
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive electrospun membrane with enhanced wettability
Anupama Sargur Ranganath ,
V. Anand Ganesh,
Kostiantyn Sopiha,
Rahul Sahay and
Avinash Baji*
,
V. Anand Ganesh,
Kostiantyn Sopiha,
Rahul Sahay and
Avinash Baji*
Division of Engineering Product Development, Singapore University of Technology and Design (SUTD), Singapore – 487372. E-mail: Avinash_Baji@sutd.edu.sg; Tel: +65 6499 4502
First published on 5th April 2017
Abstract
This study investigated the switchable wettability behavior of poly-N-(isopropyl acrylamide) (PNIPAM) and poly-(vinylidene fluoride) (PVDF) blend membranes that were fabricated using electrospinning. The wettability of the membranes was determined by water contact angle measurements. The wettability of the membranes was controlled by varying the concentration of PNIPAM in the blend. The results demonstrated that the addition of PVDF to the PNIPAM did not change the thermoresponsive behavior of PNIPAM. All PNIPAM/PVDF blend samples switched from hydrophilic to hydrophobic state when the temperature was increased from room temperature to 40 °C. However, blend samples with 25 wt% PNIPAM were demonstrated to be more hydrophilic and hydrophobic compared to other samples at room temperature and elevated temperature respectively. This was attributed to the phase separation of the polymers that resulted in domains with drastically different surface energies on the surface of the fibers.
1 Introduction
Stimuli-responsive materials such as poly(N-isopropylacrylamide) (PNIPAM) have been extensively investigated in recent years as wettability of these materials can be controlled with the help of external stimuli such as temperature and/or pH. The change in wettability of PNIPAM due to reversible hydrogen bonding with water molecules makes them attractive for applications such as drug delivery systems,1 cell attachment,2 controlling bio-fouling sensors,3 and actuators,4,5 etc. The swelling and de-swelling kinetics of PNIPAM is known to depend on the sample's surface area as well as the chemical composition. Below the lower critical solution temperature (LCST), the amide groups of PNIPAM interact with water molecules via hydrogen bonding. However, above the LCST, the chain conformation changes from the linear expanded chain conformation to compact, dehydrated conformation. Below the LCST, the linear PNIPAM is unstable and soluble in aqueous solutions.4,6,7 Considerable efforts have been made so far to improve PNIPAM's stability in aqueous solutions and to improve its mechanical properties. Common technique on improving the stability relies on chemically cross-linking PNIPAM.8–15 The degree of crosslinking and the size of the PNIPAM structures play a crucial role in the swelling and de-swelling of the PNIPAM.Crosslinking of PNIPAM is more often carried out by thermal8,12,13 or UV-based14,16 cross-linkers. Typically, polymers that have hydroxyl, carboxylic acid or amino functional groups tend to chemically react with the PNIPAM via condensation reaction initiated by thermo-curing. Similarly, polymers such as polybutadiene that have C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, polymerize by UV curing.15 Alternatively, chemical stability to the PNIPAM chains is provided by grafting PNIPAM on the surfaces of polyvinylidene fluoride (PVDF).17–20 A straightforward approach is to blend PNIPAM with PVDF to obtain temperature sensitive PVDF composites. It is known that PNIPAM chains tend to phase separate and form PNIPAM domains on the surface of the PVDF. The responsive behavior of PNIPAM due to chain conformational change also ensures that the blend displays similar temperature-sensitive behavior.21
C double bonds, polymerize by UV curing.15 Alternatively, chemical stability to the PNIPAM chains is provided by grafting PNIPAM on the surfaces of polyvinylidene fluoride (PVDF).17–20 A straightforward approach is to blend PNIPAM with PVDF to obtain temperature sensitive PVDF composites. It is known that PNIPAM chains tend to phase separate and form PNIPAM domains on the surface of the PVDF. The responsive behavior of PNIPAM due to chain conformational change also ensures that the blend displays similar temperature-sensitive behavior.21
In this study, we use electrospinning to fabricate22–26 PNIPAM/poly(vinylidene fluoride) (PVDF) fibrous blends and investigate the wettability of the samples as a function of PNIPAM concentration within the blends. A cross-linker, octaglycidyl polyhedral oligomeric silsesquioxane (OG-POSS) and an initiator, 2-ethyl-4-methylimidazole (EMI) is also added to PNIPAM.13 All the samples demonstrated switchable wettability when the temperature is increased from ambient temperature to 40 °C. Interestingly, samples containing 25 wt% PNIPAM exhibited enhanced wettability when compared with other samples. Our results also show that the hydrophilicity of the membranes can be controlled by varying the PNIPAM content in the blend proportion.
2 Experimental details
2.1 Materials
Poly(N-isopropylacrylamide) (PNIPAM, Mw = 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Scientific Polymer Products Inc. Octaglycidyl polyhedral oligomeric silsesquioxane (OG-POSS, ((C6H11O2)n(SiO1.5)n n = 8, 10, 12)) was obtained from Hybrid Plastics Inc. Poly(vinylidene fluoride) (PVDF, Mw = 534000), 2-ethyl-4-methylimidazole (EMI) and N,N-dimethylformamide (DMF, 99.5%) were obtained from Sigma-Aldrich while tetrahydrofuran (THF, 99%) was obtained from Tedia company, Inc.
000) was purchased from Scientific Polymer Products Inc. Octaglycidyl polyhedral oligomeric silsesquioxane (OG-POSS, ((C6H11O2)n(SiO1.5)n n = 8, 10, 12)) was obtained from Hybrid Plastics Inc. Poly(vinylidene fluoride) (PVDF, Mw = 534000), 2-ethyl-4-methylimidazole (EMI) and N,N-dimethylformamide (DMF, 99.5%) were obtained from Sigma-Aldrich while tetrahydrofuran (THF, 99%) was obtained from Tedia company, Inc.
2.2 Membrane fabrication
Solutions with 20 wt% polymer concentration were prepared by dissolving known concentration of PNIPAM and PVDF in DMF/THF (1:1) solvent mixture at 60 °C. The concentration of PNIPAM in the blend was 100, 75, 50, 25 and 0 wt%. OG-POSS was used as a crosslinker for PNIPAM (15 wt% of PNIPAM concentration) with EMI (0.3 wt% of PNIPAM concentration) as suggested by Wang et al., in their article.13 The solutions were mechanically stirred for about 8 h until the polymer was completely dissolved. For comparison purpose, all the polymeric solutions, including pristine PVDF contained OG-POSS. This enabled us to isolate the effect of OG-POSS and investigate the influence of PVDF on the thermoresponsive behavior of PNIPAM/PVDF composites. However, it should be noted that, PVDF–OG-POSS is referred to as PVDF alone in the text. Thereafter, the solution was loaded into a 5 ml syringe and electrospun using a standard electrospinning machine (Nanospinner 24 – Inovenso). Electrospinning was conducted at 17 kV with the feed rate of 0.8 ml h−1. The distance between the needle and the collector was adjusted to 10 cm. The residual solvent from the obtained fibers was removed by drying the fibers at 60 °C for 8 h in an oven. Following this, the temperature was increased to 150 °C and maintained for 8 h to promote crosslinking.
