
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning a thiophene-fused benzoxadizole as an acceptor to build a narrow bandgap polymer for all-polymer solar cells†
Yang Yanga,
Jiacheng Wanga,
Xiaowei Zhan *b and
Xingguo Chen*a
*b and
Xingguo Chen*a
aHubei Key Laboratory on Organic and Polymeric Opto-electronic Materials, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: xgchen@whu.edu.cn
bDepartment of Materials Science and Engineering, College of Engineering, Peking University, Beijing 100871, China. E-mail: xwzhan@pku.edu.cn
First published on 5th April 2017
Abstract
In this work, a thiophene-fused benzoxadizole (BXT) unit was designed as a new acceptor and synthesized for the first time to build a D–A conjugated polymer (PBXT-IDT) with 4,4,9,9-tetrakis(4-hexylphenyl)-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene (IDT) for all polymer solar cells (all-PSCs). Due to the strong electron-withdrawing ability of the BXT unit, PBXT-IDT exhibited a very narrow optical bang gap of 1.43 eV and very strong ICT absorption in the range of 500–850 nm that could be complementary with poly(3-hexylthiophene) (P3TH) in the visible absorption region. Moreover, PBXT-IDT showed relatively low HOMO–LUMO energy levels of −5.33 eV and −3.64 eV, respectively, which can be act as an electron-accepting material to match with P3HT as an electron-donating material for all-PSCs. Therefore, the all-PSC device with a blend of PBXT-IDT and P3HT as the active layer was fabricated and the photovoltaic performances were investigated. The device showed a PCE of 1.09% with a high Voc of 0.84 V and a relatively low energy loss (Eloss) of 0.59 V. This indicates that reasonable structural modification of benzoxadizole (BX) can pave a new way to design a polymer as an electron-accepting material in all-PSCs.
Introduction
Polymer solar cells (PSCs) have received much attention due to their many advantages, including low-cost processing, flexibility, visible transparency and low weight.1 Benefiting from the development of new electron donors, dramatic developments have been achieved in conventional PSCs with fullerene derivative PCBM (such as PC61BM and PC71BM) as the electron-accepting material.2 However, fullerene derivatives also have some insufficiencies to be overcome, such as a difficulty in enhancing the absorption in the visible solar spectrum region and tuning the energy levels.3 Thus, some non-fullerene accepting materials have been developed including some small molecules such as ITIC or polymers such as N2200 to replace PCBM.Recently, all polymer solar cells (all-PSCs) that contains two or more different polymers act as the electron-accepting material and the electron-donating material, respectively, have attracted much attention due to the advantages such as easily solution-processibility, possibility of a vast number of combinations of donor/acceptor pairs, tenability for absorption complementation in the visible and near-infrared regions of the solar spectrum and the better phase separation stability than that of conventional PSCs with PCBM as an accepting material.4,5 For example, some perylenediimide (PDI) or naphthalenediimide (NDI) based polymers as typical n-type materials showed strong light absorption and high electron transport properties, so they were widely used as the polymer accepting materials to construct all-PSCs.6 Much recently, Li and Zhang et al. built an all-PSCs with N2200 (poly((N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl)-alt-5,5′-(2,2′-bithiophene))) as the accepting material and difluoro-benzotriazole based polymer as the donating material to achieve the excellent photoelectric conversion efficiency as high as 8.27%.7 Yan et al. also reported a novel polymer accepting material based on the PDI and vinylene unit that achieved an efficiency up to 7.57% in the all-PSCs.8 In addition, a number of electron-withdrawing units including diketopyrrolopyrrole (DPP),9 2,1,3-benzothiadiazole (BT),10 thieno[3,4-c]pyrrole-4,6-dione (TPD)11 and B←N bridged bipyridine (BNBP)12 were also used as the building blocks in designing the polymer accepting materials.
