
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-delivery of hydrophilic and hydrophobic anticancer drugs using biocompatible pH-sensitive lipid-based nano-carriers for multidrug-resistant cancers
Samira Naderinezhad a,
Ghasem Amoabediny*a and
Fateme Haghiralsadatb
a,
Ghasem Amoabediny*a and
Fateme Haghiralsadatb
aDepartment of Biotechnology and Pharmaceutical Engineering, Faculty of Chemical Engineering, School of Engineering, University of Tehran, Enghelab Av. Postal code: 1439957131, Tehran, Iran. E-mail: amoabedini@ut.ac.ir; Tel: +98 9124486374
bDepartment of Life Science Engineering, Faculty of New Sciences & Technologies, University of Tehran, Tehran, Iran
First published on 9th June 2017
Abstract
For decades, multi-drug resistance (MDR) to chemotherapeutic drugs has been a serious challenge for researchers and has limited the use of anticancer drugs in malignancy treatment. Combination therapy has been considered as one of the most promising methods to address this problem. In the current study, we optimized niosome nanoparticles containing chemotherapeutic agent doxorubicin and chemosensitizer curcumin in term of surfactant content. Then, a new biocompatible structure (LipoNiosome, combination of niosome and liposome) containing Tween 60: cholesterol: DPPC (at 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:15:3) with 3% DSPE-mPEG was designed and developed to serve as a model for selective co-delivery of hydrophilic and hydrophobic drugs to cancerous cells. The proposed formulation provided potential benefits, including pH-sensitive sustained release, smooth globular surface morphology, high entrapment efficiency (∼80% for both therapeutic agents) and small diameter (42 nm). Exposure of cancer cells to LipoNiosome-doxorubicin–curcumin has shown an excellent performance of specific cellular internalization and synergistic toxic effect (>40%; as compared to free drugs and >23% when compared to single doxorubicin delivery) against Saos-2, MG-63 and KG-1 cell lines. A new cationic formulation (zeta potential: +35.26 mV; diameter: 52.2 nm) was also designed for co-delivery of above-mentioned drugs and gene as well. Finely, we suggested a kinetic model (Korsmeyer–Peppa with R2 = 93% near cancer cells) for in vitro drug release of the co-delivery system. The presently formulated nano-based systems would provide researchers with a more obvious understanding of new LipoNiosome formulation as a successful lipid-based nano-carriers for co-delivery of doxorubicin, curcumin and other anticancer agents.
:30:15:3) with 3% DSPE-mPEG was designed and developed to serve as a model for selective co-delivery of hydrophilic and hydrophobic drugs to cancerous cells. The proposed formulation provided potential benefits, including pH-sensitive sustained release, smooth globular surface morphology, high entrapment efficiency (∼80% for both therapeutic agents) and small diameter (42 nm). Exposure of cancer cells to LipoNiosome-doxorubicin–curcumin has shown an excellent performance of specific cellular internalization and synergistic toxic effect (>40%; as compared to free drugs and >23% when compared to single doxorubicin delivery) against Saos-2, MG-63 and KG-1 cell lines. A new cationic formulation (zeta potential: +35.26 mV; diameter: 52.2 nm) was also designed for co-delivery of above-mentioned drugs and gene as well. Finely, we suggested a kinetic model (Korsmeyer–Peppa with R2 = 93% near cancer cells) for in vitro drug release of the co-delivery system. The presently formulated nano-based systems would provide researchers with a more obvious understanding of new LipoNiosome formulation as a successful lipid-based nano-carriers for co-delivery of doxorubicin, curcumin and other anticancer agents.
1. Introduction
Cancer is basically uncontrolled cell proliferation that aggressively invades other parts of the body. According to the International Agency for Research on Cancer (IARC), 14.1 million new cases of cancer were estimated to occur in 2012, with almost 8.2 million mortality cases. Based on IARC estimates, cancer is the 2nd most common cause of death in both economically developing and developed countries. About 13% of global cancer cases are estimated to have occurred in southwestern of Asia.1 The main cancer treatment modality, chemotherapy, has limitations that including various side effects. To overcome the previously-mentioned impediments of current cancer treatments, nanotechnology can pave the way.Multidrug resistance (MDR) in cancers, defined as resistance of tumor to the cytotoxic effects of several drugs, has been considered as a major obstacle to the clinical cancer treatment. Resistance against anticancer drug has been attributed to increased drug efflux, decreased drug uptake, activation of DNA repair process along with the activation of detoxifying systems.2 In order to treat drug-resistant tumors, combinations of multiple anticancer agents, including chemosensitizers such as, small interfering RNAs (siRNAs) and herbal medicine like curcumin (CUR) and classic antineoplastic drugs have been applied. For example, combination of antileukemic drug and chemosensitizers has been demonstrated to effectively modulate multiple signaling pathways by inactivation of MDR-related mRNAs.3–5 In our previous works, formulation of siRNA and doxorubicin (DOX) delivery system were optimized and a new YSA-targeted liposomal DOX-siRNA was developed to exert a synergistic effect to overcome MDR in osteosarcoma.6,7 Another challenge for researchers is that single-drug therapy reinforces alternative molecular pathways in cancer cells due to drug-resistance mutations. Co-delivery of multiple anticancer agents via a nanocarrier has been considered as an approach to overcome MDR.8
Anthracycline (antibiotics) are the most commonly used agents, which block the topoisomerase function, thereby inhibiting cell proliferation. Doxorubicin (Adriamycin®), an anthracycline antibiotic, is a potent FDA-approved chemotherapy drug.9,10 For many years, it has been acknowledged that DOX could treat several cancer types, such as breast cancer, bone marrow cancer, and osteosarcoma. However, DOX's side effects and unpredictable toxicity to normal cells adversely affects the immune system, which makes patient more susceptible to infections. It also causes cardiotoxicity.11 To address this issue, numerous nanoparticle drug delivery vehicles have emerged during the past two decades.2,4
Compared to free DOX, nano-shielded DOX has less toxicity to normal cells however due to sustained release properties and higher cytotoxicity against cancer cells in short term and overcomes MDR in long term.12
Multidrug resistance raised by up-regulation of BCL-2 protein and inactivation DOX by accumulation into acidic cytoplasmic vesicles resulting in induce MDR gene expression, reduced sensitivity to DOX and decreases therapeutic efficiency of DOX for osteosarcoma, bone marrow cancers and etc.13–17
Curcumin (CUR), a chemosensitizers, was used to fight cancer governed by blocking NFκB signaling pathway, down-regulation overexpression of P-glycoprotein and Bcl-2.18 Therefore, CUR is currently being co-administered with DOX and other cytotoxic drugs to reduce the efflux of drugs through upregulated efflux transporters in resistant hepatocellular carcinoma in mice.19
CUR has shown a number of benefits for patients with colorectal and pancreatic cancer. In addition, neither human nor animal studies have found any side effect or toxicity of CUR.20,21 Despite many beneficial pharmacological activities, such as anti-oxidant, antiviral and anti-inflammatory properties and the safety of CUR, its retention time in the body is restricted due to low serum bioavailability, hepatic elimination and poor absorption resulted by hydrophobicity.22 Tsai et al. (2011) have reported that CUR may prevent cancer in the colon, skin and stomach after oral administration while no dose-limiting toxicity was observed. However, they showed that it has avid metabolism, resulting in mean plasma concentration of 31.5 mg mL−1 after 8 g ingestion per day.23 Great efforts have been made by researchers to enhance CUR solubility and protection of CUR against inactivation by hydrolysis.24 Nano-biotechnology has been introduced as a promising strategy in the development of drug delivery systems for hydrophobic drugs such as CUR. Xi Yang et al. designed and characterized curcumin-encapsulated polymeric micelles to improve the stability of CUR. They also studied the anti-cancer efficiency of the prepared nano-stabilized aqueous curcumin for colon cancer treatment.25
During the past decade, niosomes and liposomes have been extensively applied due to their stability and biocompatibility, respectively. The performance of single encapsulation of DOX or CUR into the liposome or niosome has already been well investigated.17–19,26–28 However, little is known about stability and biocompatibility of their combined form in liposome and niosome.
The purpose of this research was to investigate the hypothesis whether or not co-delivery of DOX–CUR could show synergistic effects in different cancer cell lines and could overcome against DOX resistance. Moreover, it was hypothesized that this co-delivery could diminish drugs' side effects in normal cells. Various co-formulations were formulated to achieve high entrapment efficiency, small size, and controlled release behavior. For better safety, the doses of both drugs were decreased and the cationic LipoNio formulations were developed. Mathematical model was developed to estimate drug release behavior. Furthermore, co-delivery of DOX and CUR was carried out to estimate cytotoxicity against MG-63, KG-1 and SaOs-2 cell lines.
