DOI:
10.1039/C7RA02237A
(Paper)
RSC Adv., 2017,
7, 22699-22705
Design and synthesis of uracil urea derivatives as potent and selective fatty acid amide hydrolase inhibitors†
Received
23rd February 2017
, Accepted 17th April 2017
First published on 26th April 2017
Abstract
Fatty acid amide hydrolase (FAAH) is one of the key enzymes involved in the biological degradation of endocannabinoids, especially anandamide. Pharmacological blockage of FAAH restores the levels of endocannabinoids, providing therapeutic benefits in the management of inflammation, depression and multiple sclerosis. In this study, a series of uracil urea derivatives as FAAH inhibitors were designed and synthesized. Structural modifications at the C5 position and side chain of N-hexyl-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-carboxamide (1a) led to FAAH inhibitors with improved potency and selectivity. Structure–activity relationship (SAR) studies indicated that C5 electron-withdrawing substituents were preferred for optimal potency but not for selectivity, whereas replacement of the alkyl chain with phenylalkyl moieties or biphenyl groups significantly improved both inhibitory potency and selectivity towards FAAH. Two highly potent picomolar FAAH inhibitors (4c, IC50 = 0.3 ± 0.05 nM; 4d, IC50 = 0.8 ± 0.1 nM) were developed. Compound 4c inhibited FAAH in a rapid, selective, noncompetitive, and irreversible pattern. This study provides several highly potent and selective FAAH inhibitors and an optimized chemical scaffold for the development of FAAH inhibitors. We anticipate that these FAAH inhibitors will enable new possibilities in understanding FAAH functions and development of therapeutics for pain and inflammatory diseases.
1. Introduction
Fatty acid amide hydrolase (FAAH) is a member of the amidase signature family, responsible for the inactivation of endogenous cannabinoids, especially anandamide (AEA).1–3 AEA modulates a wide range of physiological processes through the interaction with G protein-coupled CB1 and CB2 cannabinoid receptors. Genetic and pharmacological blocking of FAAH leads to increased endogenous AEA levels without the undesirable “cannabinoid effects” caused by cannabinoid receptor agonists.4–6 Therefore, restoring the endogenous AEA by FAAH inhibition has become an alternative therapeutic strategy for inflammation, cancer, and neuropathic diseases.5,7–11