2.3 Characterization
The fibrous membranes were sputter coated with gold (30 mA, 90 seconds) and then examined using a scanning electron microscope ((SEM) JEOL, JSM-6700F) at an accelerating voltage of 5 kV.A state of the art 3-in-1 integrated microscope, OLS4500 (atomic force microscopy (AFM), phase mode) from Shimadzu was used to obtain phase images of the fiber surface.
Fourier transformation infrared spectroscopy (FTIR) spectra of the samples were collected using a Bruker Alpha spectrometer. FTIR spectra (ATR mode) were recorded at a resolution of 4 cm−1 with an average of 64 scans at room temperature.
Thermal degradation behavior of the blend samples were characterized using a thermogravimetric analyzer (TGA, (TAQ90, TA Instruments)). The samples were heated at the rate of 5 °C min−1 at nitrogen atmosphere to prevent oxidation. The samples were further characterized for their melting behavior and degree of crystallinity by using a differential scanning calorimeter (DSC). The samples were heated at the rate of 5 °C min−1. Melting enthalpy, ΔHf of PVDF and blends is measured from DSC profile and for pure crystalline PVDF,  = 104.7 J g−1 from literature.27 The degree of crystallinity is calculated as below
= 104.7 J g−1 from literature.27 The degree of crystallinity is calculated as below
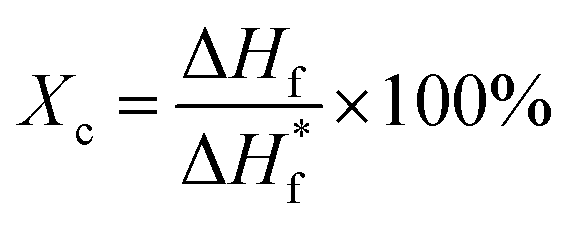 | (1) |
The static contact angle was determined using the bubble drop method with a KINO-optical contact angle and interface tension meter. The temperature response of the samples was investigated by taking the contact angle measurements at room temperature and above the LCST temperature of PNIPAM. To measure the contact angle above the LCST temperature of PNIPAM, the sample was heated at 40 °C in a temperature controlled chamber associated with the goniometer. In order to maintain the temperature, the chamber is enclosed by all sides with a small opening at the top for the dozing unit. The transparent glass on the sides walls of the chamber enables the light to pass through to take pictures of the water drop for CA measurements. The reported surface contact angle value averages at least five measurements taken from different regions of the fibrous membrane.
Hygroscopic studies on the sample were performed using the setup as explained in our previous work.24 The schematic of the setup is as illustrated in Fig. 1. The TGA in the setup is used to measure the dynamic change in the weight of the sample as a function of humidity. The sample was allowed to equilibrate for 90 minutes at every humidity step. The experimental trials were carried on PNIPAM samples for 300 minutes. 90 minutes for each RH step was chosen as the weight of sample reasonably stabilized after 90 minutes. Humidity was varied by controlling the proportion of dry and wet air supplied to the TGA. The RH of the supplied air was measured using a separate humidity sensor (Sensirion (SHT2x)).
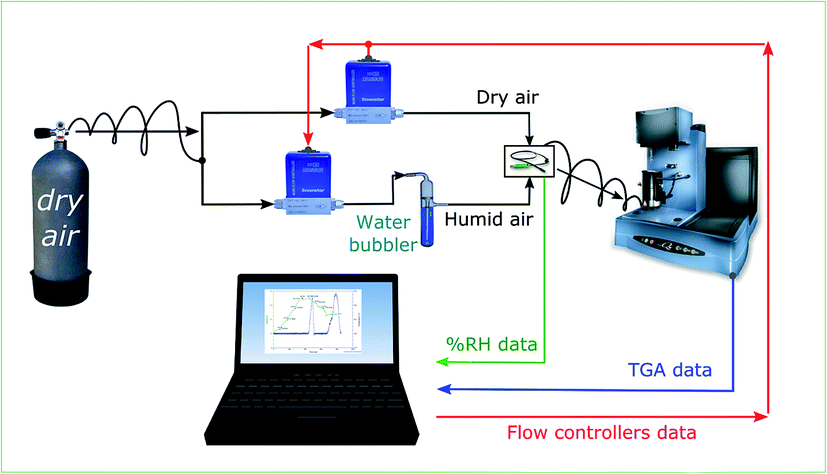 | ||
| Fig. 1 Schematic of the setup used for hygroscopic measurement. Ratio of dry and humid air is varied to control the humidity of the supplied air to TGA, where the sample's weight is measured as a function of relative humidity (% RH). | ||
3 Results and discussion
The stability of PNIPAM based membranes in aqueous media is important for their applications. In this study, the stability of PNIPAM is achieved by cross-linking PNIPAM with OG-POSS. For example, Wang et.al. crosslinked PNIPAM with octaglycidyl polyhedral oligomeric silsesquioxane.13 Similarly, Slemming-Adamsen et.al. crosslinked PNIPAM/gelatin nanofibers by electrospinning PNIPAM-NHS (ester terminated) and gelatin with 1-ethyl-3-(3-dimethyl-aminopropyl)-1-carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS).14 In this study, OG-POSS is used as a cross-linker to improve the stability of PNIPAM in aqueous media. PNIPAM along with OG-POSS and EMI are electrospun and then thermally treated to obtain highly cross-linked PNIPAM membrane. To demonstrate the stability of these membranes, the fibers are immersed in turbulent water stirred at 600 rpm using a hotplate. SEM images (not shown here) of rigorously washed membranes demonstrated that the fibers maintained their geometry and morphology, and thus their stability in aqueous media. Although this membrane is stable in aqueous media, crosslinking process makes the membrane stiff and brittle. Increase in stiffness is attributed to the formation of random network of cross-links within and across the fiber (at