2. Experimental
2.1. Materials
Doxorubicin HCl (DOX) and curcumin (purity ≥ 95%) were obtained from Ebewe Pharma (Austria) and Sigma-Aldrich (St Louis, MO, USA), respectively. Surfactants, Tween 60 and Span 60, were purchased from DaeJung Chemicals & Metals (South Korea). Tween 80 and Tween 20 were obtained from Merck (Darmstadt, Germany). The distearoyl phosphoethanolamine, polyethylene glycol (Lipoid PE 18:0/18:0 – PEG2000, DSPE-mPEG 2000), DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine phospholipid) and SPC80 (soybean phospholipids with 75% phosphatidylcholine) were obtained from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol and DOTAP (1,2-dioleoyl-3-trimethylammonium-propane) were supplied by Sigma-Aldrich (St. Louis, MO, USA) and Avanti Polar Lipids (AL, USA), respectively. Ammonium sulfate salt and NaHCO3 were obtained from Merck (Darmstadt, Germany). PBS tablets, dialysis bag (MW = 12 kDa), DMSO (dimethyl sulfoxide), HEPES buffer, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) and paraformaldehyde solution were obtained from Sigma-Aldrich (St. Louis, MO, USA). DAPI (4′,6-diamidino-2-phenylindole) was supplied from Thermo Fisher Scientific (Massachusetts, USA). All other chemicals, solvents and salts were of the analytical grade and used without further purification unless specified.
2.2. Cell lines and preparation of biological samples
Human primary (short-term culture) osteoblasts, osteosarcoma cell line MG-63 and SaOs-2, bone marrow acute myeloblastic leukemia (AML) cell line KG-1 and human bone marrow fibroblast-like HBMF-SPH cell line were supplied from the Pasteur Institute of Iran (Tehran, Iran). Human primary osteoblasts SaOs-2 and MG-63 were cultured in DMEM medium (Gibco, Grand Island, USA). HBMF-SPH and KG-1 cells were cultured in RPMI-1640 medium (Gibco, Grand Island, USA). All cells were supplemented with 10% FBS (fetal bovine serum) (Gibco, Grand Island, USA) with penicillin-streptomycin (Gibco, Grand Island, USA) under standard condition (37 °C and 5% CO2 in a humidified incubator).2.3. Preparation of drug-loaded nanoparticles
DOX- and CUR-loaded nanoparticles were synthesized and screened for particle size, controlled release and high entrapment efficiency parameters. In order to optimize the results and determine the optimal conditions following experiments were performed:• Evaluation of the influence of different types of surfactants with various hydrophobic tail and hydrophilic heads and in different molar ratios (0, 15% and 35%) was carried out on dual-drug delivery system.
• The preparation of LipoNiosome was investigated by addition of 15% synthetic and natural phospholipids (SPC80, DPPC).
• Suggested hydration methods for preparing drug-carrier vesicles, including thin film and pH gradient were evaluated.
• After having optimized synthetic conditions, cationic formulations for dual drug/gene delivery were prepared.
Nanoparticles, containing hydrophilic and hydrophobic drugs, were prepared by thin film and pH-gradient technique as fully described elsewhere.12 Fig. 1 shows the schematic view of research steps. Briefly, the surfactants in these formulations were 70% (w/w) in all, and as can be seen in Table 1, various kinds of surfactants with different concentration ranges were dissolved in chloroform as an organic phase in the presence of 3% DSPE-mPEG and 30% cholesterol with or without phospholipids (Table 2). CUR stock solution was dissolved in methanol (1 mg mL−1) and added to the mixture of surfactant and lipids. The thin films were dried to remove chloroform and methanol, using rotary evaporator (Heidolph, Germany) at 45 °C under reduced pressure. Hydration of dry lipid film was carried out by adding 1300 μL diluted DOX solution at 63 °C for 60 min. After completion of hydration, ultra-sonication was applied for 45 min (15 seconds on and 10 seconds off, amplitude of 70 at 100 watts) to minimize particle aggregation using ultrasonic homogenizer (model UP200St, Hielscher Ultrasonics GmbH, Germany). For pH-gradient method, the dried films of CUR, surfactants and lipids were hydrated with 1300 μL of ammonium sulfate (pH = 4) at 63 °C for 47 min. Then, nanoparticles were sonicated over an ice bath to produce small vesicles. Afterwards, ammonium sulfate was removed by replacing the medium with fresh PBS (pH = 7.4) using a dialysis cellulose membrane for 2 h at 25 °C. Finally, the diluted DOX with sterile water for injection, was added at 54 °C. The dose of both drugs was 0.5 mg mL−1 for all of the formulations and the L/D ratios were kept at 20 and 10 for thin film and pH-gradient methods, respectively.
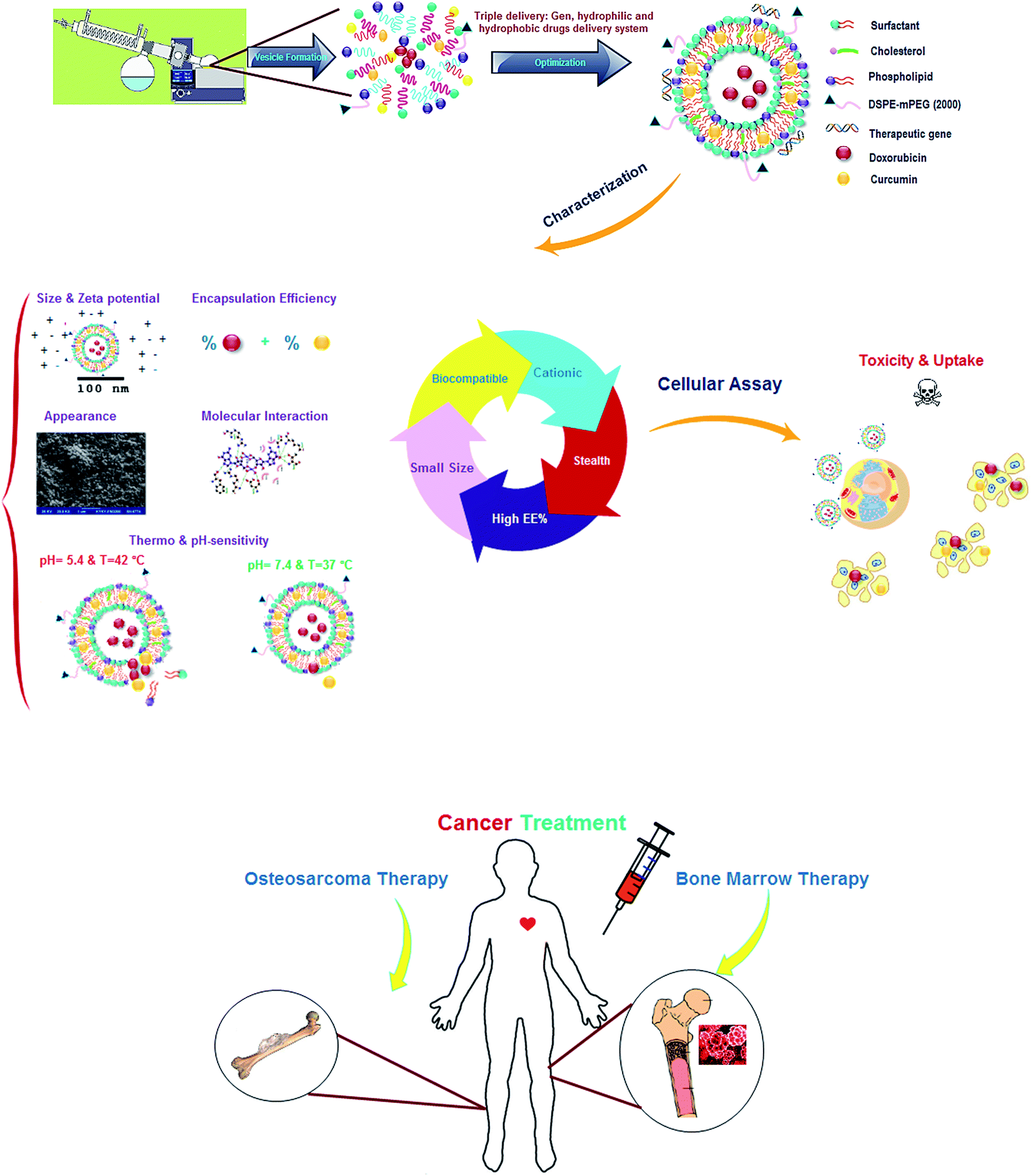 | ||
| Fig. 1 The schematic representation experimental design of the study. | ||
| Code | Type of surfactant | Mole of surfactants (%) | EE% | Size (nm) | % R (8 h) | % R (24 h) | % R (48 h) | % R (96 h) |
|---|---|---|---|---|---|---|---|---|
| a EE: entrapment efficiency. | ||||||||
| F1 | Span 60 | 70 | EE% DOX = 25.01 | 185.9 | % R DOX = 25.42 | % R DOX = 33.94 | % R DOX = 39.28 | % R DOX = 45.6 |
| EE% CUR = 50.21 | % R CUR = 20.16 | % R CUR = 26.88 | % R CUR = 29.75 | % R CUR = 34.64 | ||||
| F2 | Tween 60 | 70 | EE% DOX = 85.12 | 85.5 | % R DOX = 38.42 | % R DOX = 46.38 | % R DOX = 51.67 | % R DOX = 61.76 |