In previous reports, benzoxadizole (BX) as an electron-deficient unit had been used to build D–A polymer donating materials in the PSCs, and high Voc was obtained due to its much lower HOMO energy level compared with it analogous polymers based on benzothiadiazole (BT) unit.13 However, as far as we know, the BX unit has never been used to design the polymer as an electron-accepting material. Much recently, our group had demonstrated that the fusion of a thiophene ring onto the BT unit was an effective way to modulate the HOMO–LUMO energy levels and broaden absorption spectrum of the polymer,14a so the polymer based on thieno[2,3-f]-2,1,3-benzothiadiazole-6-carboxylate (BTT) unit showed smaller optical bandgap (Eg) than that of polymer based on the BT unit. In this work, we designed a new electron-deficient unit by fusing a thiophene ring onto the benzoxadizole (BX) unit, namely, thieno[2′,3′:4,5]benzo[1,2-c][1,2,5]oxadiazole-6-carboxylate (BXT). Then we applied it to construct a new narrow bandgap polymer (PBXT-IDT) with indacenodithienothiophene (IDT) donor. Herein, the photophysical and electrochemical properties of PBXT-IDT were studied, and the photovoltaic performances of all-PSCs based on PBXT-IDT as an electron-accepting material and P3TH as an electron-donating material were investigated in detail.
Experimental
Synthetic procedures
6-(Ethoxycarbonyl)thieno[2′,3′:4,5]benzo[1,2-c][1,2,5]oxadiazole 1-oxide (3). To a 250 mL round-bottomed flask were added 50 mL concentrated hydrochloric acid and 1 (756 mg, 2.8 mmol). NaNO2 (193 mg, 2.8 mmol) was added to the flask slowly at 0 °C. After reacting for 30 minutes, the NaN3 (182 mg, 2.8 mmol) was added slowly. After stirring at room temperature for 3 h, the reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4. Compound 2 was obtained by removing the solvent without further purification. The crude product (2) was dissolved in 20 mL toluene. The reaction solution was heated to reflux for 2 h. After removing the solvent under vacuum, the crude product was purified by column chromatography (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, petroleum ether:dichloromethane) to yield 3 (266 mg, 36%) as yellow powder. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.918 (s, 2H), 7.828 (s, 1H), 4.395–4.466 (m, 2H), 1.429 (t, J = 6.9 Hz, 3H). 13C-NMR (75 MHz, CDCl3, δ ppm): 161.15, 145.19, 143.03, 140.39, 128.72, 110.41, 110.25, 108.34, 62.43, 14.11. EI-MS: calcd for [C11H8N2O4S]+: 264.02; found: 264.02. (Note: sodium azide and organic azide are toxic and potentially explosive hazard, so they should be handled with great care!).
:1, petroleum ether:dichloromethane) to yield 3 (266 mg, 36%) as yellow powder. 1H-NMR (300 MHz, CDCl3, δ ppm): 7.918 (s, 2H), 7.828 (s, 1H), 4.395–4.466 (m, 2H), 1.429 (t, J = 6.9 Hz, 3H). 13C-NMR (75 MHz, CDCl3, δ ppm): 161.15, 145.19, 143.03, 140.39, 128.72, 110.41, 110.25, 108.34, 62.43, 14.11. EI-MS: calcd for [C11H8N2O4S]+: 264.02; found: 264.02. (Note: sodium azide and organic azide are toxic and potentially explosive hazard, so they should be handled with great care!).
Ethyl thieno[2′,3′:4,5]benzo[1,2-c][1,2,5]oxadiazole-6-carboxylate (4):. 3 (266 mg, 1.0 mmol), PPh3 (528 mg, 2.0 mmol) and 10 mL THF were added to a 100 mL round-bottomed flask. After stirring for 2 h, the reaction mixture was poured into water and extracted with chloroform. The mixture was purified by column chromatography (1
:1, petroleum ether: chloroform) to yield 4 (223 mg, 90%) as yellow powder. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.345 (s, 1H), 8.261 (s, 1H), 7.916 (s, 1H), 4.410–4.480 (m, 2H), 1.440 (t, J = 6.9 Hz, 3H). 13C-NMR (75 MHz, CDCl3, δ ppm): 161.63, 160.26, 148.40, 145.62, 144.63, 140.71, 129.26, 112.56, 108.92, 62.73, 14.46. EI-MS: calcd for [C11H8N2O3S]+: 248.03; found: 248.00.