So far, there are two main categories of FAAH inhibitors that have been developed.12–14 First, reversible inhibitors, such as the α-ketoheterocycle-based inhibitors, bind to the active serine residue of FAAH with a reversible hemiketal bond. OL-135 is the most well-studied potent and reversible FAAH inhibitor.4,15,16 Second, irreversible inhibitors, including aryl carbamates,17–20 ureas21–25 and sulfonyl fluoride analogues,26 which form covalent binding with the catalytic site of FAAH, e.g., URB597.27 Administration of URB597 to rats elevated endogenous AEA levels and exhibited anxiolytic effects in animal models.5,28 Additionally, benzothiazole,29 β-lactam,30 (thio)hydantoins31 and some nonsteroidal anti-inflammatory drugs (NSAIDs)32–34 also have been reported to inhibit FAAH.
Although the overall physiological role of FAAH has been reported, it is difficult to distinguish the function of FAAH in each specific tissue and organ. The reason is the wide distribution of FAAH in the whole body and lack of suitable topical FAAH inhibitors. Most of current FAAH inhibitors have low blood clearance and readily distributed and inhibit FAAH throughout the body after treatment. They are suitable for the systemic inhibition of FAAH, but it's difficult to obtain targeted FAAH inhibition in the specific tissues without inhibiting FAAH in other non-targeted sites. Given the critical pharmacological properties of FAAH, understanding the function of FAAH in different tissues is of great significance. FAAH inhibitors suitable for the topical administration is highly desired to explore functions of FAAH in different tissues and organs.
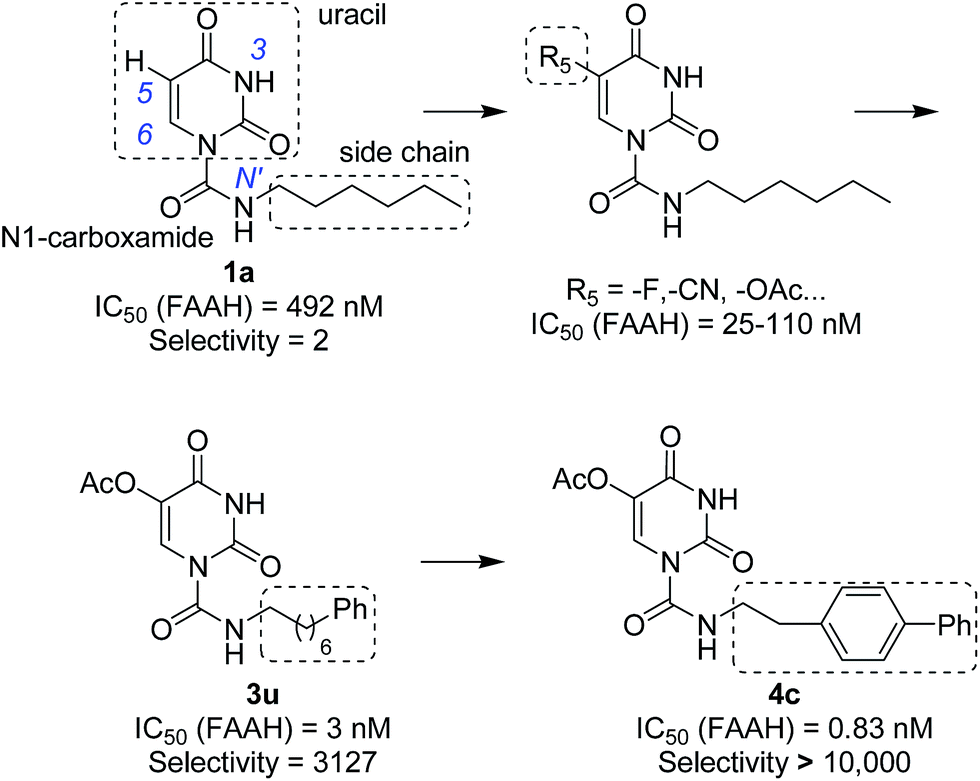
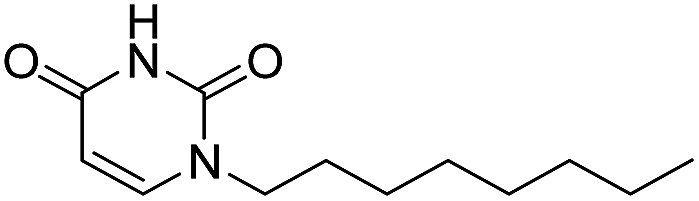
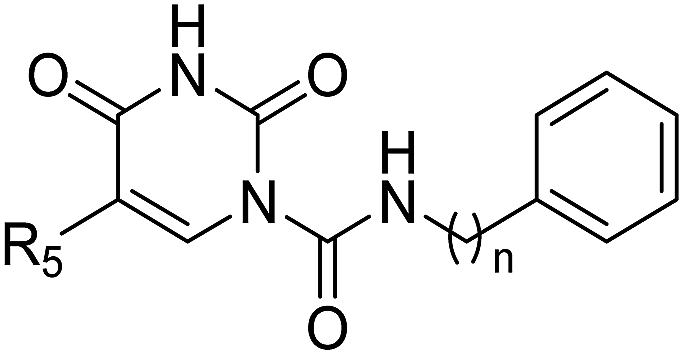
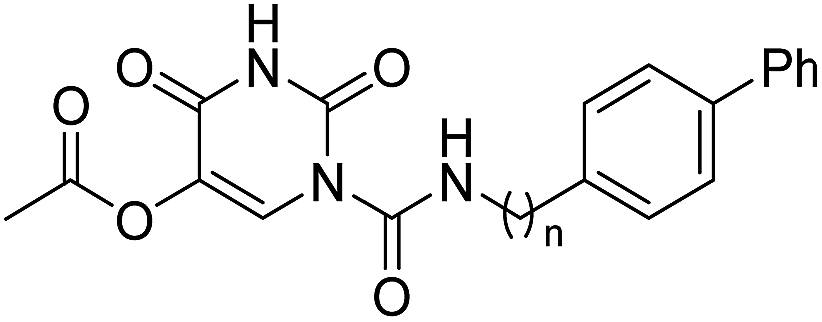
In our previous study,35 we found that uracil urea 1a (Fig. 1), an acid ceramidase (AC) inhibitor (IC50 = 426 ± 104 nM),36 was also a potent FAAH (IC50 = 492 ± 53 nM) and N-acylethanolamine-hydrolysing acid amidase (NAAA) inhibitor (IC50 = 990 ± 170 nM). To develop FAAH inhibitors with properties suitable for topical FAAH inhibition and improve the selectivity of 1a towards FAAH, we modified the uracil electronic properties and the side chain conformation of 1a. A series of uracil urea derivatives designed as selective FAAH inhibitors were synthesized and characterized. The results showed that modification of 1a targeting the uracil ring improved the inhibition potency towards FAAH, and the side chain replacement with phenylalkyl moieties or biphenyl groups enhanced both potency and selectivity (Fig. 1). Several highly potent and selective FAAH inhibitors were reported, including two picomolar inhibitor, 4c (IC50 = 0.3 ± 0.05 nM) and 4d (IC50 = 0.8 ± 0.1 nM), a series of nanomolar inhibitors, e.g., 3f (IC50 = 4 ± 0.9 nM), 3u (IC50 = 3 ± 0.4 nM), 4a (IC50 = 1.1 ± 0.2 nM) and 4b (IC50 = 8.3 ± 1.2 nM).
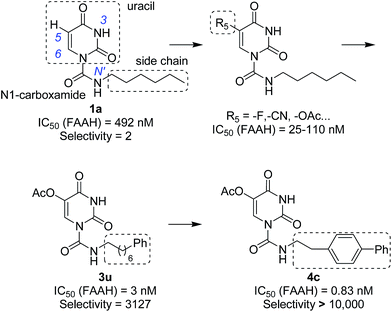 |
| | Fig. 1 The design strategies of uracil urea derivatives as highly potent and selective FAAH inhibitors. The selectivity represents the selectivity of uracil urea derivatives towards FAAH over NAAA. | |