| EE% CUR = 95.11 | % R CUR = 27.16 | % R CUR = 31.96 | % R CUR = 37.14 | % R CUR = 43.78 | ||||
| F3 | Tween 80 | 70 | EE% DOX = 62.02 | 60.9 | % R DOX = 50.63 | % R DOX = 61.66 | % R DOX = 66.9 | % R DOX = 77.69 |
| EE% CUR = 87.70 | % R CUR = 38.1 | % R CUR = 43.82 | % R CUR = 46.39 | % R CUR = 54.67 | ||||
| F4 | Span 60:Tween 20 |
55:15 |
EE% DOX = 66.51 | 54.2 | % R DOX = 35.28 | % R DOX = 45.06 | % R DOX = 51.13 | % R DOX = 61.05 |
| EE% CUR = 62.13 | % R CUR = 30.96 | % R CUR = 37.98 | % R CUR = 41.81 | % R CUR = 52.06 | ||||
| F5 | Span 60:Tween 60 |
55:15 |
EE% DOX = 42.02 | 127.4 | % R DOX = 42.48 | % R DOX = 47.97 | % R DOX = 53.79 | % R DOX = 61.26 |
| EE% CUR = 68.05 | % R CUR = 25.05 | % R CUR = 30.06 | % R CUR = 34.55 | % R CUR = 37.07 | ||||
| F6 | Span 60:Tween 80 |
55:15 |
EE% DOX = 48.03 | 95.9 | % R DOX = 39.02 | % R DOX = 49.50 | % R DOX = 53.43 | % R DOX = 59.46 |
| EE% CUR = 71.11 | % R CUR = 34.89 | % R CUR = 40.64 | % R CUR = 44.99 | % R CUR = 55.24 | ||||
| F7 | Span 60:Tween 20 |
35:35 |
EE% DOX = 76.21 | 48 | % R DOX = 41.39 | % R DOX = 50.81 | % R DOX = 56.66 | % R DOX = 62.84 |
| EE% CUR = 91.03 | % R CUR = 30.38 | % R CUR = 37.73 | % R CUR = 41.83 | % R CUR = 53.59 | ||||
| F8 | Span 60:Tween 60 |
35:35 |
EE% DOX = 81.32 | 97.1 | % R DOX = 41.07 | % R DOX = 48.53 | % R DOX = 52.81 | % R DOX = 62.62 |
| EE% CUR = 81.51 | % R CUR = 22.95 | % R CUR = 31.28 | % R CUR = 34.56 | % R CUR = 43.49 | ||||
| F9 | Span 60:Tween 80 |
35:35 |
EE% DOX = 70.43 | 64.5 | % R DOX = 41.65 | % R DOX = 51.70 | % R DOX = 55.45 | % R DOX = 62.84 |
| EE% CUR = 78.91 | % R CUR = 32.25 | % R CUR = 40.91 | % R CUR = 45.09 | % R CUR = 61.4 | ||||
| Code | Type of phospholipid | EE% | Size (nm) | % Release (8 h) | Zeta potential (mV) | PDI |
|---|---|---|---|---|---|---|
| F10 | SPC80 | EE% DOX = 93.02 | 65.1 | % R DOX = 71.07 | −34 | 0.283 |
| EE% CUR = 98.01 | % R CUR = 73.38 | |||||
| F11 | DPPC | EE% DOX = 88.23 | 42 | % R DOX = 45.29 | −37 | 0.324 |
| EE% CUR = 77.11 | % R CUR = 30.63 | |||||
| F12 | DOTAP | EE% DOX = 85.02 | 52.2 | — | +35.26 | 0.302 |
| EE% CUR = 94.03 |
2.4. Characterization of DOX/CUR-NPs
:20, 1:100 and 1:75 (v/v) to lyse the membranes and rapid shed of entrapped drugs.Dialysis method is one of the methods to determine the concentration of DOX/CUR in PBS solution and subsequently, the drug release behavior of nanoparticles. At the first step, the sample of nanoparticles DOX–CUR was transferred into a dialysis tube and the release of both drugs was monitored in 7 mL of PBS solution (at 37 and 42 °C, pH = 7.4, 6.5 and 5.4) in shaking water bath at 75 rpm for 96 h. Then, in order to determine drug release rate by applying UV/visible spectroscopy (model T80+, PG Instruments, United Kingdom), 1200 μL of the sample was collected at specific time intervals and then substituted with an equal volume of fresh PBS to maintain the condition and ensure a constant initial volume. Finally, the amount of drug released was measured and the drug release (R) behavior was described by a semi-empirical mathematical model named Korsmeyer–Peppa.29
2.5. Cytotoxicity study
To study anti-proliferative activity of free drugs (DOX & CUR), blank niosome, blank LipoNio (biocompatible niosome), LipoNio-DOX (5, 12.5 and 25 μg mL−1), LipoNio-CUR (12.5, 25 and 50 μg mL−1), as well as co-administration of LipoNio-DOX-LipoNio-CUR (5, 12.5 and 25 μg mL−1), and co-delivery of LipoNio-DOX–CUR (5, 12.5 and 25 μg mL−1), were incubated 72 h with 104 cells in a 96-well plate prior to assessment with the colorimetric MTT assay. After 72 h after cell seeding in medium, the control wells and samples were removed and washed with PBS and then incubated with 20 μL of 5 mg mL−1 MTT in PBS for 3 h. The resultant formazan crystals were dissolved in DMSO. The absorbance of resulting samples was measured using EPOCH microplate spectrophotometer (synergy HTX, Bio-Tek, USA) at 570 nm.2.6. Nano-LipoNisomal DOX–CUR localization assay
To determine distribution of the DOX and CUR in the nucleus, the DOX and CUR fluorescence intensity was detected. In short, MG-63, KG-1 and SaOs-2 cells (5 × 105 per well) were seeded in 30 mm dishes, then treated with LipoNio-DOX–CUR, free CUR and free DOX at the concentration of 10 μg mL−1. After 3 and 8 h of incubation, cells were washed thrice with PBS (pH 7.4). All floating cells were collected using centrifuge (at 1200 rpm for 10 minutes) before washing with PBS. The nucleus of cells was counterstained with DAPI (0.125 μg mL−1) for 15 min. Images were obtained by fluorescence microscopy (Olympus, Japan).302.7. Statistical analysis
Statistical data analyses were performed via Student t-test to compare the differences between groups and P < 5% was considered significant. Experiments were done in triplicate and average values were reported. The quality of fitting was evaluated by R2 (ref. 31) and non-linear regression analysis was performed using MATLAB software (version 7.8). The relative standard deviation was calculated to show the precision.To determine the amount of drug entrapment and drug release, standard solutions were prepared over a concentration range, and analyzed by UV spectroscopy at the characteristic value of λmax with support by the standard curve of drugs measured in a similar condition.