crossover points). To reduce its brittleness and improve its flexibility, a polymer with a low glass transition temperature (Tg) such as, PVDF is added to the electrospinning solution. PVDF has been chosen as it exhibits high hydrophobicity due to the presence of two fluorine atoms in the polymer repeat unit.28,29Representative SEM images of the fabricated PNIPAM/PVDF blend samples are shown in Fig. 2. The average fiber diameter of the blends with 100, 75, 50, 25 and 0 wt% PNIPAM samples are determined to be 1.44 ± 0.41, 1.54 ± 0.05, 1.65 ± 0.18, 1.13 ± 0.24 and 1.46 ± 0.96 μm respectively. As evident from Fig. 2, neat PNIPAM fibers and fibers of PNIPAM/PVDF blend have a smooth morphology consistent with other published work.30 On the contrary, neat PVDF fibers are found to have a rough surface morphology. The presence of micro/nanoscale wrinkles on the surface of PVDF fibers is attributed to low surface tension and electrical repulsion of solution jet containing OG-POSS during fast phase separation.31 We used atomic force microscopy (AFM) to confirm this and further examine the microstructure of PVDF fibers.32 Fig. 3a shows the AFM image of PVDF fibers. It is clear from the AFM image that the surface of the fiber is rough and consists of two distinct phases with aggregates of OG-POSS (darker regions). Thus, the AFM image also confirms the phase separation between OG-POSS and PVDF. This phase separation is caused by a rapid migration of OG-POSS to the surface (see Fig. 3a). The migration is due to the low surface tension of OG-POSS and the reduced cohesive energy between OG-POSS and polymer chain compared to the cohesive energy between polymer chains.33–35
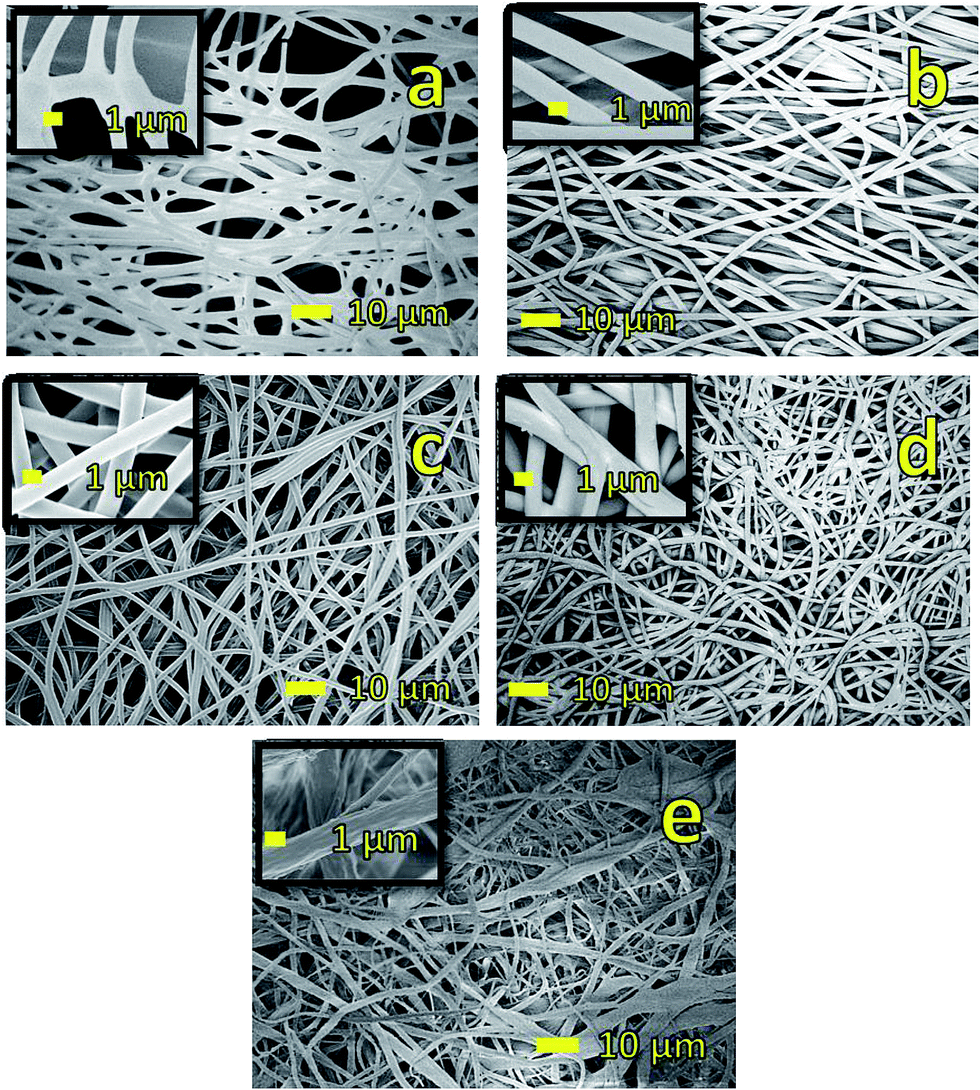 | ||
| Fig. 2 Representative SEM micrographs of the membranes (a) neat PNIPAM fibers. (b) 75/25 (wt/wt)% PNIPAM/PVDF fibers (c) 50/50 (wt/wt)% PNIPAM/PVDF fibers (d) 25/75 (wt/wt)% PNIPAM/PVDF fibers, and (e) neat PVDF–OG-POSS fibers. SEM image in (a) shows fusing of fibers seen after the heat treatment. Figures (b) to (d) shows that fiber have smooth surface morphology and image in (e) shows fibers with rough surface morphology. | ||
 | ||
| Fig. 3 AFM phase images of (a) PVDF fiber; (b) 50 wt% of PNIPAM in PNIPAM/PVDF blend and (c) 25 wt% of PNIPAM in PNIPAM/PVDF blend. Black colored arrow shows the orientation of the fiber length, red arrow points to the dark domains, which in figure (a) is OG-POSS agglomeration; in figures (b) and (c) is PVDF as this domain increases with increasing PVDF content. Green arrow points to the lighter domains, which in figure (a) is PVDF; in figures (b) and (c) is PNIPAM. | ||
Following this, the thermoresponsive behavior of the membrane is investigated by measuring the wettability at room temperature and above the LCST of PNIPAM. For this purpose, the samples are placed in a temperature controlled chamber, that is mounted on the sample stage in front of the high speed camera. A 5 μl water droplet is placed on the surface of the membrane. The image of this droplet is captured at a speed of 2 frames per second (FPS) at room temperature and 0.5 FPS at an elevated temperature (40 °C). The contact angle (CA) is measured using an open source software, FIJI ImageJ (dropcast plugin) and is plotted as a function of time as shown in Fig. 4. Fig. 4a shows the CA measurements at ambient temperature, while Fig. 4b shows the CA measurements at an elevated temperature (40 °C). Fig. 4a and b show that neat PNIPAM membranes are hydrophilic at room temperature and relatively hydrophobic at elevated temperature, as expected of a thermoresponsive membrane.24 For example, contact angle measured on neat PNIPAM after 30 seconds time interval at room temperature and elevated temperature is ∼30° and ∼81° respectively. This CA is lower than what we reported in our earlier work on PNIPAM membranes that are obtained without the addition of cross-linking agent. In our previous work, high CA of 132 °C is attributed to the large surface area of the fibrous membrane.24,36 In the present work, measurements are performed on crosslinked PNIPAM. The reason for reduced CA is attributed to the decrease in surface roughness resulting from the fusing of fibers at the cross-over points in a membrane (Fig. 2a). The fiber fusing occurred due to crosslinking process. This is because the crosslinking temperature (150 °C) is higher than the glass transition temperature of PNIPAM (141.45 °C).37
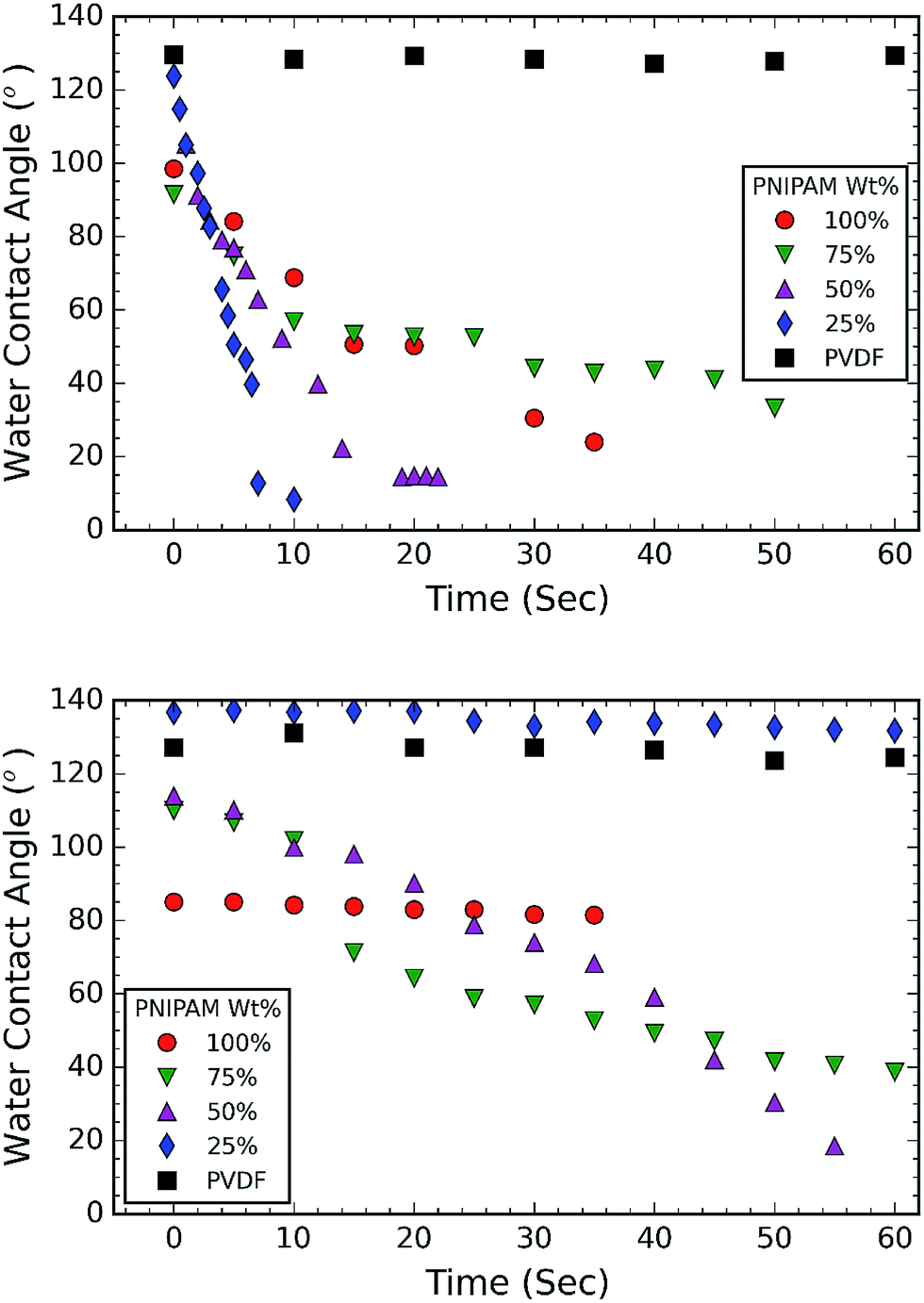 | ||