2. Chemistry
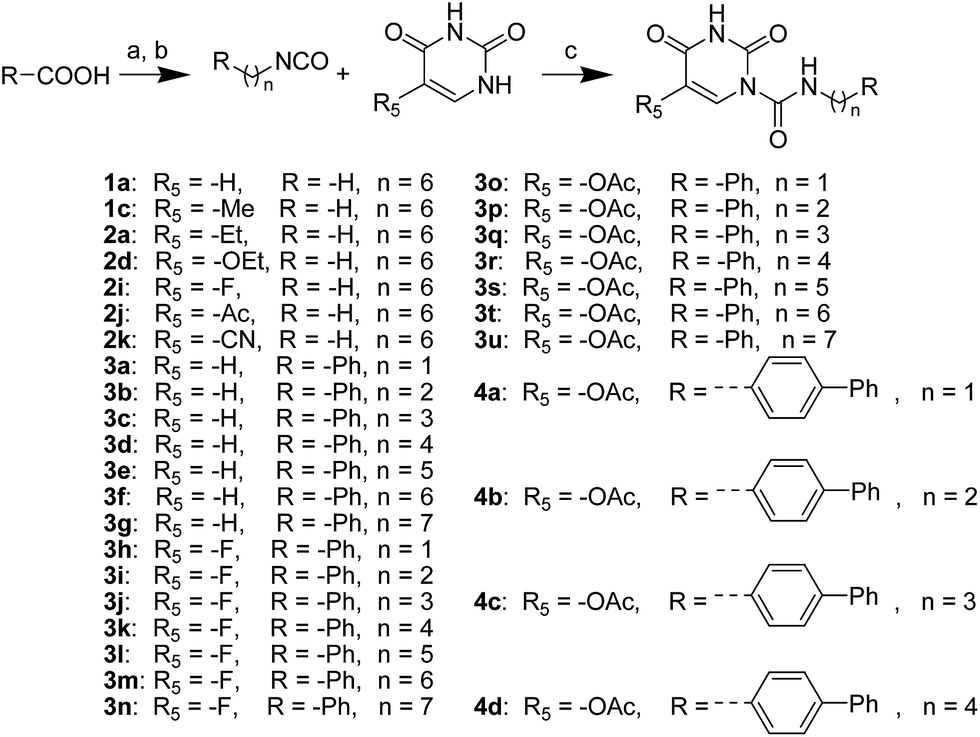
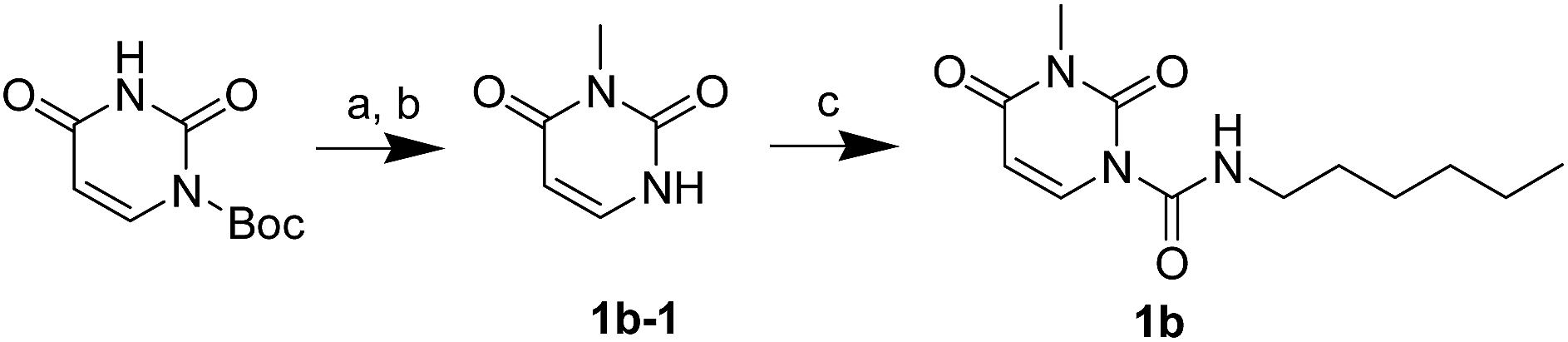
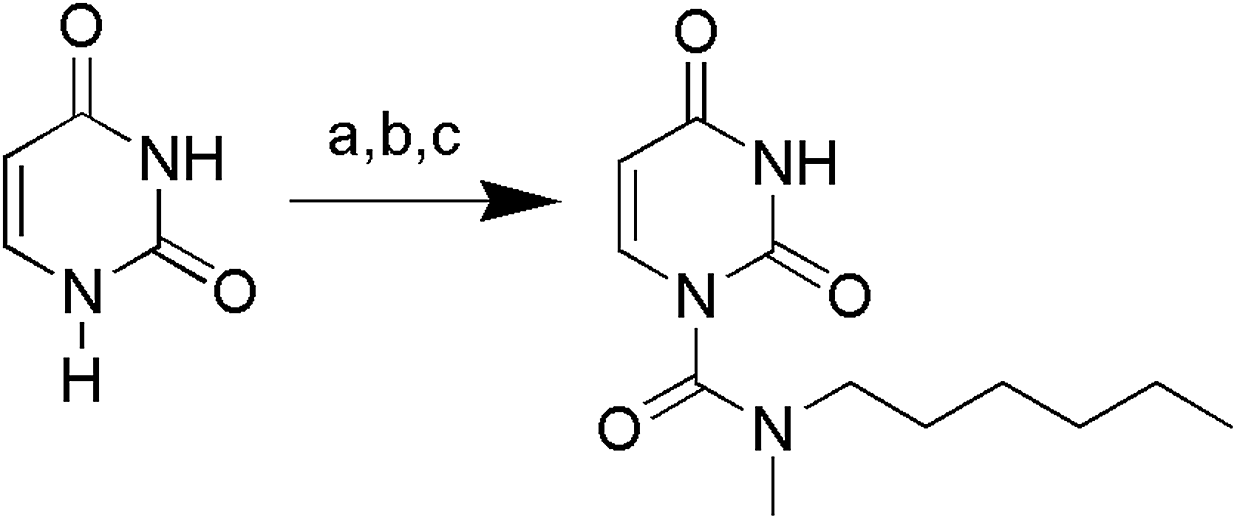
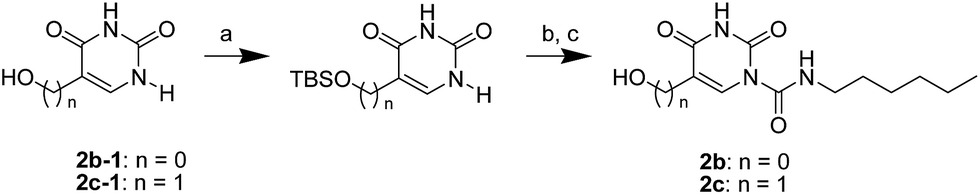
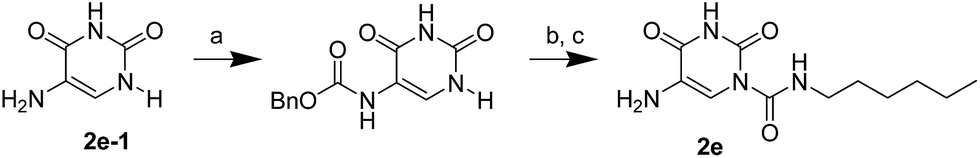
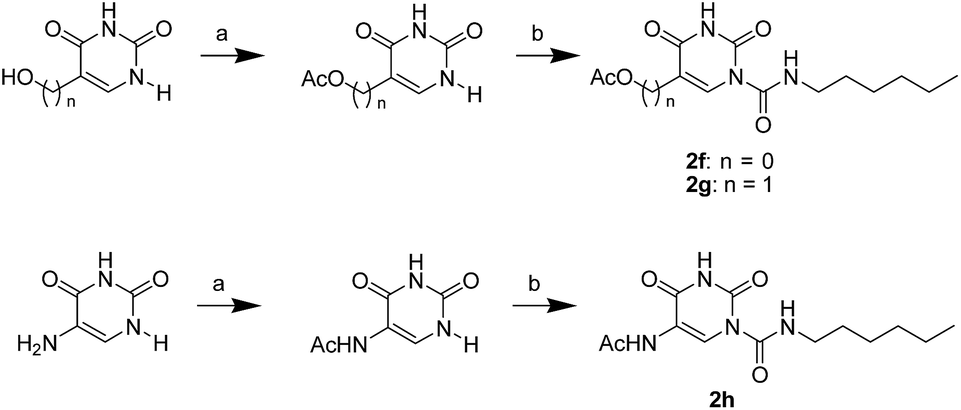
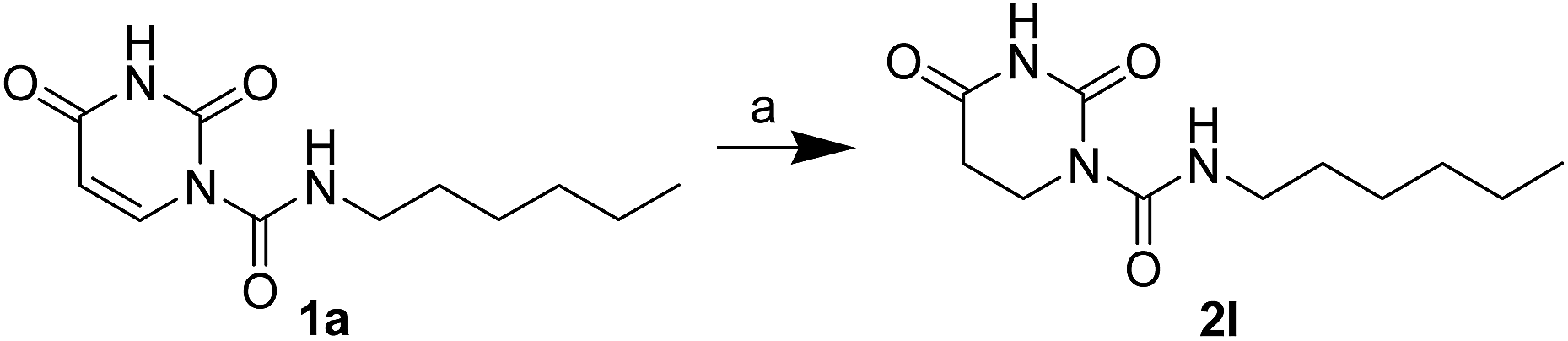
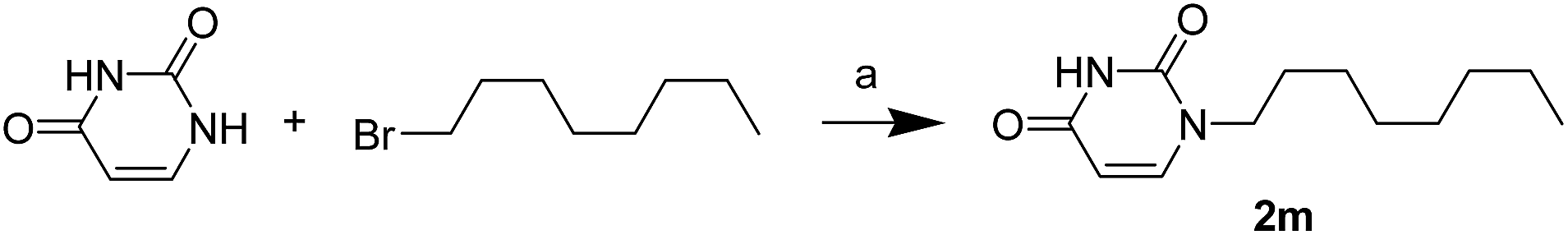
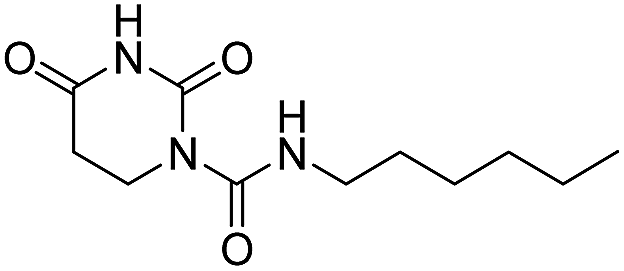
The uracil ureas 1a, 1c, 1d, 2a, 2d, 2i–2k, 3a–3u, 4a–4d were prepared by the reaction of isocyanates with 5-substituted uracils following a reported procedures with minor modification.36 Isocyanates were synthesized via Curtius rearrangement of appropriate acylazides prepared from successive reactions of the corresponding acid with oxalyl chloride ((COCl)2) and sodium azide (NaN3) (Scheme 1).20 1b and 1e were synthesized following a literature procedure.36 1b was prepared by the reaction of hexyl isocyanate with 3-methyl substituted uracil 1b-1. 1b-1 was obtained by reaction of N-Boc uracil and iodomethane (MeI), and then deprotection with HCl in MeOH (Scheme 2). 1e was synthesized by reaction of uracil with triphosgene followed by treatment with N-methylhexanamine (Scheme 3). 2b and 2c were prepared with 3-step procedure starting from 2b-1 and 2c-1. Compound 2b-1 and 2c-1 were protected by t-butyl di-methylsilyl chloride (TBSCl) and directly used in the carbamoylation reaction, followed by deprotection with tetrabutylammonium fluoride (TBAF) (Scheme 4). 5-Amino uracil (2e-1) protected with benzyl carbonochloridate (CbzCl) in pyridine was directly used in the carbamoylation reaction, and followed by deprotection with trimethylsilyl iodide (TMSI) to afford 2e (Scheme 5). 2f–2h were synthesized by treatment of the uracil derivatives with acetic anhydride (Ac2O) followed by reaction with isocyanates (Scheme 6). 2l was prepared by catalytic hydrogenation of 1a (Scheme 7). 2m was obtained by reaction of 1-bromooctane with uracil (Scheme 8).
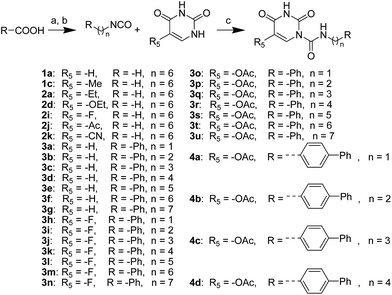 |
| | Scheme 1 The synthesis of compounds 1a, 1c, 2a, 2d, 2i–2k, 3a–3u, 4b–4d. Reagents and conditions: (a) (COCl)2, cat. DMF, CH2Cl2, then NaN3 in H2O, 0 °C; (b) toluene, reflux; (c) DMAP, pyridine, 60 °C. | |
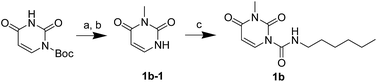 |
| | Scheme 2 The synthesis of compound 1b. Reagents and conditions: (a) MeI, Cs2CO3, THF, room temperature; (b) HCl, CH3OH, room temperature; (c) hexyl isocyanate, DMAP, pyridine, 60 °C. | |
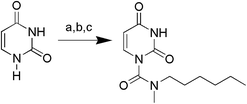 |
| | Scheme 3 The synthesis of compound 1e. Reagents and conditions: (a) NaH, triphosgene, THF, room temperature; (b) HCl, CH3OH, room temperature; (c) DMAP, pyridine, N-methylhexylamine, room temperature. | |
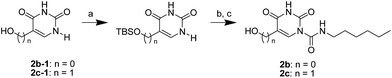 |