3. Results and discussion
3.1. The effect of surfactant on DOX/CUR niosome formula
In order to determine the optimal formulation for achieving high entrapment efficiency, controlled-release (at 37 °C and pH = 7) and small vesicle size, various niosomal CUR/DOX formulations were evaluated (Table 1). Comparing surfactants used in formulation (F1 vs. F2), including Span 60 and Tween 60, indicated that the hydrophobic chain of Tween 60 is longer than Span 60 resulting in higher CUR entrapment. Moreover, the results indicated that hydrophilic end of Tween 60 played a more predominant role in higher DOX entrapment over that of Span 60. In addition, acyl chain of Tween 20 (F4 vs. F1) had little effect on the enhancement of CUR release due to weaker interaction between CUR and the hydrophobic acyl chain. Although the hydrophilic and hydrophobic parts of Tween 60 and Tween 80 were similar to each other, but the presence of a double bond in the alkyl chain of Tween 80, formation of momentary polar functional groups and the presence of electron cloud have made it more unsaturated and mobile. This phenomenon increased drug leakage during preparation step and subsequently caused a reduction in final entrapment efficiency (F2 vs. F3 and F8 vs. F9). In addition, it increased drug release in Tween 80 formulation. The higher phase transition temperature (Tm) of Span 60 (F1 vs. F2 and F3) resulted in sustained release pattern and less drug entrapment in hydration step of preparation, as well as slow drug release, and subsequently brought the more stable system.Also, with the addition of 15% Tween (20, 60 and 80) (F4, F5 and F6) to Span 60 formulations, DOX and CUR entrapment into nanoparticles were constantly increased due to increasing aqueous and non-aqueous space of vesicles provided by the long chain of Tween. Since in F5 and F6, the dominant content of formulations is contained span 60, a little addition of Tween 60/80 (15%) increases entrapment efficiency and drug release independent to type of Tween. Since the chains of Tween became entangled in those of Span 60, it led to improved entrapment. However, the effective role of Tween 20 (F7 and F4) in improving entrapment of DOX into niosome can be attributed to the dominance of hydrophilic part compared to the total volume of the molecule. Thanks to flexible chains, Tween 80 molecules (F6) could compact themselves among Span 60 chains and became smaller in size as compared to Tween 60 (F5). Formulation prepared with Tween 20 (F4) was also smaller in size compared to F5 due to smaller size of alkyl length. This implies that the enhancement of entrapment efficiency and reduced diameter of Tween formulation can also be explained by the presence of PEG molecule tagged to Tween molecule in addition to DSPE-mPEG in all formulations. The Presence of DSPE-mPEG (2000) in formulations has made niosome smaller, less aggregated and stable in vivo and in vitro. PEGylation also improved drug entrapments.7,32,33
Examining the entrapment efficiency in different drug systems indicated better performance of dual-drug system compared to single drug one. These findings suggest that delivering multiple drugs simultaneously (a hydrophobic and a hydrophilic drug) restores balance followed by synergistic co-entrapment effect in the system, and also provides stability.
In order to improve drug penetration into vesicles, controlled drug release and make LipoNio formulation more stable and biocompatible for in vivo and in vitro application, 30% cholesterol was applied in all of our formulations.12,34 Zeta potential and PDI of all formulations ranged from −27 to −58 and 0.06 to 0.21, respectively. Based on sustained drug release, small diameter, high entrapment efficiency and simplicity of the formulation F2 with Tween 60 as surfactant has chosen as the formulation for further studies.
3.2. Effect of phospholipid on DOX/CUR LipoNio formulations
For induction biocompatibility to niosome, various LipoNiosomal formulations were compared based on the type of phospholipids in terms of entrapment efficiency, mean diameter, zeta potential and percentage of release during 8, 24, 48 and 96 hours (Table 2). F10 and F11 were composed of natural and synthetic phospholipids, respectively. According to the results, the LipoNio formula containing SPC80 showed higher drug entrapment and larger diameter compared to F11. However, the percentage of drug release was higher for F10 compared to F11. These results were consistent with those of our previous work.12 The hydrophobic alkyl chains were approximately equal in length for both SPC80 and DPPC, but unsaturation of SPC80 has made it more mobile and flexible. Thus, the flexibility of binding was improved by the addition of SPC80; however, it made the drug release fairly rapid (burst drug release, contrary to the purpose of sustained-drug release) and also resulted in nanoparticles with larger diameter. It can also be attributed to the fact that Tm of SPC80, unlike DPPC, was lower than body temperature, which resulted in its instability.35 Another explanation would be that the higher entrapment efficiency of F10 which led to increase of release rate due to concentration gradient between both sides of the niosomal membrane.The entrapment efficiency of CUR for F11 is lower than to F2, due to loss of free space in biliary of vesicle, between Tween 60 chains, after filling with phospholipid chains.
To prepare formulation for co-drugs-gene delivery, F12 was synthesized with the cationic phospholipid (DOTAP). As can be seen, zeta potential has made particles extremely positively -charged with the addition of 15% DOTAP with the entrapment efficiency slightly increased. The PDI (∼0.3 for all) indicated that no agglomeration occurred which can be attributed to mutually repellent force between the particles with the same-sign charge in the suspension.
3.3. Effect of preparation method
The LipoNiosomal formulation was prepared using pH gradient and thin film methods. Table 3 shows the effect of preparation methods on characteristics of vesicles. It was found out that the LipoNiosomal formulation prepared by the pH gradient method led to formation of particles with larger diameter and with a little higher encapsulation efficiency of CUR. It also showed a +10 mV change in zeta potential that resulted in prevention of agglomeration due to repulsive electrostatic forces. Although it confirms our previous findings for the single delivery of DOX, there is some concern about the formation of a complex between PBS solution and CUR during buffer replacement step. As described in literature, CUR was decomposed after 30 min of incubation in PBS.36 In fact, the hydration step was performed in thin film method by injection of acidic water (sterile water for injection) with pH value of 5, while for pH gradient method a PBS solution (at pH = 7.4) was used for buffer replacement step. So, the stability of CUR depends on the pH, i.e., improved with decreased pH. These results can be attributed to the fact that the proton was removed from the phenolic group in CUR, leading to decomposition of CUR.| Code | Preparation method | EE% | Size (nm) | Zeta potential (mV) | PDI |
|---|---|---|---|---|---|
| F11 | Thin-film | EE% DOX = 88.23 | 42 | −37 | 0.324 |
| EE% CUR = 77.11 | |||||
| F13 | pH-Gradient | EE% DOX = 88.01 | 158.4 | −23 | 0.15 |
| EE% CUR = 95.1 |
3.4. Characterization of optimized formulation
 | ||
| Fig. 2 Scanning electron microscopy (SEM) of (A) blank LipoNiosome; and (B) dual drug-loaded LipoNiosome (DOX–CUR-LipoNiosome). | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the ester group and –CH2 bending in lipids and surfactant, respectively. The characteristic peaks centered at 1348.8 cm−1 and 1250.8 cm−1 belonged to alkane C–H rock and alkyl C–N stretch (for DPPC and DSPE-mPEG), respectively. The band which appeared at 1096.3 cm−1 was attributed to C–O stretch in ether and ester groups (Tween 60 and DSPE-mPEG). All peaks were repeated in the FTIR spectrum of blank LipoNios, LipoNio-DOX and LipoNio-CUR, which clearly confirmed that there were no additional peaks and no chemical interaction between drugs and other components in the formulation. The FTIR spectrum of DOX and CUR shows several characteristic peaks centered at 1274 cm−1 for CUR and 1616.4 cm−1 which was characteristic of DOX.
O stretching of the ester group and –CH2 bending in lipids and surfactant, respectively. The characteristic peaks centered at 1348.8 cm−1 and 1250.8 cm−1 belonged to alkane C–H rock and alkyl C–N stretch (for DPPC and DSPE-mPEG), respectively. The band which appeared at 1096.3 cm−1 was attributed to C–O stretch in ether and ester groups (Tween 60 and DSPE-mPEG). All peaks were repeated in the FTIR spectrum of blank LipoNios, LipoNio-DOX and LipoNio-CUR, which clearly confirmed that there were no additional peaks and no chemical interaction between drugs and other components in the formulation. The FTIR spectrum of DOX and CUR shows several characteristic peaks centered at 1274 cm−1 for CUR and 1616.4 cm−1 which was characteristic of DOX.