| Fig. 4 Water contact angle (WCA) trend of membranes at (a) room temperature; and (b) elevated temperature (40 °C). The plots show that water spreads faster with increase in PVDF content during first 10 seconds. At elevated temperature, PNIPAM is hydrophobic and hence there is an increase in WCA. Membrane consisting of 25 wt% PNIPAM shows improved thermoresponsive wettability. | ||
The CA measurements at ambient temperature (Fig. 4a) suggest that all the membranes with PNIPAM are hydrophilic (CA < 90°). It is evident from Fig. 4a that 50 wt% and 25 wt% PNIPAM are more hydrophilic compared to other samples. Firstly, 25 wt% PNIPAM sample is super hydrophilic takes only 10 seconds to absorb a water drop. The contact angle on 50 wt% and 75 wt% PNIPAM samples is measured to be 52.16° and 56.9° respectively. The contact angle PNIPAM after 10 seconds is measured to be 68.76°. The CA measurements at elevated temperature (Fig. 4b) also show the same trend for 75 wt% and 50 wt% PNIPAM, albeit at a slower rate. After a period of 10 seconds, both 75 wt% and 50 wt% PNIPAM samples exhibit a CA of 100°. On the contrary, 25 wt% PNIPAM is stable around 136°.
The CA measurements reveal that PNIPAM/PVDF composite fibrous membranes show thermoresponsive behavior. This is attributed to the surface segregation of PNIPAM. Similar segregation effect of PNIPAM is reported in the literature. For example, Chen et.al. used electrospinning to fabricate PNIPAM/polycaprolactone blends and demonstrated that the thermoresponsive property is due to migration of the PNIPAM towards the surface of the fibers.38 The kinetic factor governs the mobility of polymeric chains in order to attain the state of minimum energy. Herein, the lower density polymer (PNIPAM) migrates towards the region of high shear, i.e. towards the surface of the fibers.38,39 Our AFM images also confirm this and demonstrate that presence of different phases on the surface of the fibers (Fig. 3).
TGA and DSC are used to investigate the phase separation of PVDF and PNIPAM in the samples. TGA and DSC scans of the samples are shown in Fig. 5 and 6 respectively. It is evident from Fig. 5a that the onset degradation temperature of neat PNIPAM and neat PVDF membrane is 349.4 °C and 438.6 °C respectively. However, the temperature at which maximum rate of degradation occurs is obtained from the derivative of the weight with temperature (Fig. 5b). It shows that maximum rate of degradation for neat PNIPAM and neat PVDF occurs at 398.7 °C and 449.04 °C respectively. 25 wt% PNIPAM shows two-step degradation, and the samples with 50 wt% and 75 wt% PNIPAM shows single step. The first degradation peak matches with the degradation peak of neat PNIPAM sample, while the other degradation peak in case of 25 wt% PNIPAM is closer to the degradation peak of neat PVDF. This indicates phase separation at the molecular level for 25 wt% PNIPAM.40 To confirm this behavior, samples with lower PNIPAM content, such as 20 and 10 wt% were tested for thermal degradation (not shown here). These samples also showed a two-step degradation. Thus, TGA profile of PNIPAM/PVDF polymeric blend suggests that above 25 wt% of PNIPAM, the PVDF and PNIPAM are miscible at the molecular level. DSC is also used to investigate the effect of PNIPAM on the melting temperature of PVDF. The melting peak of PVDF is determined to be 161.6 °C. The degree of crystallinity of PVDF as calculated from eqn (1) reduces with increasing PNIPAM content in the composite membrane (Fig. 6b). Therefore, atactic nature of PNIPAM interferes with close packing and semi crystalline nature of PVDF.41 This interference with crystallization of PVDF also explains that the degradation of blends occurring at a relatively lower temperature than pristine PVDF. Thus, the evidence from DSC and TGA suggests that, PNIPAM and PVDF are miscible when the PNIPAM concentration is 50 or 75 wt%. Whereas PVDF and PNIPAM are partially miscible when PNIPAM concentration is 25 wt% or less. Yet another factor that changes with increasing PNIPAM is the amount of moisture a membrane can absorb. This is depicted by the first broad peak less than 100 °C and is attributed to the moisture loss (non-liquid form) from the sample. Moisture loss is confirmed by the absence of first broad peak during the second run on DSC (curve not shown here), while maintaining the melting peak around 160 °C. The moisture sorption property of the composite membranes is analyzed in detail and are discussed during the later sections of this paper.