Ethyl 4,8-dibromothieno[2′,3′:4,5]benzo[1,2-c][1,2,5]oxadiazole -6-carboxylate (BXT):. 4 (223 mg, 0.9 mmol), Br2 (1.44 g, 9 mmol) and 30 mL chloroform were added to a 100 mL round-bottomed flask. The solution was heated to reflux for 12 h. After cooling to room temperature, the mixture was poured into water and extracted with chloroform and purified by column chromatography (1
:1, petroleum ether: chloroform) to yield BXT (292 mg, 80%) as yellow powder. 1H-NMR (300 MHz, CDCl3, δ ppm): 8.109 (s, 1H), 4.432–4.503 (m, 2H), 1.462 (t, J = 6.9 Hz, 3H). 13C-NMR (75 MHz, CDCl3, δ ppm): 161.12, 148.91, 148.03, 147.09, 143.75, 141.33, 129.47, 104.97, 100.47, 63.17, 14.48. EI-MS: calcd for [C11H6Br2N2O3S]+: 405.84; found: 405.83.
Results and discussion
Synthesis and characterization
In our previous work,14 it was found that the fusion of a thiophene ring at the side of benzothiadiazole (BT) or benzotriazole (BTAZ) unit can stabilize the quinoid population of the conjugated backbone because of the aromaticity of the side-fused thiophene ring. This structural modification not only strengthens the intramolecular charge transfer (ICT), but it also reduces the optical bandgap, thus to effectively harvest the sunlight. As known, benzoxadizole (BX) unit is a very strong electron-withdrawing group to be applied to build D–A polymers as electron-donating materials for PSCs.13 In order to further broaden the absorption spectra and regulate the energy levels of benzoxadizole (BX) based D–A polymers, a thiophene ring has been fused onto BX unit to build a new acceptor of thiophene-fused benzoxadizole (BXT) unit. It can be expected that the fused thiophene ring can also stabilize the quinoid structure of the conjugated backbone and strengthen the ICT. In addition, another weak electron-withdrawing group of alkoxycarbonyl group has been introduced at the α-position of fused thiophene unit, which can further reduce the HOMO–LUMO energy levels. It can be expected that the deep HOMO–LUMO energy levels of the corresponding BXT-based copolymer can make it as an electron-accepting material for PSCs.The synthetic route for electron-deficient unit (BXT) and the polymer PBXT-IDT was outlined in Scheme 1. Ethyl 5-amino-6-nitrobenzo[b]thiophene-2-carboxylate (1) was synthetized according to the method reported by our group.14a Compound 2 was prepared by the diazotization of 1 and then the substitution by sodium azide carefully. Compound 3 could be easily obtained by heating 2 in toluene. Finally, the thiophene-fused benzoxadizole (BXT) as a new electron acceptor was obtained by the reduction of 3 with PPh3. The monomer, di-Br-BXT was synthesized by the bromination of BXT. The corresponding polymer of PBXT-IDT was prepared via the microwave-assisted Stille cross-coupling reaction of di-Br-BXT with IDT. The number average molecular weight (Mn) of the polymer PBXT-IDT was about 7.5 kDa with PDI = 1.6, which was determined by GPC (the Gel permeation chromatography) using tetrahydrofuran as the eluent. The thermal stability of the polymer PBXT-IDT was measured by the thermogravimetric analysis (TGA). It showed good thermal stability with decomposition temperature beyond 332 °C (see Fig. S1 in ESI†).