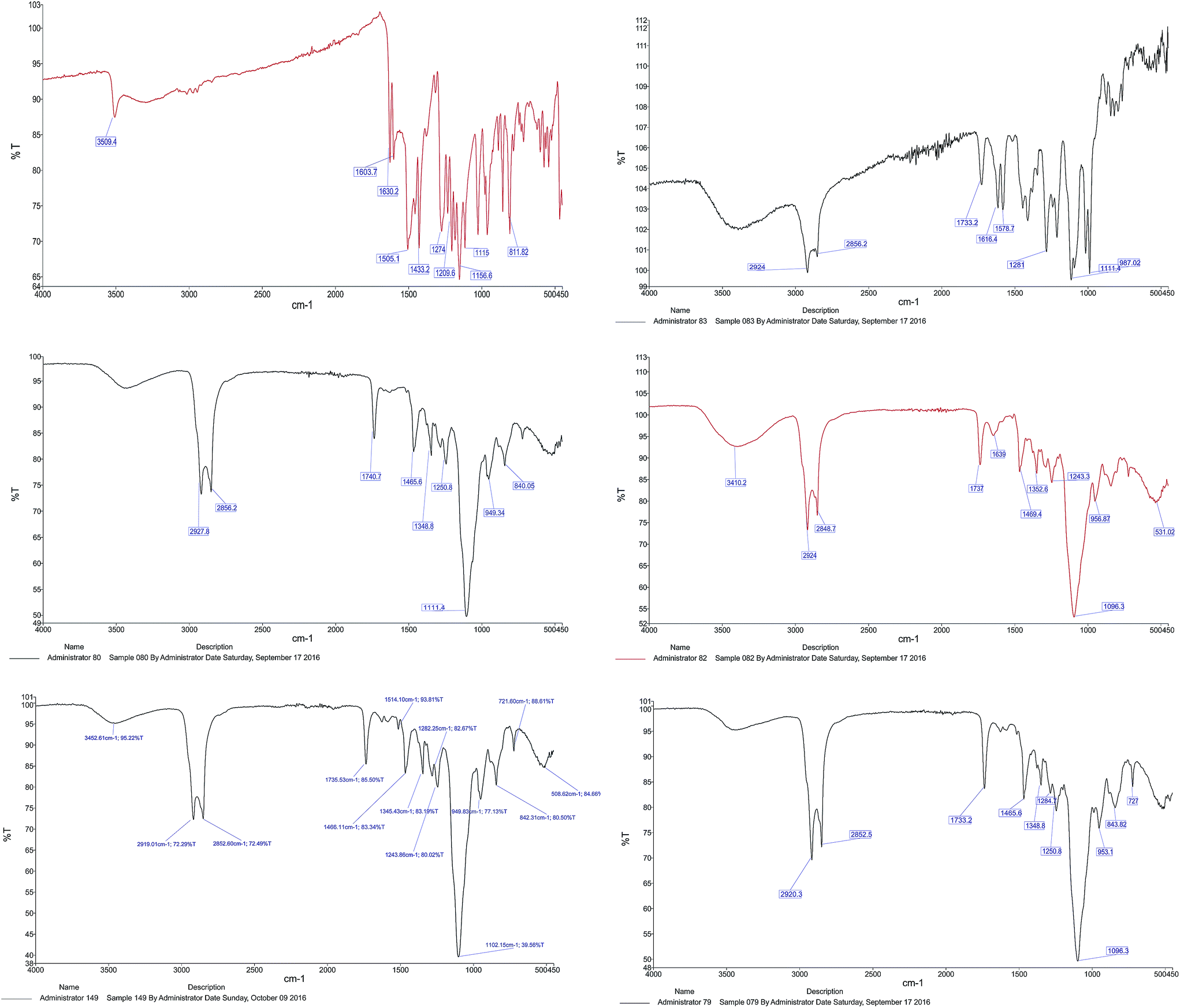 | ||
| Fig. 3 FTIR spectra of CUR FTIR, DOX FTIR, blank LipoNio FTIR, LipoNio-DOX FTIR, LipoNio-CUR FTIR, LipoNio-DOX–CUR FTIR. | ||
Some peaks got broader or were slightly shifted to another wave number; resulted by intermolecular forces, e.g., hydrogen bonding in Tween 60. The results also confirmed that both drugs encapsulated by the LipoNio were stable during formulation. All peaks were also observed in the spectrum of the single drug encapsulation of DOX or CUR; it shows that co-encapsulation of drugs has kept the integrity of drugs and carrier formulation.
000 new cases per year in USA.37 Osteosarcoma is the most common malignant bone tumor among children. Classic treatment of osteosarcoma needs the children to endure painful chemotherapy.38 As shown in Fig. 4A, the cell viability efficacy was improved when the cell lines were exposed to blank LipoNiosome instead of blank noisome (∼14.75%↑). In order to verify the enhanced anti-proliferative activity of the LipoNio-DOX–CUR compared to free DOX/CUR and LipoNio-DOX–CUR, the cytotoxicity of our dual-drugs delivery system was tested against MG-63, KG-1, primary bone and HBMF-SPH cells. Free DOX, free CUR, co-administration of LipoNio-DOX + LipoNio-CUR as well as single delivery of LipoNio-DOX or CUR were used as the controls (Fig. 4B and C). The cytotoxic capacity of LipoNio-DOX–CUR in all concentrations was higher than other under study group (Fig. 4B–E). The proliferation inhibition by drugs was strongly dose-dependent, and it was more effective for entrapment drugs due to slow internalization of drugs.
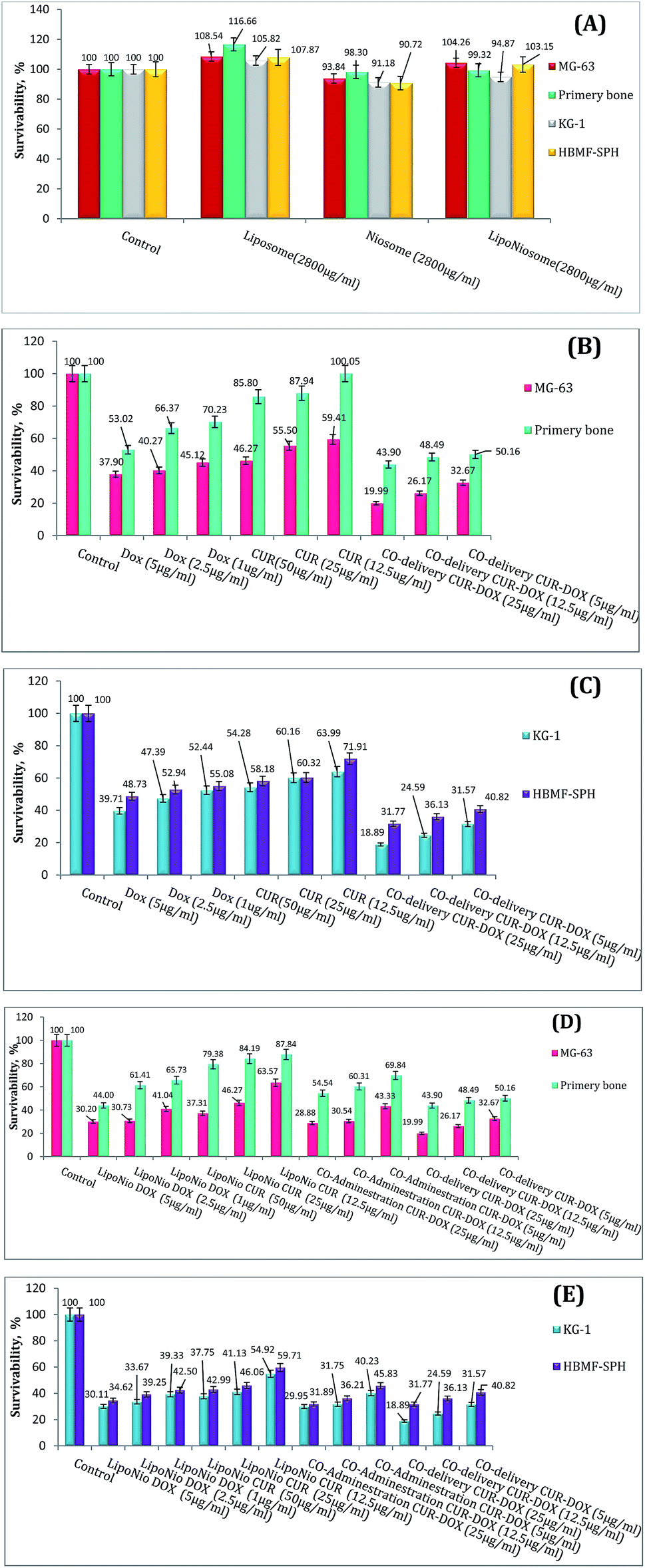 | ||
| Fig. 4 Survival analysis of MG-63, primary bone cell, KG-1 and HBMF-SPH in various drug concentrations (1, 2.5, 5, 12.5, 25 and 50 μg mL−1): biocompatibility comparison of liposome, niosome and LipoNio (A); comparison between toxicity of free drugs and LipoNio-DOX–CUR in various concentrations for OS (B); for AML (C); comparison between cytotoxicity of single drug carrier, co-administration of both single carriers (LipoNio-CUR + LipoNio-DOX), and co-delivery system (LipoNio-DOX–CUR) for OS (D) and for AML (E). | ||
Free CUR was less toxic compared to free DOX in normal cells. Furthermore, the highest concentrations of free CUR and DOX decreased the % survival in other cell lines treated with DOX up to 45% and up to 50–60% when the cells were treated with CUR.