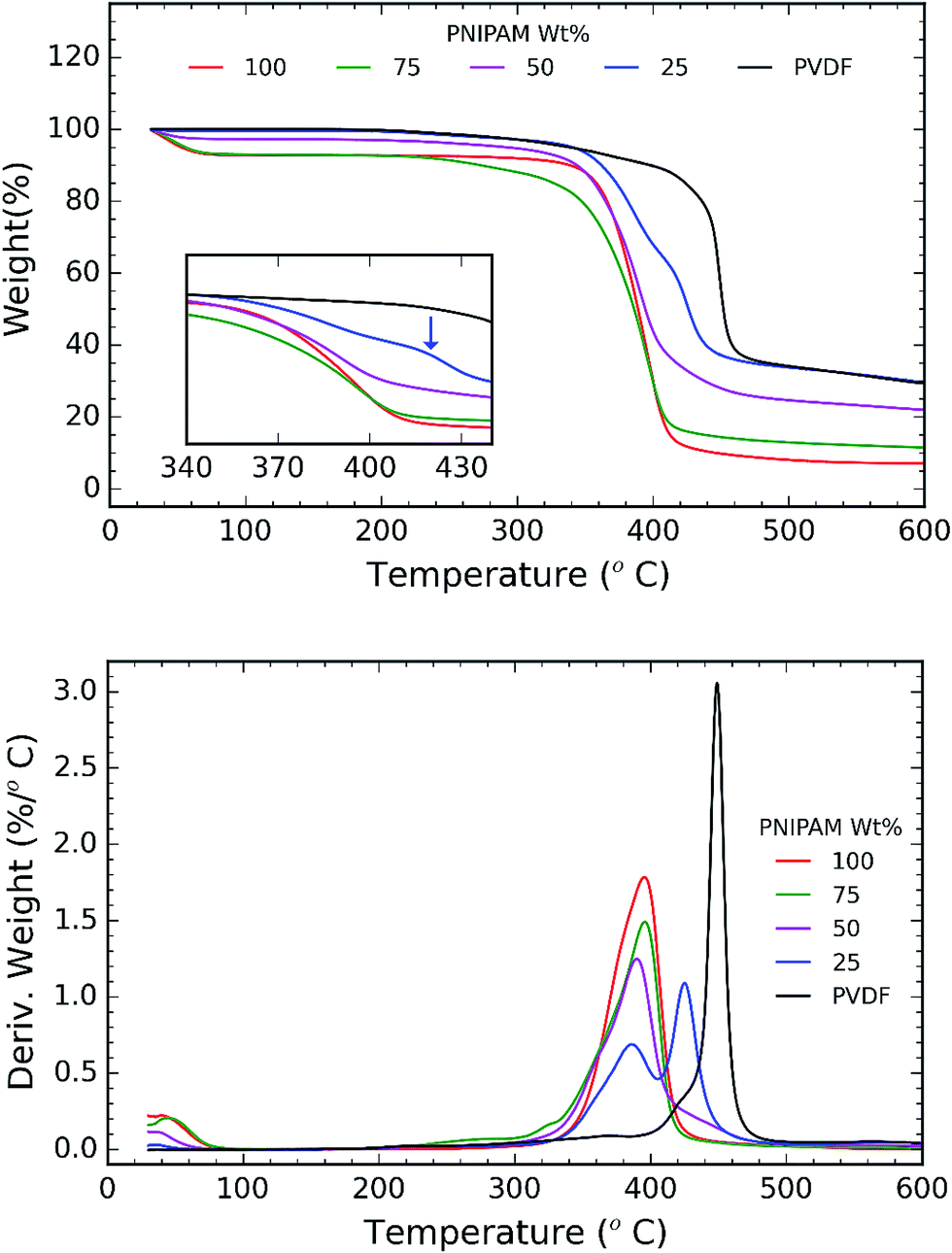 | ||
| Fig. 5 (a) TGA plot of weight% vs. temperature. Inset shows a two step degradation and is indicated by a blue arrow; and (b) TGA plot of derivative of weight (%/°C) vs. temperature. The plot shows two step degradation for samples consisting of 25 wt% PNIPAM indicating phase separation. | ||
 | ||
| Fig. 6 (a) DSC heating scans recorded for samples as a function of PNIPAM content (run 1); and (b) plot of crystallinity% vs. PNIPAM content in the samples. | ||
Exceptional wettability of 25 wt% PNIPAM sample can be explained using the results from DSC and TGA. Molecular interaction between the PVDF and PNIPAM phases is understood using DSC and TGA results. The presence of two peaks in the plot of weight derivation vs. temperature for 25 wt% PNIPAM samples indicates phase separation between PVDF and PNIPAM (see Fig. 5b). This results in the presence of distinct large domains of PNIPAM and PVDF. The AFM phase image further confirms the presence of these domains (Fig. 3b and c). The phase image of 25 wt% sample shows larger domain of dark regions when compared with the 50 wt% PNIPAM sample. The push pull effect by PVDF and PNIPAM domains cause the water drop to spread faster at room temperature. However, at elevated temperature the PNIPAM domains resist wettability due to LCST behavior and is simultaneously supported by hydrophobicity of PVDF.