 | ||
| Scheme 1 Synthetic routes for BXT unit and the corresponding polymer PBXT-IDT. | ||
Photophysical and electrochemical properties
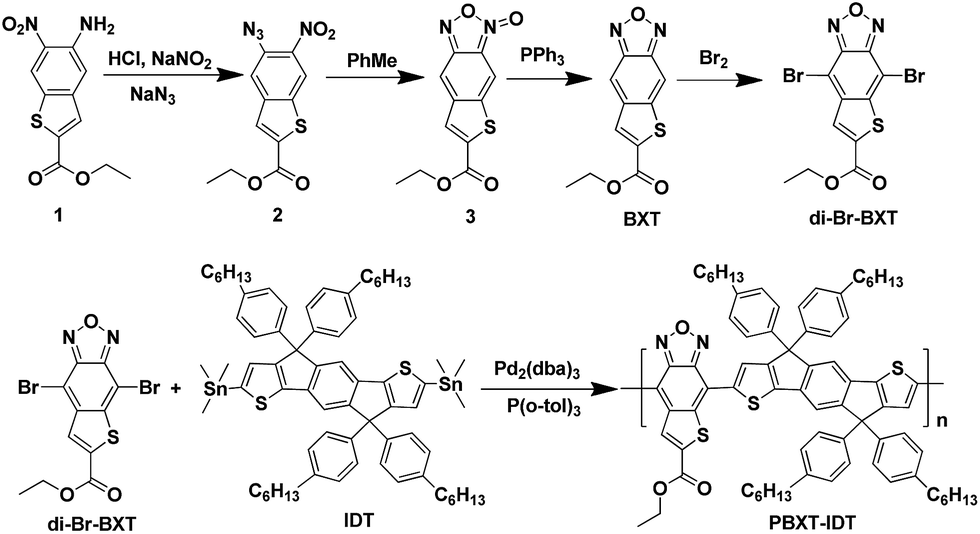
The UV-vis absorption spectra of PBXT-IDT in diluted chlorobenzene solution and in thin film were shown in Fig. 1, and corresponding data were listed in Table 1. As can be seen, in solution it displayed two absorption bands (band I 350–450 nm and band II 550–850 nm). The high energy band of 350–450 nm was correlated to the local π–π* transition. The broad and strong low energy band in the region of 550–850 nm was attributed to the strong intramolecular charge transfer (ICT) effect. Compared with the solution, the polymer exhibited obvious redshift absorption spectra in the thin film state, and its ICT absorption was extended to about 900 nm. From the absorption onset edge of the thin film, the optical bandgap of PBXT-IDT was estimated to be about 1.43 eV, implying that fusing a thiophene ring onto BX unit was an effective way to design the low bandgap polymer.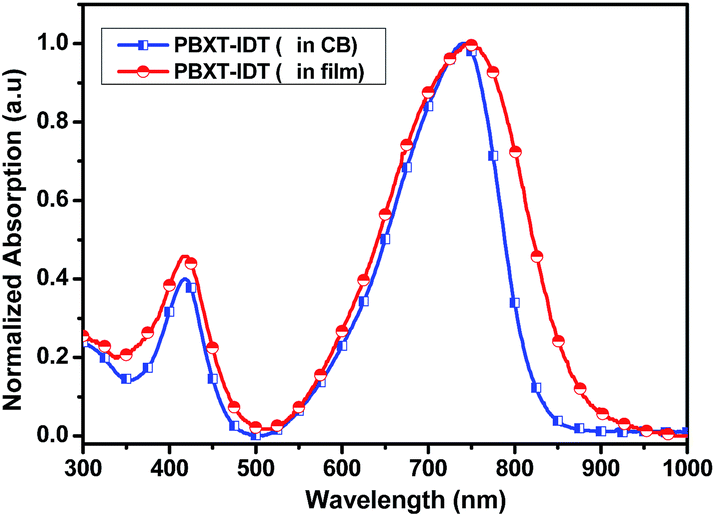 | ||
| Fig. 1 Absorption spectra of PBXT-IDT in chlorobenzene (CB) and in thin film for comparison. | ||
Fig. 2 showed the absorption spectra of PBXT-IDT, P3HT and the blend of P3HT:PBXT-IDT (4:1, p/n) in thin films. It can be seen that PBXT-IDT and P3HT showed good complementary absorption in the visible region of 300–900 nm, indicating that the blend of PBXT-IDT and P3HT in the film is much favorable for the light harvesting.7,15 The complementary absorption can also be seen from the absorption coefficient in thin film as shown in Fig. S3.† The polymer donor P3HT shown strong absorption in the region of 350–650 nm with the absorption coefficient of 1.2 × 105 cm−1 at 526 and 546 nm and the polymer acceptor PBXT-IDT shown strong absorption in the region of 600–900 nm with the relatively low absorption coefficient of 5.7 × 104 cm−1 at 748 nm.