Our findings indicate that the anti-proliferative effect of CUR entrapped inside LipoNiosome was enhanced which can be due to increased contact surface and solubility. Because of toxic nature of DOX and concerns about increased cytotoxicity of the current formulations resulted from sustained drug release, it is possible that the formulation could also result in increased cytotoxicity to primary bone cells. LipoNio-DOX–CUR showed significant higher cytotoxicity (p > 0.1) compared to the primary bone cells and HBMF-SPH cells.
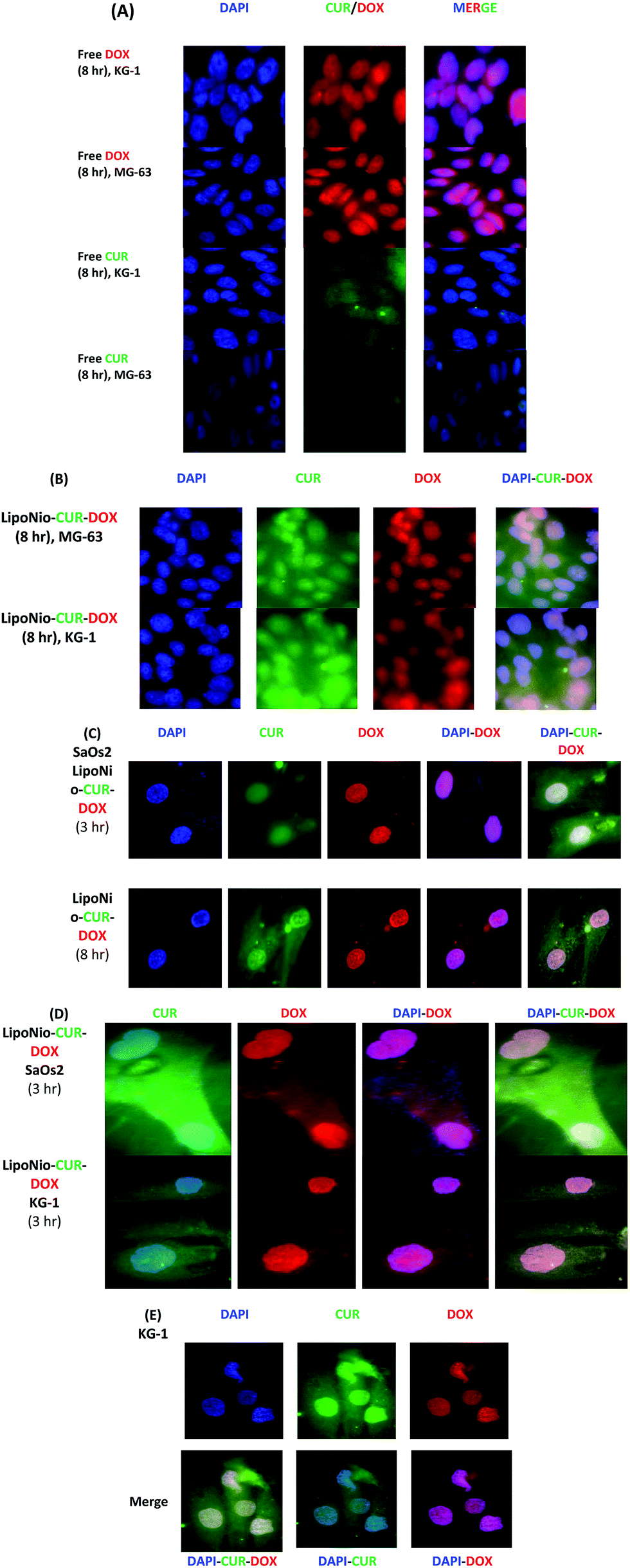 | ||
| Fig. 5 Cellular uptake images of MG-63, KG-1 and SaOs2 cells, treated with free DOX, free CUR and drugs entrapped into LipoNio vesicles. DAPI was used for labeling the nuclei. DOX and CUR have intrinsic red and green fluorescence, respectively. (A) Comparison between the cellular uptake of free drugs after 8 h-treatment in KG-1 and MG-63 cell line; (B) cellular uptake of MG-63 and KG-1 cells treated with LipoNio-DOX–CUR; (C) cellular uptake of SaOs2 cells treated with LipoNio-DOX–CUR for 3 and 8 h; (D) cellular uptake of SaOs2 and KG-1 cells treated with LipoNio-DOX–CUR for 3 h; (E) typical details of merging. | ||
The MG-63 cell line successfully absorbed the entrapped DOX. The prepared LipoNio-DOX–CUR formulation could effectively enter the cancerous cells, mostly into the nucleus whereas free DOX was predominately distributed in the cytoplasmic region. The accumulation of DOX in nucleus could induce apoptosis and inhibit DNA replication.39 Furthermore, CUR with its antioxidant activity can neutralize free radicals and increase activity of the topoisomerase II, DNA cleavage enzyme.28,40,41 Concentrating CUR within cancerous cells via carrier effectively enhanced anticancer activity of DOX. The prepared formulation served as a sustained-release carrier for both drugs so that most of the purple and turquoise blue colors were resulted by drugs transfected via carrier or previously released drug in acidic compartments. The release of the drug was increased in the cells due to the low pH value of the lysosome, leading to loosening the carrier membrane and enhanced drug release.
Comparing MG-63 and KG-1 cell lines indicated that more intense and widespread purple and turquoise blue fluorescence was observed for KG-1, and the fluorescence intensity of drug within cells was significantly stronger than that in osteosarcoma cell line.
Interestingly, cell viability results also confirmed that KG-1 cell line was more sensitive for both entrapped and free DOX. Zhang et al. recently studied the localization of DOX and CUR entrapped in PEG nanoparticles after 2, 4 and 8 h exposure of HepG2 cells and found successful drug internalization after 8 h.28 On the contrary, our results showed high drug accumulation after 3 h treatment. This may be due to smaller size of nanoparticles in our study that led to an increase in their diffusion. Also, our new nano LipoNiosomal formulation was more sensitive to changes in pH than other studies,26 i.e., it resulted in significant improvement in nuclear transfection of DOX in cancerous cells (increased release of DOX in lysosome with pH = 4). The negatively charged surface of LipoNio-DOX–CUR contradicted the assumption that the interaction between nanoparticles and a negatively-charged cell membrane resulted in the improvement of charge-dependent cellular transfection. Thus, it is hypothesized that electrostatic force could not have played specific role in the enhancement of cellular transfection of nanoparticles. In addition, electrostatic force reduced the side effects resulted by undesirable uptake of DOX into normal cells. After 8 h of incubation, it should be noted that the fluorescence intensity of LipoNio-DOX–CUR was significantly enhanced. We also checked the cellular uptake of SaOs2 cells treated with LipoNio-DOX–CUR as an external model validation.