Fig. 7a shows the FTIR spectra recorded for the samples. FTIR spectrum of pure PVDF nanofiber membranes shows the stretching in beta phase at 510 and 840 cm−1. The band at 1166 cm−1 is associated with symmetrical stretching of CF2 group. The band at 1236 cm−1 is attributed to the gamma phase, while the band at 1400 cm−1 is assigned to in-plane bending or scissoring of the CH2 group. Incorporation of PNIPAM with the composites results in the merging of the FTIR peaks of both the polymers.21 FTIR spectrum of pure PNIPAM and PNIPAM/PVDF blend membranes show the characteristic peaks associated with PNIPAM. The peak at 3285 cm−1 is attributed to the secondary amide N–H stretching, while the peak at 2974 cm−1 is associated with –CH3 asymmetric stretching. The peaks at 1635 cm−1 and 1540 cm−1 are associated with secondary amide carbonyl stretching amide I and amide II respectively. The isopropyl stretching absorption is associated with the peak seen around 1380 cm−1. Furthermore, the absorption intensities of CO stretching and N–H stretching decreases as the amount of PVDF in the mixture increases, confirming that the PNIPAM increases with increasing ratio of PNIPAM/PVDF in the mixture. Fig. 7b shows the effect of crosslinking in PNIPAM. The disappearance of the peak at 910 cm−1, which is attributed to the opening of the carbonyl ring in OG-POSS during crosslinking.13
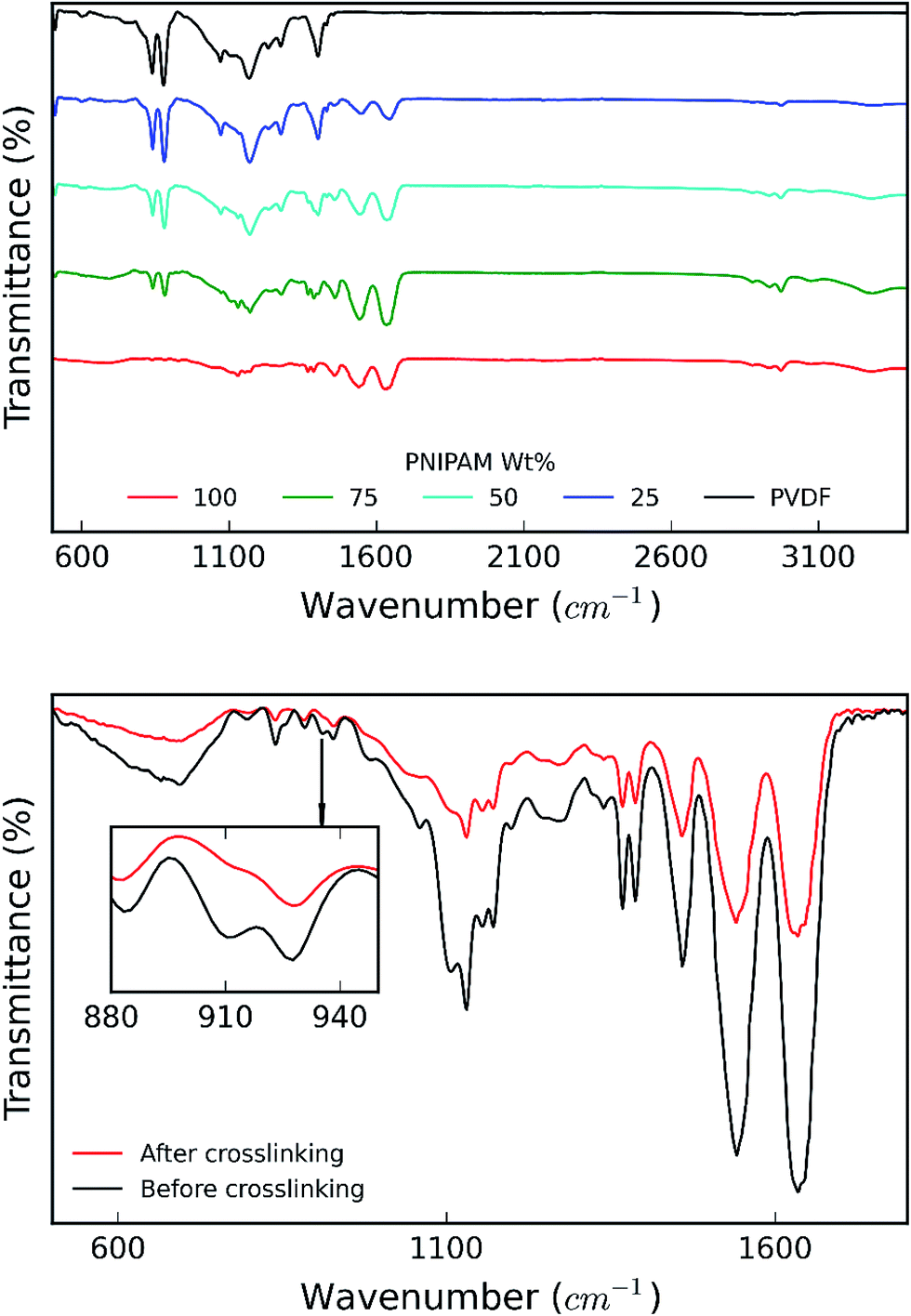 | ||
| Fig. 7 (a) IR spectra obtained for samples as a function of PNIPAM content in the samples; and (b) IR spectra for PNIPAM before and after crosslinking. | ||
In the next step, we investigated the ability of the membranes to absorb moisture from humid atmosphere. For this purpose, the samples are subjected to air with controlled humidity using the setup as shown in Fig. 1. The relative humidity (RH) is controlled by varying the ratio of dry and humid air while maintaining constant air flow of 60 sccm. We observed that the weight of the membranes changes with humidity due to moisture absorption. The supplied RH and corresponding relative weight of the PNIPAM membrane are illustrated in Fig. 8a and b, respectively. The membranes with 50 and 25 wt% of PNIPAM were studied under the same conditions. Fig. 9a and b respectively compares the desorption and absorption behavior of the blend and pristine membranes. We found that the water absorption ability of the membrane increases with PNIPAM content, reaching a maximum value of 10% for the pristine PNIPAM. This can be explained by the hydrophilicity of PNIPAM which governs the overall moisture absorption ability of the polymeric blend. At the same time, we see that the pristine PVDF absorbs less than one percent of moisture as expected of a hydrophobic polymer.28,29 Furthermore, the close packing of semi-crystalline regions of PVDF prevents diffusion of the water vapor molecules in the fiber.42 Therefore, PVDF provides a stable framework to support PNIPAM to exhibit its sorption properties. This also suggests that by controlling the PNIPAM/PVDF ratio in the blend, the moisture absorption of a blend membrane can be tuned for the targeted applications.