 | ||
| Fig. 2 Absorption spectra of PBXT-IDT, P3HT and P3HT/PBXT-IDT (4:1, p/n) in thin films. | ||
The HOMO and LUMO energy levels of PBXT-IDT were investigated by the cyclic voltammetry (CV) measurement as shown in Fig. S4,† and the electrochemical data were also summarized in Table 1. Its onset oxidation and reduction potential were about 0.53 V and −1.16 V, respectively, which were applied to estimate the HOMO level of −5.33 eV and LUMO level of −3.64 eV. The HOMO–LUMO energy levels of PBXT-IDT is at least 0.3 eV lower than those of P3TH (HOMO = −4,76 eV and LUMO = −2.74 eV), indicating that this polymer can be acted as an electron-accepting material to be well-matched with P3HT as the polymer donating material for all-PSCs. In addition, because of the higher LUMO energy level of PBXT-IDT than that of PC61BM,18 an enhanced open circuit voltage (Voc) of the PSC can be expected to apply PBXT-IDT as the polymer accepting material and P3HT as the polymer donating matreial.15–18 As shown in Fig. 3, the P3HT/PBXT-IDT blend in thin film exhibited effective photoluminescence (PL) quenching (90%) indicating that the exciton can split efficiently in the blend.
 | ||
| Fig. 3 Photoluminescence spectra of P3HT and P3HT/PBXT-IDT blend in thin film. | ||
Photovoltaic performances
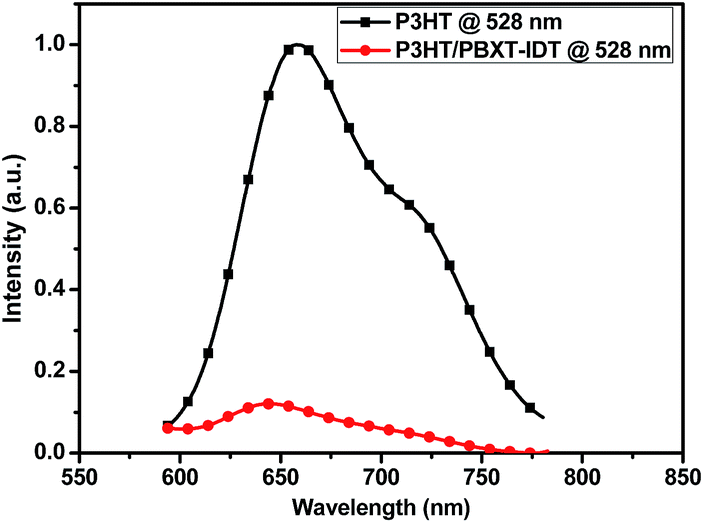
All-PSC devices using the configuration of ITO/PEDOT:PSS/P3HT:PBXT-IDT/Ca/Al were fabricated to investigate the photovoltaic performance of the polymer PBXT-IDT as an electron-accepting material to build active layer with P3TH as an electron-donating material. The mixture of P3HT/PBXT-IDT in chlorobenzene (CB) solvent (14 mg mL−1 in total) was spin-coated on a PEDOT/PSS layer to form an active layer. The open-circuit voltage (Voc), short circuit current density (Jsc), fill factor (FF), and power conversion efficiency (PCE) of the devices were listed in Table 2, and the relevant current density–voltage (J–V) and external quantum efficiency (EQE) curves were shown in Fig. 4 and 5, respectively. As can be seen in Table 2, these devices showed the higher Voc than the devices using P3HT as the donating material and PC61BM as the accepting material because of the higher LUMO energy level of PBXT-IDT. By changing the weight ratio of P3HT:PBXT-IDT, all-PSC devices with 4:1 blend ratio of P3HT:PBXT-IDT exhibited the best power conversion efficiency (PCE) of 0.49 ± 0.05% with Jsc of 1.34 ± 0.11 mA cm−2, FF of 42.14 ± 1.97%, and Voc of 0.86 ± 0.01 V. Thermal treatment at 100 °C could improve the PCE of the devices due to the increased Jsc (from 1.34 ± 0.11 to 2.08 ± 0.12 mA cm−2). To further improve the PCE of the device, variant amount of chloroform (CF) was added into CB solution for building the active layer. At optimized volume ratio of CB/CF (10:1), the Jsc was increased from 2.08 ± 0.12 to 2.54 ± 0.21 mA cm−2. Thus, the PCE was further increased from 0.79 ± 0.05% to 0.99 ± 0.06%. The best PCE of the all-PSC devices was 1.09%, with Voc of 0.84 V. The photon energy loss (Eloss) of the P3HT/PBXT-IDT system was about 0.59 V that was quantified by Eloss = Eg/q − Voc.