| | Scheme 4 The synthesis of compounds 2b and 2c. Reagents and conditions: (a) pyridine, TBSCl, room temperature; (b) hexyl isocyanate, DMAP, pyridine, 60 °C; (c) TBAF, THF, room temperature. | |
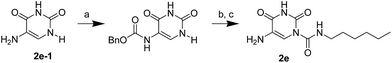 |
| | Scheme 5 The synthesis of compound 2e. Reagents and conditions: (a) pyridine, CbzCl, room temperature; (b) hexyl isocyanate, DMAP, pyridine, 60 °C; (c) TMSI, THF, room temperature. | |
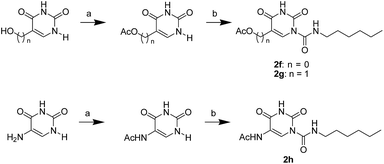 |
| | Scheme 6 The synthesis of compounds 2f, 2g and 2h. Reagents and conditions: (a) pyridine, Ac2O, room temperature; (b) hexyl isocyanate, DMAP, pyridine, 60 °C. | |
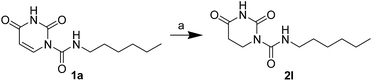 |
| | Scheme 7 The synthesis of compound 2l. Reagents and conditions: (a) 10% Pd/C, H2, CH3OH, room temperature. | |
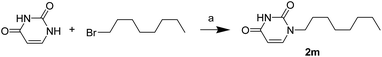 |
| | Scheme 8 The synthesis of compound 2m. Reagents and conditions: (a) K2CO3, DMF, room temperature. | |
3. Enzyme assay
The inhibition activity of new compounds against rat FAAH (rFAAH) and rat NAAA (rNAAA) was tested, using [3H]-AEA and d4-PEA, respectively, as substrates. The residual hydrolysis products of the substrates were determined by liquid chromatography-mass spectrometer (LC-MS).
4. Results and discussion
4.1. SAR study
It has been reported that carbamate and urea FAAH inhibitors interact with the nucleophilic Ser241 of FAAH through the electrophilic carbonyl group, and most of these inhibitors contain O-aryl or N-aryl moiety as leaving group. We speculated that the activity of uracil urea inhibitors may dependent on the presence of N1-carboxamide as the electrophilic carbonyl group and uracil as leaving group. Accordingly, two different chemical modifications were used to optimize the potency and selectivity of uracil urea derivatives. First, inspired by existed urea FAAH inhibitors, we replaced the uracil ring with chemical groups with different electronic properties to increase the leaving group ability of uracil. Second, we modified the side chain with phenyl-containing groups that can mimic the conformation and π-unsaturation of double bonds of anandamide, and provided better drug-like characteristics and the opportunity for structure exploration (Fig. 1).
In preliminary SAR study, analogues (1a–1e, Table 1) bearing a methyl were prepared to define positions available for substitution without adversely affecting the inhibitor potency. The results showed that substitutions of position N3 (1b), C6 (1d) and N′ (1e) of 1a resulted in a greatly diminished or complete loss in inhibition toward FAAH and NAAA, while substitution of position C5 (1c) slightly decreased the inhibition against both FAAH and NAAA. Therefore, we focused on the modification of C5 in the following studies.
Table 1 Inhibitory effects of compounds 1a–1e on rFAAH and rNAAA activitiesa
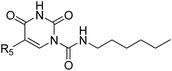
Next, we did systematic substitution of 1a at C5 (1c, 2a–2k) to investigate the effect of electronic properties of ring substitution on the inhibitory activity of uracil urea derivatives toward FAAH. Some analogues (2a, 2d, 2i, 2k) have been described by other groups were also synthesized and tested as control compounds.36,37 The results suggested that the electronic properties of C5 substituents were of great significance for the inhibition towards both FAAH and NAAA (Table 2). Compared to parent inhibitor 1a, the electron-withdrawing substituents (2i–2k) exhibited 2–5 folds enhanced inhibition against FAAH. Among compounds 2a–2k, 2i–2k were the three most potent inhibitors, being equipotent with URB597 tested under the same conditions. On the contrary, the electron-donating substituents (1c, 2a–2e) showed slightly decreased inhibitory activity against FAAH. We then tested whether further modulation of the electron-donating C5 substituents with electron-withdrawing group can restore the potency. The results showed that the addition of acetyl to the compounds 2f–2h resulted in a modest (3–7 folds) increase of inhibitory potency compared to corresponding derivatives (2b, 2c and 2e). These results suggested that the electron-withdrawing C5 substitution was critical for potent FAAH inhibition. The reason might be such modulation increases the leaving group ability of uracil, and makes molecules more sensitive to nucleophilic attack of Ser241 of FAAH. To examine this hypothesis, two 1a derivative was synthesized and tested. Derivative 2l is the saturated derivative of 1a without the leaving group uracil. Derivative 2m without electrophilic carbonyl group was obtained by replacement of N1-carboxamide with methylene. The data showed that inhibitory activity of 2l and 2m against FAAH was completely eliminated. These results confirmed that uracil leaving group and N1-carboxamide group are critical for FAAH inhibition.
Table 2 Inhibitory effects of compounds 1a, 1c, 2a–2m on rFAAH and rNAAA activitiesa
|
| |
R5 |
IC50 of FAAH (nM) |
IC50 of NAAA (nM) |
Selectivity |
| Data are presented as IC50 ± standard error of the mean. All experiments were performed in triplicate. |
| 1a |
–H |
492 ± 53 |
990 ± 170 |
2 |
| 1c |
–Me |
1026 ± 205 |
3650 ± 840 |
3.6 |
| 2a |
–Et |
543 ± 94 |
1837 ± 428 |
3.4 |
| 2b |
–OH |
693 ± 117 |
5049 ± 1103 |
7.3 |
| 2c |
–CH2OH |
425 ± 75 |
3265 ± 519 |
7.6 |
| 2d |
–OEt |
703 ± 136 |
1048 ± 168 |
1.5 |
| 2e |
–NH2 |
1520 ± 280 |
5265 ± 864 |
3.5 |
| 2f |
–OAc |
110 ± 10 |
190 ± 20 |
1.7 |
| 2g |
–CH2OAc |
120 ± 31 |
271 ± 25 |
2.2 |
| 2h |
–NHAc |
221 ± 44 |
423 ± 78 |
1.9 |
| 2i |
–F |
38 ± 9 |
250 ± 50 |
6.6 |
| 2j |
–Ac |
42 ± 11 |
486 ± 89 |
11.5 |
| 2k |
–CN |
25 ± 5 |
560 ± 42 |
22.4 |
| URB597 |
|
26 ± 5 |
>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
|
| Structure |
IC50 of FAAH (nM) |
IC50 of NAAA (nM) |
| 2l |
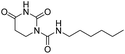 |
>10000 |
>10000 |
| 2m |
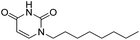 |
>10000 |
>10000 |
Except from enhancing inhibition potency towards FAAH, electron-withdrawing C5 substitution also increased the inhibition potency of uracil urea derivatives toward NAAA. For instance, acetyl substituents 2f–2h enhanced NAAA inhibition by 12–27 folds compared to the unsubstituted derivatives (2b, 2c and 2e). Electron-withdrawing C5 substituents exhibited weak selectivity towards FAAH over NAAA. The electron-withdrawing substituents failed to improve the selectivity towards FAAH over acid ceramidase. The IC50 values of electron-withdrawing substituents towards acid ceramidase (2i–2k) was an order of magnitude lower than 1a.36 These results suggested that electron-withdrawing substituents of uracil ring exhibited enhanced the inhibition potency, but their selectivity towards FAAH over acid ceramidase and NAAA was still not ideal. Further modification was required to enable high selectivity for FAAH over other competitive enzymes.
Finally, we further modified the side chain to improve the selectivity towards FAAH. FAAH hydrolyzes unsaturated substrates (e.g., anandamide) faster than saturated FAEs, while NAAA and acid ceramidase strongly prefer saturated substrates (e.g., PEA or ceramide).38 Replacement of the hexyl chain with an unsaturated group might mimic the π-characteristics and conformation of the anandamide, leading to binding affinity preference towards FAAH. Therefore, we optimized the chain terminus of C5 uracil substituents (–H, –F, –OAc) with a phenyl ring, a chemical moiety that can mimic the conformation and π-unsaturation of the Δ8,9/Δ11,12 double bonds of anandamide. Three representative series where R5 = H, F, OAc were designed and synthesized. As shown in Table 3, most of the analogous with an alkylphenyl group exhibited dramatically increased inhibitory potency against FAAH. The inhibitory potency was progressively improved by prolonging the linker length of the alkylphenyl. Interestingly, the inhibition activity of these substituents was independent of electron effect of C5 group, which was previously expected to significantly modulate the binding affinity of these derivatives. The results suggested that van der Waals interactions, strongly related to the closeness between inhibitors and protein surfaces, might be the most significant contribution to binding affinity.
Table 3 Inhibitory effects of compounds 3a–3u on rFAAH and rNAAA activitiesa
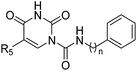
|
| |
R5 |
n |
IC50 of FAAH (nM) |
IC50 of NAAA (nM) |
Selectivity |
| Data are presented as IC50 ± standard error of the mean. All experiments were performed in triplicate. |
| 3a |
–H |
1 |
155 ± 23 |
8020 ± 1290 |
52 |
| 3b |
2 |
333 ± 105 |
6340 ± 1080 |
19 |
| 3c |
3 |
19 ± 7 |
1572 ± 326 |
83 |
| 3d |
4 |
13 ± 4 |
305 ± 57 |
23 |
| 3e |
5 |
11 ± 1 |
435 ± 63 |
40 |
| 3f |
6 |
4 ± 0.9 |
2490 ± 372 |
623 |
| 3g |
7 |
3 ± 0.8 |
4768 ± 541 |
1589 |
| 3h |
–F |
1 |
122 ± 10 |
4655 ± 482 |
38 |
| 3i |
2 |
98 ± 12 |
4510 ± 371 |
9.4 |
| 3j |
3 |
21 ± 3 |
440 ± 96 |
20 |
| 3k |
4 |
16 ± 2 |
481 ± 35 |
30 |
| 3l |
5 |
13 ± 3 |
105 ± 11 |
8 |
| 3m |
6 |
4 ± 1 |
290 ± 30 |
73 |
| 3n |
7 |
3 ± 0.8 |
822 ± 85 |
274 |
| 3o |
–OAc |
1 |
51 ± 10 |
5880 ± 647 |
115 |
| 3p |
2 |
96 ± 12 |
5440 ± 1023 |
57 |
| 3q |
3 |
32 ± 6 |
810 ± 100 |
25 |
| 3r |
4 |
18 ± 3 |
220 ± 25 |
12 |
| 3s |
5 |
10 ± 1 |
3545 ± 641 |
355 |
| 3t |
6 |
5 ± 1.1 |
7908 ± 855 |
1648 |
| 3u |
7 |
3 ± 0.4 |
9382 ± 1425 |
3127 |
These uracil urea derivatives exhibited different inhibition pattern against NAAA (Table 3). Most compounds with alkylphenyl replacement were less active than the parent hexyl derivatives. The optimal potency was observed with the linker length of C5 (for 5-F series) or C4 (for 5-H and 5-OAc series) methylene spacer, and the inhibition potency progressively decreased with lengthened or shortened linker. The tight binding pocket of NAAA, may explain the loss of NAAA inhibitory and gain in FAAH selectivity by bulkier structures. Most of the alkylphenyl derivatives were more selective for FAAH compared to parent inhibitors (1a, 2f and 2i). Among these alkylphenyl derivatives 3g, 3t and 3u were the most promising inhibitors with high potency and selectivity towards FAAH. We further substituted the phenylalkyl chain of 3t and 3u with biphenyl group and tested whether biphenyl modification will further enhance the potency. As showed in Table 4, the biphenyl derivatives (4a, 4c and 4d) were 3–16 folds more potent or similar potent (4b) compared to the parent inhibitors (3t and 3u). Moreover, the selectivity of biphenyl substituents towards FAAH was significantly enhanced. 4a–4d exhibited profound selectivity (>3000 folds) towards FAAH over NAAA.
Table 4 Inhibitory effects of compounds 4a–4d on rFAAH and rNAAA activitiesa
4.2. Selectivity evaluation
NAAA, acid ceramidase (AC) and endocannabinoid hydrolase monoacylglycerol lipase (MAGL) are three competitive targets for the uracil urea derivatives. Representative inhibitors (2f, 2i, 3u and 4c) were selected and their selectivity towards FAAH was examined. As shown in Table 5, the inhibitors were highly selective for FAAH over MAGL. Hexyl derivatives 2f and 2i exhibited inhibitory effects against all the three enzymes. Phenyl ring substitutions at the terminus of the hexyl side chain enhanced the binding affinity of the inhibitors to FAAH over NAAA (2f, 1.7 folds; 2i, 6.5 folds; 3u and 4c, >3000 folds), but it did not decrease their inhibition potency towards AC. Notably, biphenyl group substitution dramatically increased both potency and selectivity of the derivatives towards FAAH. The most potent inhibitor 4c was more than 3000 folds selective for FAAH over NAAA, and modestly selective for FAAH over AC (Table 5).
Table 5 Selectivity screening of compounds 2f, 2i, 3u and 4ca
| |
IC50 of rFAAH (nM) |
IC50 of rNAAA (nM) |
IC50 of rAC (nM) |
IC50 of rMAGL (nM) |
| Data are presented as IC50 ± standard error of the mean. All experiments were performed in triplicate. |
| 2f |
110 ± 10 |
190 ± 20 |
102 ± 23 |
>10000 |
| 2i |
38 ± 9 |
250 ± 50 |
45 ± 9 |
>10000 |
| 3u |
3 ± 0.4 |
9382 ± 1425 |
68 ± 10 |
>10000 |
| 4c |
0.3 ± 0.05 |
>10000 |
360 ± 74 |
>10000 |
The potency and selectivity of inhibitors were depended on the compound reactivity as well as the molecular shape and flexibility. The inhibition profile of uracil urea derivatives was modulated by electron properties of the C5 substituents and the conformation of the side chain. Electron-withdrawing substituent enhances both potency and selectivity for FAAH over NAAA, but failed to improve the selectivity towards FAAH over AC.36 Electron-withdrawing substitution turning 1a to a dual inhibitor of FAAH–AC. On the other hand, the side chain is the major factor contributing to the selectivity of uracil urea derivatives. Incorporation of phenyl-containing group into the side chain significantly enhanced their inhibition potency and selectivity towards FAAH over NAAA and AC, turning the uracil urea scaffold from a multi-target inhibitor to a FAAH-selective inhibitor.
4.3. Investigations of the inhibition mechanism
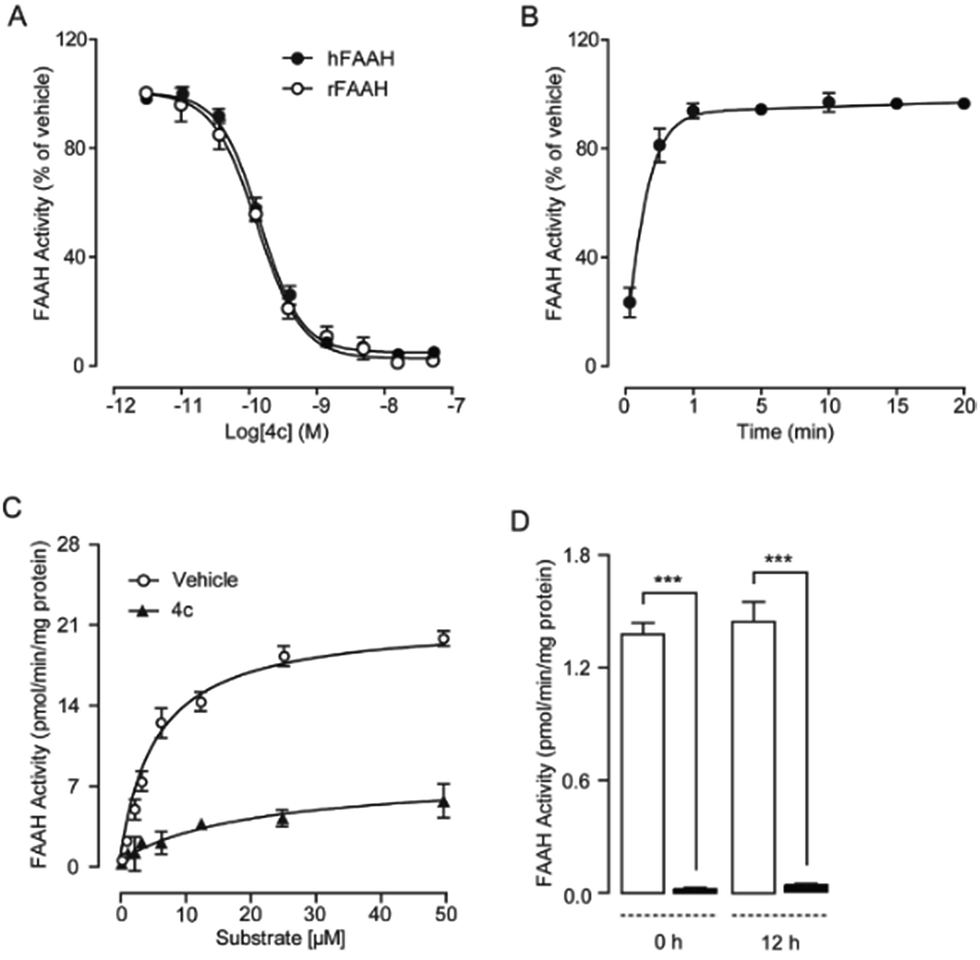
To explore the pharmacological properties of uracil urea derivatives, we analysed their interaction pattern with FAAH. Representative inhibitor 4c was selected in these studies. As well as its significant inhibition towards recombinant rat FAAH protein (IC50 = 0.3 ± 0.05 nM, Table 4), 4c displayed a profound inhibition toward recombinant human FAAH (IC50 = 0.6 ± 0.1 nM) (Fig. 2A). Kinetic analysis revealed that 4c rapidly inhibited rFAAH (t1/2 < 1 minute) (Fig. 2B), and changed the maximal catalytic velocity (Vmax) of rFAAH (Vmax in pmol per min per mg protein, vehicle: 2056 ± 107; and 4c, 50 nM: 698 ± 84), but it did not affect Michaelis–Menten constant Km (Km in μM, vehicle: 9.8 ± 0.3; and 4c, 50 nM: 10.4 ± 0.6) (Fig. 2C). No recovery of the rFAAH activity was observed by dialysis of the 4c–rFAAH interaction complex even after 12 hours (Fig. 2C). These results indicated that 4c was an irreversible and non-competitive FAAH inhibitor, thereby providing strong evidence for the formation of covalent bonds between compound and enzyme.
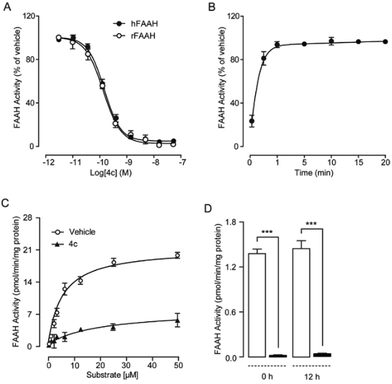 |
| | Fig. 2 Effect of 4c on FAAH activity in vitro. (A) Dose-dependent inhibition of recombinant rat FAAH (rFAAH) and recombinant human FAAH (hFAAH) activity by 4c. (B) Time course of rFAAH inhibition by 4c (50 nM). (C) Michaelis–Menten analysis of the FAAH reaction in the presence of vehicle (open circles, 1% DMSO) or 4c (closed triangles, 50 nM). (D) FAAH activity in the presence of vehicle (open bars 1% DMSO) or 4c (closed bars, 50 nM) before dialysis and 12 hours after dialysis. Results are expressed as mean ± SEM (n = 4–6). ***P < 0.001. | |
4.4. Biological stability evaluation
The hydrolysis of compounds (2f, 2i and 4c) was studied in 80% (v/v) rat plasma at 37 °C. The results showed that more than 60% of compounds were hydrolyzed after 10 min incubation (Table 6). Analogues 2f and 2i containing less sterically hindered hexyl chain were cleaved more rapidly than the bulkier compound 4c. LC-MS analysis of degradation products of inhibitors (2f, 2i and 4c) showed the formation of hexylamine and 4-biphenylpropylamine, which resulted from the hydrolysis of the N1-carboxamide (data not shown). FAAH is broadly distributed throughout the body, involved in a complex anandamide-based signalling system. Most of current FAAH inhibitors, e.g. PF-04457845, are distributed throughout all tissues and organs after administration. They are suitable for the systemic application, but probably not optimal agents to investigate the function of FAAH in specific organ or tissue. The inhibitors we developed exert acute FAAH inhibitory in the target site and rapidly inactivated to prevent unwanted systemic effects. Our findings may offer a new topically applied tool to investigate the function of FAAH at a selected tissue or organ.
Table 6 Percent of compounds 2f, 2i and 4c remaining after incubation in 80% (v/v) rat plasmaa
| |
2f |
2i |
4c |
| Compounds were measured at 10 μM in 80% (v/v) rat plasma with 2.5% DMSO. |
| Remain inhibitors |
0 min |
100% |
100% |
100% |
| 5 min |
15% |
21% |
56% |
| 10 min |
7% |
11% |
33% |
| 15 min |
n.a. |
n.a. |
8% |
5. Conclusions
Here, we report the synthesis and characterization of a series of uracil urea derivatives, designed as FAAH inhibitors by progressive modification of 1a. SAR studies suggested that electron-withdrawing substituents in the C5 position enhanced the potency of FAAH inhibition and the replacement of the side chain with phenylalkyl moieties or biphenyl groups significantly improved both the inhibitory potency and selectivity towards FAAH. The two most potent inhibitors, 4c (IC50 = 0.3 ± 0.05 nM) and 4d (IC50 = 0.8 ± 0.1 nM) showed moderate to high selectivity towards FAAH over other endocannabinoid-metabolizing enzymes. Rapid dilution and kinetic analysis indicate that 4c is a selective, irreversible and non-competitive FAAH inhibitor. This study provides us some promising FAAH inhibitors and an optimized scaffold for the development of strategies to investigate tropical FAAH function, and to explore new therapeutics for inflammation, cancer, and neuropathic diseases.
Acknowledgements
We thank Dr Daniele Piomelli at the University of California, Irvine for the kind gifts of the HEK293–rNAAA cells and the HEK293–rFAAH cells. This study was supported by grants from National Natural Sciences Foundation of China (No. 81373273 to QY, No. 81602974 to YL), Key Research Program of Frontier Science, CAS, Grant No. QYZDJ-SSW-SLH033, The Major State Basic Research Development Program of China (grant number 2014CB965101), and China Postdoctoral Science Foundation Funded Project (2016M592876XB) to YL.
References
- D. K. Giang and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2238 CrossRef CAS.
- S. Shin, T. H. Lee, N. C. Ha, H. M. Koo, S. Y. Kim, H. S. Lee, Y. S. Kim and B. H. Oh, EMBO J., 2002, 21, 2509 CrossRef CAS PubMed.
- M. H. Bracey, M. A. Hanson, K. R. Masuda, R. C. Stevens and B. F. Cravatt, Science, 2002, 298, 1793 CrossRef CAS PubMed.
- A. H. Lichtman, D. Leung, C. C. Shelton, A. Saghatelian, C. Hardouin, D. L. Boger and B. F. Cravatt, J. Pharmacol. Exp. Ther., 2004, 311, 441 CrossRef CAS PubMed.
- S. Kathuria, S. Gaetani, D. Fegley, F. Valino, A. Duranti, A. Tontini, M. Mor, G. Tarzia, G. La Rana, A. Calignano, A. Giustino, M. Tattoli, M. Palmery, V. Cuomo and D. Piomelli, Nat. Med., 2003, 9, 76 CrossRef CAS PubMed.
- B. F. Cravatt, K. Demarest, M. P. Patricelli, M. H. Bracey, D. K. Giang, B. R. Martin and A. H. Lichtman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9371 CrossRef CAS PubMed.
- C. J. Fowler, K. O. Jonsson and G. Tiger, Biochem. Pharmacol., 2001, 62, 517 CrossRef CAS PubMed.
- H. S. Hansen, B. Moesgaard, H. H. Hansen and G. Petersen, Chem. Phys. Lipids, 2000, 108, 135 CrossRef CAS PubMed.
- H. S. Hansen, Exp. Neurol., 2010, 224, 48 CrossRef CAS PubMed.
- H. H. Schmid, P. C. Schmid and V. Natarajan, Prog. Lipid Res., 1990, 29, 1 CrossRef CAS PubMed.
- J. M. Walker, S. M. Huang, N. M. Strangman, K. Tsou and M. C. Sanudo-Pena, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12198 CrossRef CAS.
- M. Salaga, M. Sobczak and J. Fichna, Eur. J. Pharm. Sci., 2014, 52, 173 CrossRef CAS PubMed.
- K. Otrubova, C. Ezzili and D. L. Boger, Bioorg. Med. Chem. Lett., 2011, 21, 4674 CrossRef CAS PubMed.
- M. Seierstad and J. G. Breitenbucher, J. Med. Chem., 2008, 51, 7327 CrossRef CAS PubMed.
- D. L. Boger, H. Miyauchi, W. Du, C. Hardouin, R. A. Fecik, H. Cheng, I. Hwang, M. P. Hedrick, D. Leung, O. Acevedo, C. R. Guimaraes, W. L. Jorgensen and B. F. Cravatt, J. Med. Chem., 2005, 48, 1849 CrossRef CAS PubMed.
- L. Chang, L. Luo, J. A. Palmer, S. Sutton, S. J. Wilson, A. J. Barbier, J. G. Breitenbucher, S. R. Chaplan and M. Webb, Br. J. Pharmacol., 2006, 148, 102 CrossRef CAS PubMed.
- G. Tarzia, A. Duranti, A. Tontini, G. Piersanti, M. Mor, S. Rivara, P. V. Plazzi, C. Park, S. Kathuria and D. Piomelli, J. Med. Chem., 2003, 46, 2352 CrossRef CAS PubMed.
- G. Moreno-Sanz, A. Duranti, L. Melzig, C. Fiorelli, G. F. Ruda, G. Colombano, P. Mestichelli, S. Sanchini, A. Tontini, M. Mor, T. Bandiera, R. Scarpelli, G. Tarzia and D. Piomelli, J. Med. Chem., 2013, 56, 5917 CrossRef CAS PubMed.
- M. Mor, S. Rivara, A. Lodola, P. V. Plazzi, G. Tarzia, A. Duranti, A. Tontini, G. Piersanti, S. Kathuria and D. Piomelli, J. Med. Chem., 2004, 47, 4998 CrossRef CAS PubMed.
- M. Mor, A. Lodola, S. Rivara, F. Vacondio, A. Duranti, A. Tontini, S. Sanchini, G. Piersanti, J. R. Clapper, A. R. King, G. Tarzia and D. Piomelli, J. Med. Chem., 2008, 51, 3487 CrossRef CAS PubMed.
- K. Ahn, S. E. Smith, M. B. Liimatta, D. Beidler, N. Sadagopan, D. T. Dudley, T. Young, P. Wren, Y. Zhang, S. Swaney, K. Van Becelaere, J. L. Blankman, D. K. Nomura, S. N. Bhattachar, C. Stiff, T. K. Nomanbhoy, E. Weerapana, D. S. Johnson and B. F. Cravatt, J. Pharmacol. Exp. Ther., 2011, 338, 114 CrossRef CAS PubMed.
- M. Kono, T. Matsumoto, T. Kawamura, A. Nishimura, Y. Kiyota, H. Oki, J. Miyazaki, S. Igaki, C. A. Behnke, M. Shimojo and M. Kori, Bioorg. Med. Chem., 2013, 21, 28 CrossRef CAS PubMed.
- G. L. Li, H. Winter, R. Arends, G. W. Jay, V. Le, T. Young and J. P. Huggins, Br. J. Clin. Pharmacol., 2012, 73, 706 CrossRef CAS PubMed.
- J. M. Keith, R. Apodaca, M. Tichenor, W. Xiao, W. Jones, J. Pierce, M. Seierstad, J. Palmer, M. Webb, M. Karbarz, B. Scott, S. Wilson, L. Luo, M. Wennerholm, L. Chang, S. Brown, M. Rizzolio, R. Rynberg, S. Chaplan and J. G. Breitenbucher, ACS Med. Chem. Lett., 2012, 3, 823 CrossRef CAS PubMed.
- D. S. Johnson, C. Stiff, S. E. Lazerwith, S. R. Kesten, L. K. Fay, M. Morris, D. Beidler, M. B. Liimatta, S. E. Smith, D. T. Dudley, N. Sadagopan, S. N. Bhattachar, S. J. Kesten, T. K. Nomanbhoy, B. F. Cravatt and K. Ahn, ACS Med. Chem. Lett., 2011, 2, 91 CrossRef CAS PubMed.
- S. O. Alapafuja, S. P. Nikas, I. T. Bharathan, V. G. Shukla, M. L. Nasr, A. L. Bowman, N. Zvonok, J. Li, X. Shi, J. R. Engen and A. Makriyannis, J. Med. Chem., 2012, 55, 10074 CrossRef CAS PubMed.
- A. Lodola, M. Mor, S. Rivara, C. Christov, G. Tarzia, D. Piomelli and A. J. Mulholland, Chem. Commun., 2008, 214 RSC.
- C. J. Fowler, Eur. Neuropsychopharmacol., 2015, 25, 749 CrossRef CAS PubMed.
- X. Wang, K. Sarris, K. Kage, D. Zhang, S. P. Brown, T. Kolasa, C. Surowy, O. F. El Kouhen, S. W. Muchmore, J. D. Brioni and A. O. Stewart, J. Med. Chem., 2009, 52, 170 CrossRef CAS PubMed.
- M. Feledziak, C. Michaux, A. Urbach, G. Labar, G. G. Muccioli, D. M. Lambert and J. Marchand-Brynaert, J. Med. Chem., 2009, 52, 7054 CrossRef CAS PubMed.
- G. G. Muccioli, N. Fazio, G. K. Scriba, W. Poppitz, F. Cannata, J. H. Poupaert, J. Wouters and D. M. Lambert, J. Med. Chem., 2006, 49, 417 CrossRef CAS PubMed.
- G. Palermo, A. D. Favia, M. Convertino and M. De Vivo, ChemMedChem, 2016, 11, 1252 CrossRef CAS PubMed.
- A. D. Favia, D. Habrant, R. Scarpelli, M. Migliore, C. Albani, S. M. Bertozzi, M. Dionisi, G. Tarozzo, D. Piomelli, A. Cavalli and M. De Vivo, J. Med. Chem., 2012, 55, 8807 CrossRef CAS PubMed.
- L. Bertolacci, E. Romeo, M. Veronesi, P. Magotti, C. Albani, M. Dionisi, C. Lambruschini, R. Scarpelli, A. Cavalli, M. De Vivo, D. Piomelli and G. Garau, J. Am. Chem. Soc., 2013, 135, 22 CrossRef CAS PubMed.
- Y. Qiu, Y. Zhang, Y. Li and J. Ren, Molecules, 2016, 21, 229 CrossRef PubMed.
- D. Pizzirani, C. Pagliuca, N. Realini, D. Branduardi, G. Bottegoni, M. Mor, F. Bertozzi, R. Scarpelli, D. Piomelli and T. Bandiera, J. Med. Chem., 2013, 56, 3518 CrossRef CAS PubMed.
- N. Realini, C. Solorzano, C. Pagliuca, D. Pizzirani, A. Armirotti, R. Luciani, M. P. Costi, T. Bandiera and D. Piomelli, Sci. Rep., 2013, 3, 1035 CrossRef PubMed.
- N. Ueda, K. Yamanaka and S. Yamamoto, J. Biol. Chem., 2001, 276, 35552 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra02237a |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
Yuhang Li
b and
Yuhang Li