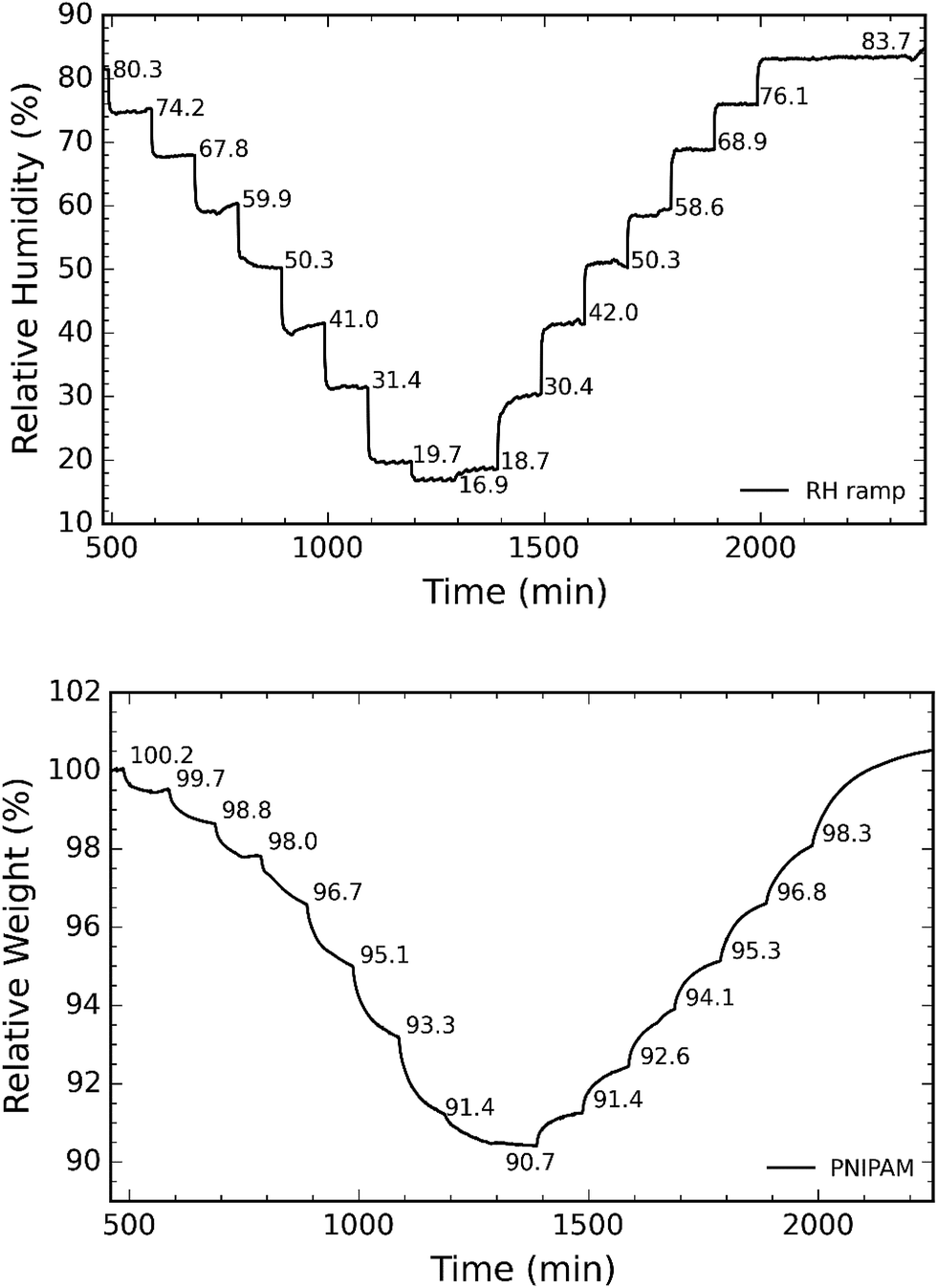 | ||
| Fig. 8 (a) Representative curve of RH as a function of time; and (b) corresponding relative weight of sample as recorded by the TGA. | ||
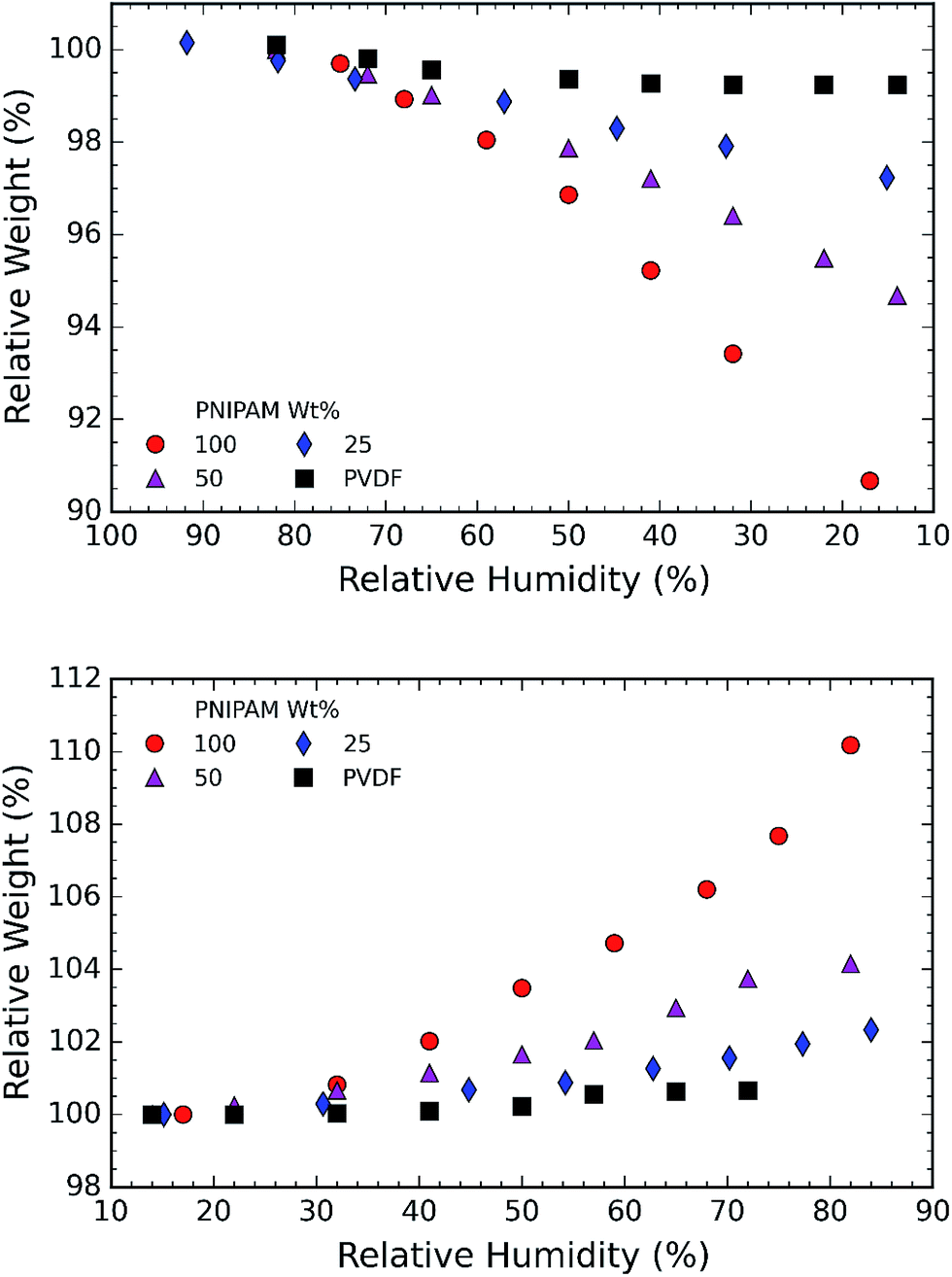 | ||
| Fig. 9 (a) Plot of relative weight% vs. relative humidity%. The plot demonstrates water desorption behavior of the membranes with the reduction in RH%; (b) plot of relative weight% vs. relative humidity%. The plot demonstrates water absorption behavior of the membranes with the increase in RH%. The water absorbed in the samples in seen to be higher for samples containing higher PNIPAM concentration. | ||
4 Conclusion
We investigated the switchable wettability behavior of poly-N-(isopropyl acrylamide) (PNIPAM) and poly-(vinylidene fluoride) (PVDF) blend membranes that were fabricated using electrospinning. The fibrous membranes were fabricated by varying the concentration of PNIPAM in the blend. The wettability results demonstrated that all the PVDF/PNIPAM blend samples switched from hydrophilic to hydrophobic state when the temperature was increased from room temperature to 40 °C. In particular, blend samples with 25 wt% PNIPAM is 25 seconds faster in absorbing water at ambient temperature than PNIPAM and 40% more hydrophobic at elevated temperatures. The exceptional behavior of the membranes with 25 wt% PNIPAM is attributed to the phase separation of the polymers. At the room temperature, the hydrophilic PNIPAM results in regions with high surface energy and PVDF regions possess low surface energy. This kind of polymer distribution prevents a water droplet from attaining stability and forces it to spread. However, at an elevated temperature, relative hydrophobicity of PNIPAM along with PVDF reduces the membrane surface energy and increases the water contact angle.The chemical analysis of the membranes using FTIR results confirms that the polymers do not react with each other. Furthermore, it is supported by hygroscopic tests which depend on the hydrophilic properties of material. The hygroscopic property of the membranes increases in proportion with PNIPAM content. Thus the blend membrane with 25 wt% of PNIPAM absorbs 2.5 wt% of moisture at 85% relative humidity and 25 °C as against 10 wt% of moisture by pristine PNIPAM. In conclusion, the blending of PNIPAM and PVDF in a ratio of 25/75 enhances the surface wettability, while maintaining the number of hydrophilic/hydrophobic sites in the bulk of the membrane.