| Blend film | Solvent | Voc (V) | Jsc (mA cm−2) | FF (%) | PCEb (PCEmax) (%) |
|---|---|---|---|---|---|
| a Annealing at 100 °C for 10 min.b Average data were obtained from 9 devices. | |||||
| CB | 0.86 ± 0.01 | 1.34 ± 0.11 | 42.14 ± 1.97 | 0.49 ± 0.05(0.54) | |
| P3HT/PBXT-IDT | CBa | 0.81 ± 0.01 | 2.08 ± 0.12 | 47.12 ± 2.75 | 0.79 ± 0.05(0.85) |
| CB/CF(10/1)a | 0.83 ± 0.01 | 2.54 ± 0.21 | 46.97 ± 2.31 | 0.99 ± 0.06(1.09) | |
| P3HT/PC61BM | o-DCB | 0.61 ± 0.01 | 8.64 ± 0.39 | 64.33 ± 2.78 | 3.39 ± 0.12(3.50) |
 | ||
| Fig. 4 The J–V curves of the all-PSCs based on P3HT/PBXT-IDT mixture. | ||
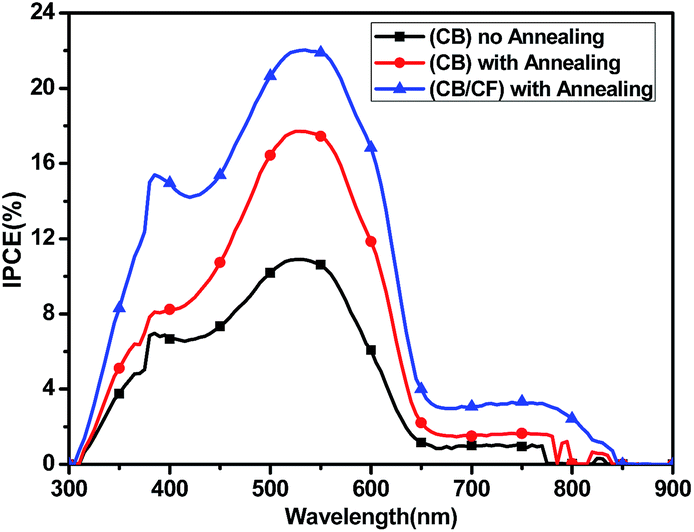 | ||
| Fig. 5 The IPCE curves of the all-PSCs based on P3HT/PBXT-IDT mixture. | ||
The increased Jsc in PSCs could be identified by the EQE curve. As shown in Fig. 5, the blend film showed the photo-response from 300–850 nm using CB as the processing solvent and its EQE was improved at annealing at 100 °C. Furthermore, the photo-response in 300–850 nm was strengthened obviously after adding CF to the blend film, so the Jsc was improved. The similarity of the spectra shape of EQE curves and absorption spectra of the blend film indicated that both the polymer accepting material PBXT-IDT and donating material P3HT contributed to the photocurrent generation. It should be noted that the photo-response in the region of 550–850 nm is much weaker than that in the region of 300–650 nm. This result is well in agreement with the absorption intensity of the blend film of P3HT:PBXT-IDT (4:1, p/n) (Fig. 2), in which relatively higher absorption coefficient and larger ratio of P3TH than PBXT-IDT in the blend film influences the light-harvesting in the region of 550–850 nm.
Atomic force microscopy (AFM) and transmission electron microscopy (TEM) were measured to examine the morphology of the P3HT:PBXT-IDT (w:w, 4:1) in the mixed solvent of CB and CF after annealing. As seen in Fig. 6a, the AFM image of the active layer showed some crystalline grains which could be attributed to the self-organization of P3HT.15 The root mean square (RMS) was 0.83 nm. On the other hand, the fibrous aggregation and bicontinuous and small nanostructure phase segregation were observed in the TEM image (Fig. 6b). The TEM image and AFM height image indicated that the two polymers were mixed well in the blend film.12c
 | ||
| Fig. 6 (a) Tapping mode AFM topography images (2 × 2 μm2) and (b) TEM image of the P3HT/PBXT-IDT device. | ||
The space-charge limited current (SCLC) method was used to determine the charge transporting properties of pure PBXT-IDT film and the blend film of P3HT/PBXT-IDT (p/n = 4:1). The corresponding curves were shown in Fig. S5–S7.† As can be seen, the electron mobility of pure PBXT-IDT film is about 4.2 × 10−6 cm2 V−1 s−1, and the hole mobility and electron mobility of the P3HT/PBXT-IDT (w:w = 4:1) in the mixed solvent of CB and CF after annealing were 1.2 × 10−4 cm2 V−1 s−1 and 2.3 × 10−5 cm2 V−1 s−1, respectively. Apparently, the relatively low electron mobility for PBXT-IDT as an electron-accepting material leads to the unbalanced hole and electron mobility of the blend film of P3HT/PBXT-IDT, that can be responsible for the poor Jsc of the PBXT-IDT based all-PSC device. Therefore, the further structural modification for BXT to build the accepting material with high and balanced electron mobility to match P3TH will improve the photovoltaic performances for the corresponding all-PSCs, which is under progress.
Conclusions
In this work, a thiophene fused benzoxadizole (BXT) unit has been designed as a new acceptor to build D–A copolymer (PBXT-IDT) for all-PSCs. By fusing a thiophene ring onto benzoxadizole, the PBXT-IDT polymer exhibited strong ICT absorption in the range of 500–850 nm with much narrow optical bang gap of 1.43 eV. The polymer showed a higher LUMO energy level than that of PCBM that was conducive to the enhancement of Voc. Using this polymer as an electron-accepting material to match P3HT as an electron donating material, the all-PSC device exhibited a PCE of 1.09% with a high Voc of 0.84 V and a relatively low energy loss (Eloss) of 0.59 V. This suggests that BXT unit can be used to construct D–A polymer as an electron-accepting material and further work should be done to tailor the BXT unit and copolymerize it with different electron donors for all-PSCs.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 51173138) for financial support.Notes and references
- (a) H. Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649 CrossRef CAS; (b) L. Chen, S. Tian and Y. Chen, Polym. Chem., 2014, 5, 4480 RSC; (c) J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709 CrossRef CAS PubMed; (d) H. L. Yip and A. K. Y. Jen, Energy Environ. Sci., 2012, 5, 5994 RSC; (e) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS.
- (a) J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS; (b) S. Shi, Q. Liao, Y. Tang, H. Guo, X. Zhou, Y. Wang, T. Yang, Y. Liang, X. Cheng, F. Liu and X. Guo, Adv. Mater., 2016, 28, 9969 CrossRef CAS PubMed.
- (a) M. Lenes, S. W. Shelton, A. B. Sieval, D. F. Kronholm, J. C. Hummelen and P. W. M. Blom, Adv. Funct. Mater., 2009, 19, 3002 CrossRef CAS; (b) R. B. Ross, C. M. Cardona, D. M. Guldi, S. G. Sankaranarayanan, M. O. Reese, N. Kopidakis, J. Peet, B. Walker, G. C. Bazan, E. V. Keuren, B. C. Holloway and M. Drees, Nat. Mater., 2009, 8, 208 CrossRef CAS PubMed; (c) Y. He, H. Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377 CrossRef CAS PubMed.
- (a) J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS; (b) T. Kim, J. H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. W. Ma, U. Jeong, T. S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed; (c) J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876 CrossRef CAS PubMed; (d) C. R. McNeill, Energy Environ. Sci., 2012, 5, 5653 RSC.
- A. Facchetti, Mater. Today, 2013, 16, 123 CrossRef CAS.
- (a) H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679 CrossRef CAS PubMed; (b) X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS PubMed; (c) J. Oh, K. Kranthiraja, C. Lee, K. Gunasekar, S. Kim, B. Ma, B. J. Kim and S. H. Jin, Adv. Mater., 2016, 28, 10016 CrossRef CAS PubMed; (d) Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganäs, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 10935 CrossRef CAS PubMed.
- L. Gao, Z. G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2015, 28, 1884 CrossRef PubMed.
- Y. Guo, Y. Li, O. Awartani, J. Zhao, H. Han, H. Ade, D. Zhao and H. Yan, Adv. Mater., 2016, 28, 8483 CrossRef CAS PubMed.
- (a) W. Li, W. S. C. Roelofs, M. Turbiez, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 3304 CrossRef CAS PubMed; (b) W. Li, Y. An, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2015, 3, 6756 RSC.
- Y. Fu, B. Wang, J. Qu, Y. Wu, W. Ma, Y. Geng, Y. Han and Z. Xie, Adv. Funct. Mater., 2016, 26, 5922 CrossRef CAS.
- S. Liu, Z. Kan, S. Thomas, F. Cruciani, J. L. Brédas and P. M. Beaujuge, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef.
- (a) X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504 CrossRef CAS PubMed; (b) C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436 CrossRef CAS PubMed; (c) X. Long, N. Wang, Z. Ding, C. Dou, J. Liu and L. Wang, J. Mater. Chem. C, 2016, 4, 9961 RSC.
- (a) J. Zhao, Y. Li, A. Hunt, J. Zhang, H. Yao, Z. Li, J. Zhang, F. Huang, H. Ade and H. Yan, Adv. Mater., 2015, 28, 1868 CrossRef PubMed; (b) J. C. Bijleveld, M. Shahid, J. Gilot, M. M. Wienk and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 3262 CrossRef CAS; (c) Z. Zhang, F. Lin, H. C. Chen, H. C. Wu, C. L. Chung, C. Lu, S. H. Liu, S. H. Tung, W. C. Chen, K. T. Wong and P. T. Chou, Energy Environ. Sci., 2015, 8, 552 RSC; (d) N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Bellete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS PubMed.
- (a) P. Zhou, Z. G. Zhang, Y. Li, X. Chen and J. Qin, Chem. Mater., 2014, 26, 3495 CrossRef CAS; (b) P. Zhou, Y. Yang, X. Chen, Z. G. Zhang and Y. Li, J. Mater. Chem. C, 2017, 5, 2951 RSC.
- Y. Lin, P. Cheng, Y. Li and X. Zhan, Chem. Commun., 2012, 48, 4773 RSC.
- Y. Wu, H. Bai, Z. Wang, P. Cheng, S. Zhu, Y. Wang, W. Ma and X. Zhan, Energy Environ. Sci., 2015, 8, 3215 CAS.
- S. Li, W. Liu, M. Shi, J. Mai, T. K. Lau, J. Wan, X. Lu, C. Z. Li and H. Chen, Energy Environ. Sci., 2016, 9, 604 CAS.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C. H. Tan, S. D. Dimitrov, Z. R. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02705b |
| This journal is © The Royal Society of Chemistry 2017 |