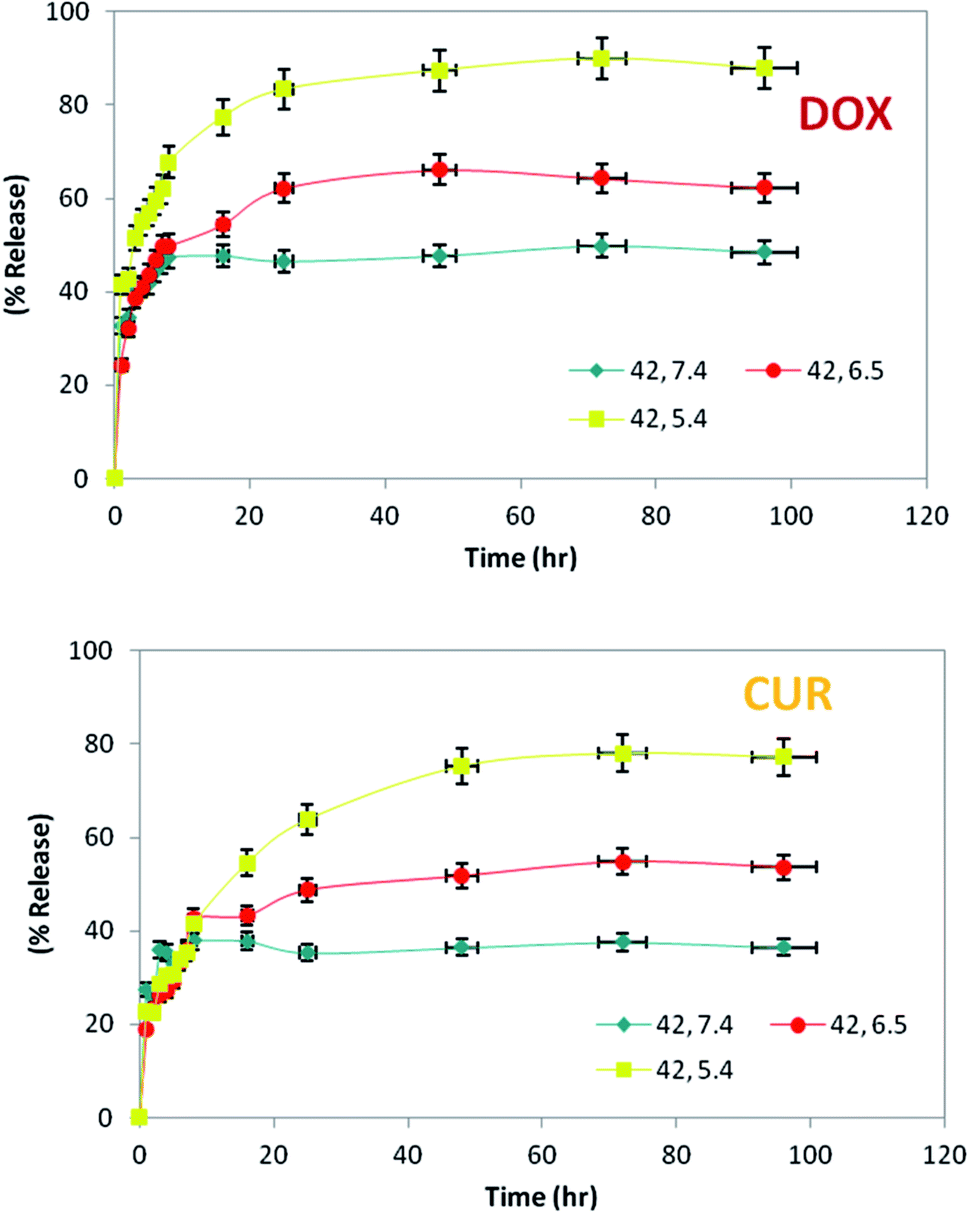 | ||
| Fig. 6 Drug release profile in various pH. | ||
Since DOX and CUR are small molecules and the permeability cut-off of the dialysis tube was 12–14 kDa, released DOX and CUR could pour easily out of the tube. Therefore, the release of drug was not limited by the dialysis tube or the size of drugs; however, the amount of drugs released was influenced by the temperature, pH of surrounding buffer and also the structure of LipoNio membrane. Three chosen pH values which represented the typical levels that nanoparticle are encountered in the physiological condition, tumor tissue and cancerous cells were 7.4, 6.5 and 5.4, respectively. Cancerous cells and tumor tissue usually have hypoxia that decreases the pH value. We used this phenomenon to make our formulation pH-sensitive to increase the toxic effect against malignant cells and subsequently reduce side effects against normal cells.
The most rapid drug release took place at 42 °C and pH = 5.4–6.5. Thus, our study indicated timing of sustained release of drugs over 4 days from the LipoNiosome. Cytotoxicity assay confirmed the pH-sensitive nature of our formulation that resulted in higher toxic effect of LipoNio-DOX–CUR on cancerous cells compared to normal cells. The release of drugs from inside the LipNio membrane to external fluid is mainly through solubility, diffusion and convection, as fully described in literature.7 Rapid release of drugs at low pH values or high temperature can be attributed to the enhancement of solubilization of aggregated drugs into vesicles, loosening of the LipoNio membrane and slightly improving temperature dependence of diffusion. Considering drug release, cell viability, and cellular uptake results, the total amount of released drug was increased over time and reached to approximately 87.86% for DOX and 77.2% for CUR (pH = 5.4, T = 42 °C) after 72 h. In other words, about 80% of LipoNio-DOX–CUR and 100% of free drugs killed 37% and 32% of the cells, respectively. It can be concluded, a lower dose of the drug created more cytotoxicity due to the fact that the LipoNio-drug particles were disassembled after uptake by the cells, and meanwhile, DOX and CUR continuously accumulated within nucleus and cytoplasm, and then played anti-cancer effect. The regression analysis was performed with the corresponding nonlinear regression correlation coefficient value (R2) equal to 0.91. The estimated model parameters of Korsmeyer–Peppa are given at Table 4. According to the results, Korsmeyer–Peppa model could sufficiently estimate the drug release behavior near the cancerous cells. The exponent value of Peppa's model was increased when LipoNio-DOX–CUR was made in contact with cancerous cells instead of normal cells, and reached about 0.5. Thus, the drug release was mainly controlled by Fician diffusion. In fact, with decreased drug release rate, it became less controlled by Fick's law. These results are in consistent with those of release experiments carried out at various pH and temperature.
| K | n | R2 | Drug | |
|---|---|---|---|---|
| Adjacent healthy cells | 32.88 | 0.0959 | 0.4599 | DOX |
| 20.4 | 0.1227 | 0.7448 | CUR | |
| Adjacent cancerous cells | 44.12 | 0.1699 | 0.9300 | DOX |
| 21.14 | 0.3053 | 0.9555 | CUR |
4. Conclusions
Our results successfully suggested a new model called LipoNiosome for hydrophobic and hydrophilic drug delivery via single nanocarrier. Phospholipid combined with surfactant was suggested as a new strategy to improve biocompatibility and characteristics of our formulations. With the addition of surfactant to nanoparticles, the drug loading capacity of carriers and the stability of the formulation were improved and particle surface tension reduced. The anti-proliferative performance of dual-drug delivery system with four different cell lines showed uptake and effectiveness of these drugs. Our findings indicated that nanocarrier-based approach adopted for delivery of CUR/DOX combinations was effective in combating cancer cells in vitro. Our long-term objective is to provide the proof-of-principle for the comprehensive model of targeted dual-drug delivery system for multidrug-resistant cancer treatment via biodegradable, biocompatible and stable carrier for simultaneous delivery of high amounts of siRNA and hydrophobic and hydrophilic drugs.Acknowledgements
The authors are grateful to Dr Fatemeh Hakimian, Department of Biophysics, Institute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran for scientific assistance during the project. The authors also thank Dr Fatemeh Montazeri, Recurrent Abortion Center, Reproductive sciences institute, Shahid Sadoughi University of Medical Science, Yazd, Iran. The authors also wish to thank the Department of Nanobiotechnology, Research Center for New Technologies in Life-Science Engineering, University of Tehran, Tehran, Iran.References
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Global cancer statistics, 2012, Ca-Cancer J. Clin., 2015, 65(2), 87–108 CrossRef PubMed.
- F. Haghiralsadat, G. Amoabediny, B. Zandieh-doulabi, P. de Boer Jantine and M. N. Helder, Treatment of osteosarcoma metastases: codelivery of siRNA and small molecule anticancer drugs based Nano-biotechnology: a review, Biotechnol. Adv., 2017 Search PubMed , submitted.
- M. Bao, Z. Cao, D. Yu, S. Fu, G. Zhang and P. Yang, et al., Columbamine suppresses the proliferation and neovascularization of metastatic osteosarcoma U2OS cells with low cytotoxicity, Toxicol. Lett., 2012, 215(3), 174–180 CrossRef CAS PubMed.
- M. Creixell and N. A. Peppas, Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance, Nano Today, 2012, 7(4), 367–379 CrossRef CAS PubMed.
- M. Saraswathy and S. Gong, Recent developments in the co-delivery of siRNA and small molecule anticancer drugs for cancer treatment, Mater. Today, 2014, 17(6), 298–306 CrossRef CAS.
- F. Haghiralsadat, G. Amoabediny, B. Zandieh-doulabi, T. Forouzanfar and M. N. Helder, Co-Delivery of doxorubicin and MAP-kinases siRNA by novel targeting pegylated cationic liposomes for overcome osteosarcoma multidrug-resistant, Int. J. Nanomed., 2017 Search PubMed , submitted.
- F. Haghiralsadat, G. Amoabediny, M. N. Helder, S. Naderinezhad, M. H. Sheikhha and T. Forouzanfar, et al., A comprehensive mathematical model of drug release kinetics from nano-liposomes, derived from optimization studies of cationic PEGylated liposomal doxorubicin formulations for drug-gene delivery, Artif. Cells, Nanomed., Biotechnol., 2017, 1–9, DOI:org/10.1080/21691401.2017.1304403.
- N. R. Patel, B. S. Pattni, A. H. Abouzeid and V. P. Torchilin, Nanopreparations to overcome multidrug resistance in cancer, Adv. Drug Delivery Rev., 2013, 65(13), 1748–1762 CrossRef CAS PubMed.
- M. Khasraw, R. Bell and C. Dang, Epirubicin: is it like doxorubicin in breast cancer? A clinical review, Breast, 2012, 21(2), 142–149 CrossRef PubMed.