Acknowledgements
A. B. would like to acknowledge the financial support from Ministry of Education (MOE – Tier 2), Singapore (Grant No. T2-MOE-1302). The authors would also like to thank Ms Shiho Moriguchi from Shimadzu Corp. Japan and Mr Tan Teck Beng from Shimadzu (Asia Pacific) Pte Ltd for their support with integrated microscope (OLS4500).References
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- A. Kikuchi, M. Okuhara, F. Karikusa, Y. Sakurai and T. Okano, J. Biomater. Sci., Polym. Ed., 1998, 9, 1331–1348 CrossRef CAS PubMed.
- L. K. Ista, V. H. Perez-Luna and G. P. Lopez, Appl. Environ. Microbiol., 1999, 65, 1603–1609 CAS.
- D. Wandera, S. R. Wickramasinghe and S. M. Husson, J. Membr. Sci., 2010, 357, 6–35 CrossRef CAS.
- F. Guo and Z. Guo, RSC Adv., 2016, 6, 36623–36641 RSC.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 2, 1441–1455 CrossRef CAS.
- H. Chen and Y. Hsieh, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6331–6339 CrossRef CAS.
- S. H. Kim, S.-H. Kim, S. Nair and E. Moore, Macromolecules, 2005, 38, 3719–3723 CrossRef CAS.
- L. Li and Y.-L. Hsieh, Nanotechnology, 2005, 16, 2852 CrossRef CAS.
- Y. Ji, K. Ghosh, B. Li, J. C. Sokolov, R. A. Clark and M. H. Rafailovich, Macromol. Biosci., 2006, 6, 811–817 CrossRef CAS PubMed.
- H. Liu, M. Zhen and R. Wu, Macromol. Chem. Phys., 2007, 208, 874–880 CrossRef CAS.
- J. Wang, A. Sutti, X. Wang and T. Lin, Soft Matter, 2011, 7, 4364–4369 RSC.
- P. Slemming-Adamsen, J. Song, M. Dong, F. Besenbacher and M. Chen, Macromol. Mater. Eng., 2015, 300, 1226–1231 CrossRef CAS.
- S. Choi, J. Hong, Y. Seo, S. Chung and C. Nah, J. Appl. Polym. Sci., 2006, 101, 2333–2337 CrossRef CAS.
- S.-R. Huang, K.-F. Lin, T.-M. Don, W.-Y. Chiu and M.-F. Lin, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2152–2162 CrossRef CAS.
- L. Ying, E. Kang, K. Neoh, K. Kato and H. Iwata, J. Membr. Sci., 2004, 243, 253–262 CrossRef CAS.
- L. Ying, E. Kang and K. Neoh, J. Membr. Sci., 2003, 224, 93–106 CrossRef CAS.
- L. Ying, E. Kang and K. Neoh, Langmuir, 2002, 18, 6416–6423 CrossRef CAS.
- Y. Li, L.-Y. Chu, J.-H. Zhu, H.-D. Wang, S.-L. Xia and W.-M. Chen, Ind. Eng. Chem. Res., 2004, 43, 2643–2649 CrossRef CAS.
- X. Chen, C. Shi, Z. Wang, Y. He, S. Bi, X. Feng and L. Chen, Polym. Compos., 2013, 34, 457–467 CrossRef CAS.
- D. H. Reneker and I. Chun, Nanotechnology, 1996, 7, 216 CrossRef CAS.
- H. Okuzaki, K. Kobayashi and H. Yan, Synth. Met., 2009, 159, 2273–2276 CrossRef CAS.
- A. Sargur Ranganath, V. Anand Ganesh, K. Sopiha, R. Sahay and A. Baji, MRS Adv., 2016, 1–6 Search PubMed.
- W. Zhou, A. Bahi, Y. Li, H. Yang and F. Ko, RSC Adv., 2013, 3, 11614–11620 RSC.
- T. He, W. Zhou, A. Bahi, H. Yang and F. Ko, Chem. Eng. J., 2014, 252, 327–336 CrossRef CAS.
- J. Liu, X. Lu and C. Wu, Membranes, 2013, 3, 389–405 CrossRef PubMed.
- A. J. Lovinger, in Poly(vinylidene fluoride), Springer, 1982, pp. 195–273 Search PubMed.
- D. W. Van Krevelen and K. Te Nijenhuis, Properties of polymers: their correlation with chemical structure; their numerical estimation and prediction from additive group contributions, Elsevier, 2009, ch. 18, pp. 689–691 Search PubMed.
- F. Song, X.-L. Wang and Y.-Z. Wang, Colloids Surf., B, 2011, 88, 749–754 CrossRef CAS PubMed.
- Y. Li, Z. Zhu, J. Yu and B. Ding, ACS Appl. Mater. Interfaces, 2015, 7, 13538–13546 CAS.
- C. Lim, E. Tan and S. Ng, Appl. Phys. Lett., 2008, 92, 141908 CrossRef.
- Y. Tang and M. Lewin, Polym. Adv. Technol., 2009, 20, 1–15 CrossRef CAS.
- S.-W. Kuo and F.-C. Chang, Prog. Polym. Sci., 2011, 36, 1649–1696 CrossRef CAS.
- A. M. M. Sousa, H. K. S. Souza, J. Uknalis, S.-C. Liu, M. P. GonÃğalves and L. Liu, Carbohydr. Polym., 2015, 115, 348–355 CrossRef CAS PubMed.
- V. A. Ganesh, A. S. Ranganath, R. Sridhar, H. K. Raut, S. Jayaraman, R. Sahay, S. Ramakrishna and A. Baji, Macromol. Rapid Commun., 2015, 36(14), 1368–1373 CrossRef CAS PubMed.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, V. K. Tiwari, B. Maiti, P. Maiti, M. Kamigaito, Y. Okamoto and B. Ray, Macromolecules, 2011, 44, 5822–5824 CrossRef CAS.
- M. Chen, M. Dong, R. Havelund, V. R. Regina, R. L. Meyer, F. Besenbacher and P. Kingshott, Chem. Mater., 2010, 22, 4214–4221 CrossRef CAS.
- Q. Shi, J. Hou, X. Xu, J. Gao, C. Li, J. Jin, S. Wong and J. Yin, Adv. Mater. Interfaces, 2015 Search PubMed.
- H. Li and H. Kim, Desalination, 2008, 234, 9–15 CrossRef CAS.
- S. O. Han, W. K. Son, D. Cho, J. H. Youk and W. H. Park, Polym. Degrad. Stab., 2004, 86, 257–262 CrossRef CAS.
- W. E. Morton and J. W. S. Hearle, in 12-Theories of moisture sorption, Woodhead Publishing, 2008, pp. 243–273 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |