa, Appl. Phys. Lett., 2009, 94, 111909 CrossRef.
- T. Gokus, R. R. Nair, A. Bonetti, M. Böhmler, A. Lombardo, K. S. Novoselov, A. K. Geim, A. C. Ferrari and A. Hartschuh, ACS Nano, 2009, 3, 3963–3968 CrossRef CAS PubMed.
- M. L. Mueller, X. Yan, J. A. McGuire and L.-s. Li, Nano Lett., 2010, 10, 2679–2682 CrossRef CAS PubMed.
- Y. Deng, D. Zhao, X. Chen, F. Wang, H. Song and D. Shen, Chem. Commun., 2013, 49, 5751–5753 RSC.
- J. Liu, Z. Yin, X. Cao, F. Zhao, A. Lin, L. Xie, Q. Fan, F. Boey, H. Zhang and W. Huang, ACS Nano, 2010, 4, 3987–3992 CrossRef CAS PubMed.
- Z. Li, Y. Chen, Y. Du, X. Wang, P. Yang and J. Zheng, Int. J. Hydrogen Energy, 2012, 37, 4880–4888 CrossRef CAS.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer Academic, New York, 2006 Search PubMed.
- M. Li, S. K. Cushing, X. Zhou, S. Guo and N. Wu, J. Mater. Chem., 2012, 22, 23374–23379 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- K. Sato, K. Shizu, K. Yoshimura, A. Kawada, H. Miyazaki and C. Adachi, Phys. Rev. Lett., 2013, 110, 247401 CrossRef PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- S. Kaniyankandy, S. N. Achary, S. Rawalekar and H. N. Ghosh, J. Phys. Chem. C, 2011, 115, 19110–19116 CAS.
- N. Liaros, S. Couris, E. Koudoumas and P. A. Loukakos, J. Phys. Chem. C, 2016, 120, 4104–4111 CAS.
- B. A. Ruzicka, L. K. Werake, H. Zhao, S. Wang and K. P. Loh, Appl. Phys. Lett., 2010, 96, 173106 CrossRef.
- D. Sun, Z.-K. Wu, C. Divin, X. Li, C. Berger, W. A. de Heer, P. N. First and T. B. Norris, Phys. Rev. Lett., 2008, 101, 157402 CrossRef PubMed.
- J. M. Dawlaty, S. Shivaraman, M. Chandrashekhar, F. Rana and M. G. Spencer, Appl. Phys. Lett., 2008, 92, 042116 CrossRef.
- S. Jingzhi, M. Lin, L. Jiewei, A. Wei, Y. Ting and G. G. Gagik, J. Phys. D: Appl. Phys., 2014, 47, 094008 CrossRef.
- Z.-B. Liu, X. Zhao, X.-L. Zhang, X.-Q. Yan, Y.-P. Wu, Y.-S. Chen and J.-G. Tian, J. Phys. Chem. Lett., 2011, 2, 1972–1977 CrossRef CAS.
- S. Kumar, M. Anija, N. Kamaraju, K. S. Vasu, K. S. Subrahmanyam, A. K. Sood and C. N. R. Rao, Appl. Phys. Lett., 2009, 95, 191911 CrossRef.
- R. Berera, R. van Grondelle and J. T. M. Kennis, Photosynth. Res., 2009, 101, 105–118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra01115f |
| This journal is © The Royal Society of Chemistry 2017 |