- X. Zhao, Q. Chen, W. Liu, Y. Li, H. Tang and X. Liu, et al., Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer, Int. J. Nanomed., 2015, 10, 257 Search PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems, J. Pharm. Pharmacol., 2013, 65(2), 157–170 CrossRef CAS PubMed.
- F. Haghiralsadat, G. Amoabediny, M. H. Sheikhha, B. Zandieh-doulabi, S. Naderinezhad and M. N. Helder, et al., New liposomal doxorubicin nanoformulation for osteosarcoma: drug release kinetic study based on thermo and pH sensitivity, Chem. Biol. Drug Des., 2017 DOI:10.1111/cbdd.12953.
- L.-C. S. Huang, H. Chuang, M. Kapoor, C.-Y. Hsieh, S.-C. Chou and H.-H. Lin, et al., Development of nordihydroguaiaretic acid derivatives as potential multidrug-resistant selective agents for cancer treatment, RSC Adv., 2015, 5(130), 107833–107838 RSC.
- N. Li, P. Zhang, C. Huang, Y. Song, S. Garg and Y. Luan, Co-delivery of doxorubicin hydrochloride and verapamil hydrochloride by pH-sensitive polymersomes for the reversal of multidrug resistance, RSC Adv., 2015, 5(95), 77986–77995 RSC.
- P. Xu, R. Wang, J. Li, J. Ouyang and B. Chen, PEG–PLGA–PLL nanoparticles in combination with gambogic acid for reversing multidrug resistance of K562/A02 cells to daunorubicin, RSC Adv., 2015, 5(75), 61051–61059 RSC.
- J. Zhang, Y. Luo, X. Zhao, X. Li, K. Li and D. Chen, et al., Co-delivery of doxorubicin and the traditional Chinese medicine quercetin using biotin–PEG 2000–DSPE modified liposomes for the treatment of multidrug resistant breast cancer, RSC Adv., 2016, 6(114), 113173–113184 RSC.
- R. Misra and S. K. Sahoo, Coformulation of doxorubicin and curcumin in poly(D,L-lactide-co-glycolide) nanoparticles suppresses the development of multidrug resistance in K562 cells, Mol. Pharm., 2011, 8(3), 852–866 CrossRef CAS PubMed.
- S. Barui, S. Saha, G. Mondal, S. Haseena and A. Chaudhuri, Simultaneous delivery of doxorubicin and curcumin encapsulated in liposomes of pegylated RGDK-lipopeptide to tumor vasculature, Biomaterials, 2014, 35(5), 1643–1656 CrossRef CAS PubMed.
- X. Zhao, Q. Chen, Y. Li, H. Tang, W. Liu and X. Yang, Doxorubicin and curcumin co-delivery by lipid nanoparticles for enhanced treatment of diethylnitrosamine-induced hepatocellular carcinoma in mice, Eur. J. Pharm. Biopharm., 2015, 93, 27–36 CrossRef CAS PubMed.
- C.-H. Hsu and A.-L. Cheng, Clinical studies with curcumin, in The Molecular Targets and Therapeutic Uses of Curcumin in Health and Disease, Springer, 2007, pp. 471–480 Search PubMed.
- J. Sanmukhani, V. Satodia, J. Trivedi, T. Patel, D. Tiwari and B. Panchal, et al., Efficacy and safety of curcumin in major depressive disorder: a randomized controlled trial, Phytother. Res., 2014, 28(4), 579–585 CrossRef CAS PubMed.
- M. Pröhl, U. S. Schubert, W. Weigand and M. Gottschaldt, Metal complexes of curcumin and curcumin derivatives for molecular imaging and anticancer therapy, Coord. Chem. Rev., 2016, 307, 32–41 CrossRef.
- Y.-M. Tsai, W.-C. Jan, C.-F. Chien, W.-C. Lee, L.-C. Lin and T.-H. Tsai, Optimised nano-formulation on the bioavailability of hydrophobic polyphenol, curcumin, in freely-moving rats, Food Chem., 2011, 127(3), 918–925 CrossRef CAS PubMed.
- A. B. Hegge, T. T. Nielsen, K. L. Larsen, E. Bruzell and H. H. Tønnesen, Impact of curcumin supersaturation in antibacterial photodynamic therapy—effect of cyclodextrin type and amount: studies on curcumin and curcuminoides XLV., J. Pharm. Sci., 2012, 101(4), 1524–1537 CrossRef CAS PubMed.
- X. Yang, Z. Li, N. Wang, L. Li, L. Song and T. He, et al., Curcumin-encapsulated polymeric micelles suppress the development of colon cancer in vitro and in vivo, Sci. Rep., 2015, 5 DOI:10.1038/srep10322.
- V. Sharma, S. Anandhakumar and M. Sasidharan, Self-degrading niosomes for encapsulation of hydrophilic and hydrophobic drugs: an efficient carrier for cancer multi-drug delivery, Mater. Sci. Eng., C, 2015, 56, 393–400 CrossRef CAS PubMed.
- J. Duan, H. M. Mansour, Y. Zhang, X. Deng, Y. Chen and J. Wang, et al., Reversion of multidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate) nanoparticles, Int. J. Pharm., 2012, 426(1), 193–201 CrossRef CAS PubMed.
- Y. Zhang, C. Yang, W. Wang, J. Liu, Q. Liu and F. Huang, et al., Co-delivery of doxorubicin and curcumin by pH-sensitive prodrug nanoparticle for combination therapy of cancer, Sci. Rep., 2016, 6 DOI:10.1038/srep21225.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Mechanisms of solute release from porous hydrophilic polymers, Int J Pharm., 1983, 15(1), 25–35 CrossRef CAS.
- F. Haghiralsadat, G. Amoabediny, M. Hasan Sheikhha, T. Forouzanfar, M. N. Helder and B. Zandieh-doulabi, A novel approach on drug delivery: investigation of new nano-formulation of liposomal doxorubicin and biological evaluation of entrapped doxorubicin on various osteosarcomas cell lines, Cell Journal (Yakhteh), Spring, 2017, vol. 19, ch. 1 Search PubMed.
- B. Vidakovic, Statistics for bioengineering sciences: with MATLAB and WinBUGS Support, Springer Science & Business Media, 2nd edn, 2011, p. 623 Search PubMed.
- J.-Y. Kim, J.-K. Kim, J.-S. Park, Y. Byun and C.-K. Kim, The use of PEGylated liposomes to prolong circulation lifetimes of tissue plasminogen activator, Biomaterials, 2009, 30(29), 5751–5756 CrossRef CAS PubMed.
- Y.-P. Li, Y.-Y. Pei, X.-Y. Zhang, Z.-H. Gu, Z.-H. Zhou and W.-F. Yuan, et al., PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats, J. Controlled Release, 2001, 71(2), 203–211 CrossRef CAS PubMed.
- E. Yitbarek, Characterization and analytical applications of dye-encapsulated zwitterionic liposomes, PhD Diss [Internet], 2010;(Ldv), http://www.lib.ncsu.edu/resolver/1840.16/5859.
- J. Li, X. Wang, T. Zhang, C. Wang, Z. Huang and X. Luo, et al., A review on phospholipids and their main applications in drug delivery systems, Asian J. Pharm. Sci., 2015, 10(2), 81–98 CrossRef.
- Y.-J. Wang, M.-H. Pan, A.-L. Cheng, L.-I. Lin, Y.-S. Ho and C.-Y. Hsieh, et al., Stability of curcumin in buffer solutions and characterization of its degradation products, J. Pharm. Biomed. Anal., 1997, 15(12), 1867–1876 CrossRef CAS PubMed.
- M. A. Sekeres, Treatment of older adults with acute myeloid leukemia: state of the art and current perspectives, Haematologica, 2008, 1769–1772 CrossRef PubMed.
- H. T. Ta, C. R. Dass, P. F. M. Choong and D. E. Dunstan, Osteosarcoma treatment: state of the art, Cancer Metastasis Rev., 2009, 28(1–2), 247–263 CrossRef PubMed.
- Y. Shi, M. Moon, S. Dawood, B. McManus and P. P. Liu, Mechanisms and management of doxorubicin cardiotoxicity, Herz, 2011, 36(4), 296–305 CrossRef CAS PubMed.
- C. López-Alarcón and A. Denicola, Evaluating the antioxidant capacity of natural products: a review on chemical and cellular-based assays, Anal. Chim. Acta, 2013, 763, 1–10 CrossRef PubMed.
- H. Zhang and R. Tsao, Dietary polyphenols, oxidative stress and antioxidant and anti-inflammatory effects, Current Opinion in Food Science, 2016, 8, 33–42 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |