
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of Ru–Pt bimetallic monolayer on nanoporous gold film electrode and its application as an ultrasensitive sensor for determination of methionine
Nahid Tavakkoli *,
Nasrin Soltani and
Elahe Khorshidi
*,
Nasrin Soltani and
Elahe Khorshidi
Chemistry Department, Payame Noor University, 19395-4697 Tehran, Islamic Republic of Iran. E-mail: Tavakkolinahid@yahoo.com; Fax: +98-313-7381002
First published on 19th April 2017
Abstract
We report here the fabrication of ruthenium/platinum (RuPt) bimetallic monolayer coated on a nanoporous gold film electrode (RuPtNPGF) by underpotential deposition of copper (UPD) with the Cu layer then replaced spontaneously by Ru and Pt. The present method could provide a very low RuPt-loading electrode and the results demonstrated that ultra thin RuPt coating behaved efficiently as a bimetallic nanostructure RuPt for electrocatalytic oxidation of methionine. The structure and morphology of the catalyst were defined by using energy dispersive X-ray spectroscopy (EDX) and scanning electron microscopy (SEM), respectively. The RuPtNPGF electrode displayed good performance along with low working potential, high selectivity, and a low detection limit for the oxidation of methionine. Whereas at the surface of the bare electrode a redox peak for methionine cannot be observed, a sharp anodic peak at a potential of 0.73 V (vs. Ag/AgCl) in pH = 7.0 is obtained using the prepared RuPtNPGF electrode. Under optimized conditions differential pulse voltammetry (DPV) of methionine showed two linear ranges for the determination of methionine: 6–105 nmol L−1 and 3–102 μmol L−1 with a detection limit of 2 nmol L−1. Finally, the proposed sensor was successfully applied for the determination of methionine in human urine, with satisfactory results.
Introduction
Noble metal nanoparticles (NPs) have a very important effect in the improvement of new biosensors on nanotechnology to fulfill the demand for highly sensitive bio-molecular diagnostics.1 Metallic nanoparticles are known to exhibit unique advantageous properties over macro (or bulk) materials, such as increasing their catalytic properties, mass transport, and surface area with high performance, when used in electrochemical applications.Recently it has been shown that bimetallic nanoparticles (BMNP), composed of two metal elements in a particle, exhibit much higher catalytic activity than single metals because of synergistic or new bifunctional effects.2 Bimetallic nanoparticles are superior to monometallic nanocrystals due to improved catalytic, electrical and optical effects.3,4 BMNP often increase the selectivity of the reaction and may provide new properties by changing the composition of metals.5 Reactions that are catalyzed in the surface are enormously sensitive to the details in the atomic level of the catalytic surface.6 Specifically, bimetallic systems based on Pt with different structures of NPs such as mixed monometallic NPs, alloyed NPs, and core–shell NPs have shown themselves to be new architectures with extremely enhanced activity in catalysis and electrocatalysis;7 and, they are widely employed as electrodes for the oxidation of methanol and reduction of oxygen8–11 in fuel cell technology. However, their application in sensor development has been more limited. Bimetallic Pt–Ru catalysts stand in a very interesting and well-studied class of materials because of their excellent catalytic effects in methanation, hydrogenolysis, and methanol fuel cells.12,13 A biosensor for glucose was reported at carbon-paste enzyme electrodes dispersed with PtRu-on-Vulcan XC-70 carbon and illustrated significantly better sensitivity relative to a dispersion of pure metals.14
Methionine (Met), (α-amino-γ-methyl mercaptobutyric acid), CH3SCH2CH(NH2)COOH, is a sulfur-bearing monocarboxylic amino acid. Met was discovered in 1921 by Mueller.15 Met is a supply of sulfur in the body with an essential responsibility in biological methylation reactions and also prevents disorders in nails and hair or skin. As an essential amino acid, methionine is not synthesized in the body; it can be simply obtained from food supplies and pharmaceuticals are commercially available. Met is significant in the formation of blood proteins, albumins and globulins. Moreover, it helps to reduce cholesterol levels by enhancing lecithin production in the liver and maintains normal growth of cells.16 It is useful in the cure of AIDS associated myelopathy17 and also cancer.18 Human fluids like blood plasma,19,20 urine and serum contain Met.21 An abnormal concentration of Met leads mainly to coronary artery disease in the human body22 and hyper methioninemia and hyperhomocysteinemia in infants.23 Moreover, the variation in Met concentration in human fluids is related to several critical diseases and, thus, the determination of Met from human fluids is required for critical disease diagnosis.24,25 Levels of Met for a healthy human were determined to be ∼30.4 mM in human plasma,19 ∼25.5 mM in human serum21 and ∼5.9 mM in human urine.21 A number of methods to detect Met have been described, including gas chromatography-mass spectrometry,19,20 HPLC detection,26 spectrophotometry27 and conductometry.28 However, limitations such as advanced devices, long time analysis and technical expertise are mentioned in connection with these methods. Whereas electrochemical methods have various benefits, such as being less expensive, more convenient and extremely sensitive and selective. So far, few reports for the measurement of methionine using modified electrodes have been noted in the literature.29–32 However, these electrodes have major limitations, including poor selectivity, low sensitivity and tedious preparation.
Here we explain the preparation of a RuPt bimetallic coated nanoporous gold film electrode for application as an electrochemical sensor for the determination of methionine. The paper involves electrode fabrication via underpotential deposition of Cu at the nanoporous gold film surface and its replacement with Ru and Pt at open circuit potential. The electrochemical activity of the RuPt bimetallic catalyst for oxidation of methionine was investigated and compared with that of Ru and Pt monometallic catalysts. Moreover, the parameters affecting the deposition of the RuPt monolayer, including different ratios of Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt and codeposition or layer by layer deposition of RuPt, were optimized for a maximum electrocatalytic effect of bimetallic RuPt for the oxidation of methionine.
:Pt and codeposition or layer by layer deposition of RuPt, were optimized for a maximum electrocatalytic effect of bimetallic RuPt for the oxidation of methionine.
Experimental
An electrochemical cell with three-electrodes including a Compact Disc (CD) with a thin film gold as the working electrode, platinum wire as the counter electrode and an Ag/AgCl electrode as the reference electrode were employed. An electrochemical instrument (Autolab PGSTAT12) was used for all electrochemical experiments. In previous articles the cell structure and use of a CD as a working electrode have been explained.33,34 The smooth gold (SG) film electrode was oxidized by applying constant potential in phosphate buffer solutions (PBS) with pH = 7.0 to prepare the nanoporous gold film electrode (NPGF). Then the layer of gold oxide was reduced to gold atoms by ascorbic acid (AA). So a gold oxide film was first prepared by anodizing the SG at 5 V versus Ag/AgCl for 180 s in 0.1 M PBS, followed by rinsing with ultrapure water, and then transferred to 1 M AA solution for 3 min at open circuit potential (OCP). The pore formation and structural evolution have been revealed by scanning electron microscopy.The Cu UPD layer was formed by holding the potential at 0.25 V for 30 s in 0.1 M H2SO4 as the supporting electrolyte including Cu ions (1 mM CuSO4). An aqueous solution containing 5 mM Pt and 20 mM Ru, as the Pt and Ru metal ion sources, was used in the Cu UPD layer replacement reactions. The Cu UPD layer coated nanoporous gold film was put in a solution containing Pt and Ru metal ions for an optimal time of 10 min to carry out the replacement reactions. Variation in the morphology of the surface of the electrode was distinguished by using scanning electron microscopy (SEM) with a Vega-Tescan model.
All reagents of analytical grade were applied as received. The methionine was purchased from Sigma.
Result and discussion
The electrochemically effective surface area of the NPGF electrode was characterized by cyclic voltammetry in 0.5 M H2SO4.35 Fig. 1 shows cyclic voltammograms (CVs) for SG and NPGF electrodes in 0.5 M H2SO4 solution at a scan rate of 10 mV s−1 from 0.5 V to 1.5 V (versus Ag/AgCl). In the CV of SG an oxide layer was formed at a positive potential of 1.2 V during the anodic scan. On the subsequent cathodic scan, the oxide layer was stripped and a sharp peak was seen at a potential of 0.86 V in the voltammogram. The increase in anodic and cathodic peak current in the CV of NPGF confirms an enhancement in the effective surface area of the NPGF electrode. The area under the reduction peak is proportional to the charge required to reduce the gold oxide monolayer and this is associated with the surface area of the gold film.36 A specific charge of 386 μC cm−2 is required for gold oxide reduction,37 and the effective surface area of the NPGF electrode exhibits a 17.5 fold increase in comparison to an SG electrode.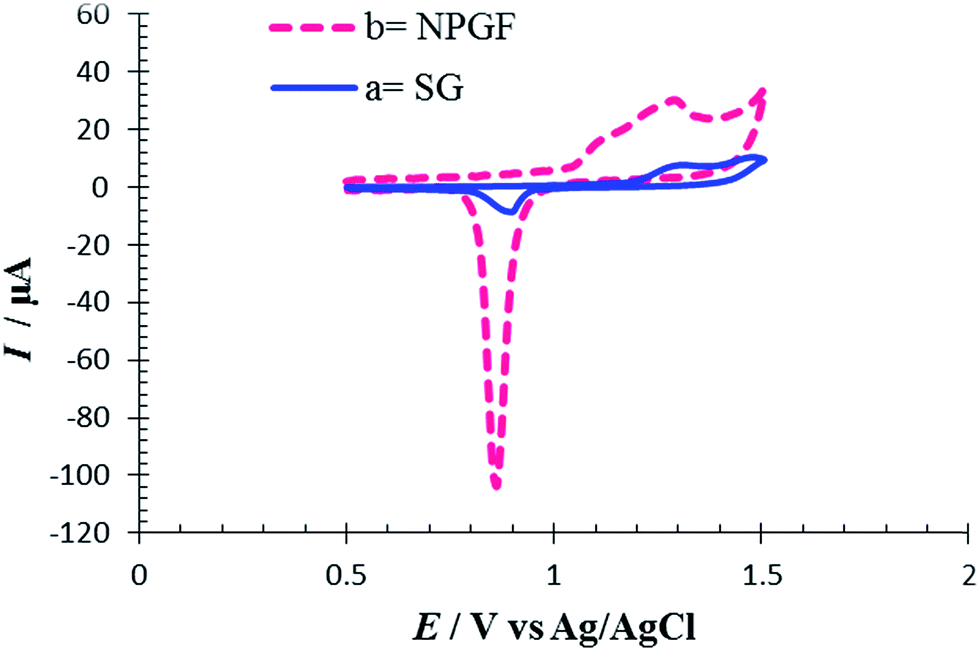 | ||
| Fig. 1 Cyclic voltammograms for (a) SG electrode and (b) a NPGF electrode in 0.5 mol L−1 H2SO4 solution. Scan rate 10 mV s−1. | ||
To perform the replacement deposition of Pt and Ru on the NPGF electrode surface, Cu as a precursor adlayer was deposited on the NPGF electrode in the UPD region. The redox replacement step occurred after the UPD process; this is a method for depositing sub-monolayer to a monolayer of metal on foreign substrates. The nanoporous gold film served as the cathode in the Cu UPD process. An ultrathin copper layer was formed by maintaining the potential at 0.25 V in a 0.1 M H2SO4 solution containing copper ions, which acts as the supporting electrolyte.
The CV of 0.5 M H2SO4 solution was recorded on an NPGF electrode after deposition of Cu to investigate the surface electrochemistry of a CuNPGF electrode (Fig. 2). As can be seen, the oxidation and reduction of Cu were observed at 0.31 V and 0.25 V versus Ag/AgCl, respectively. This evidence proves the existence of Cu on the surface of the NPGF electrode after underpotential deposition.
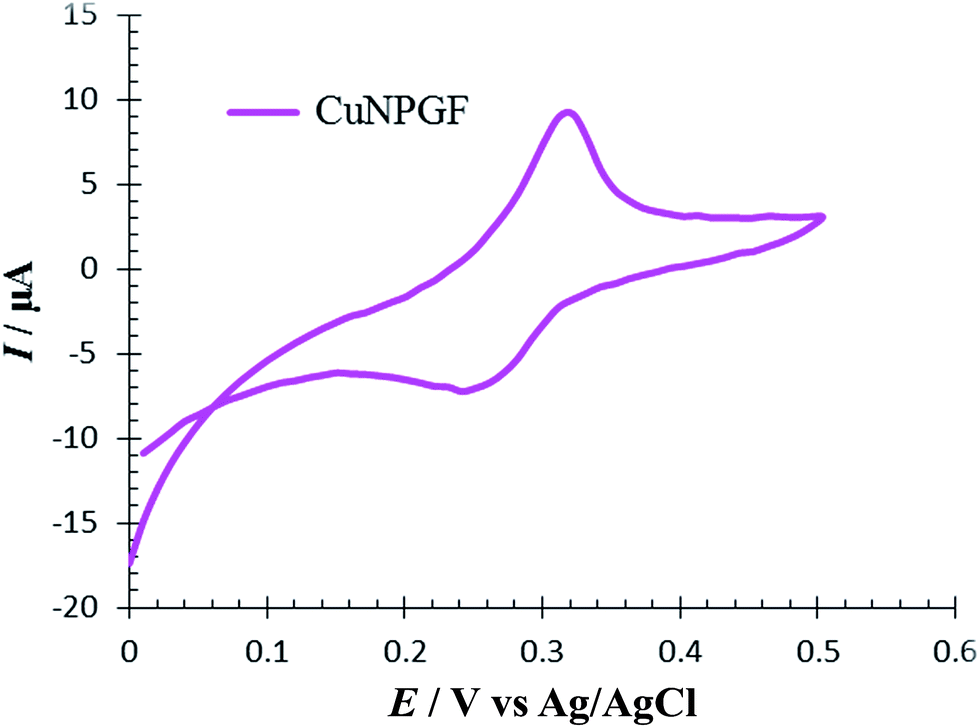 | ||
| Fig. 2 Cyclic voltammogram for CuNPGF electrode in 0.5 mol L−1 H2SO4 solution. Scan rate 10 mV s−1. | ||
To achieve RuPt deposition, the CuNPGF electrode was immersed into Pt and Ru solution to allow monoatomic surface Cu atoms to react with Pt(IV) and Ru(III) ions. The redox replacement of Cu with Pt and Ru can result in the formation of a mono- or sub-monolayer of PtRu, depending on the stoichiometry of the reaction. Assuming that all of the UPD Cu is oxidized to Cu2+, the stoichiometry of the reaction between Ru(III) and monolayer Cu is 2:3 and between Pt(IV) and Cu monolayer is 1:2, resulting in the deposition of a sub-monolayer of RuPt on the NPGF surface, defined as RuPtNPGF. Therefore, the CuNPGF electrode was immersed into a solution containing 5 mM Pt and 20 mM Ru for an optimum time of 10 min at open circuit potential. The SEM image of the RuPtNPGF electrode is shown in Fig. 3. Based on this figure, it can be concluded that even after the UPD of Cu and replacing the Cu by Ru and Pt, the electrode porosity is preserved. The relevant EDX spectra (Fig. 4) of the RuPtNPGF electrode surface qualitatively confirmed the presence of Ru and Pt (where expected) and quantitative (or close to) replacement of Cu by Pt as well as Ru during the simultaneous replacement reaction. According to the EDX results of 4:1 Ru:Pt (Table 1), it seems that the amount of deposited Ru is higher than the deposited Pt on the surface of electrode.
 | ||
| Fig. 3 SEM image of NPGF and RuPtNPGF electrodes. | ||
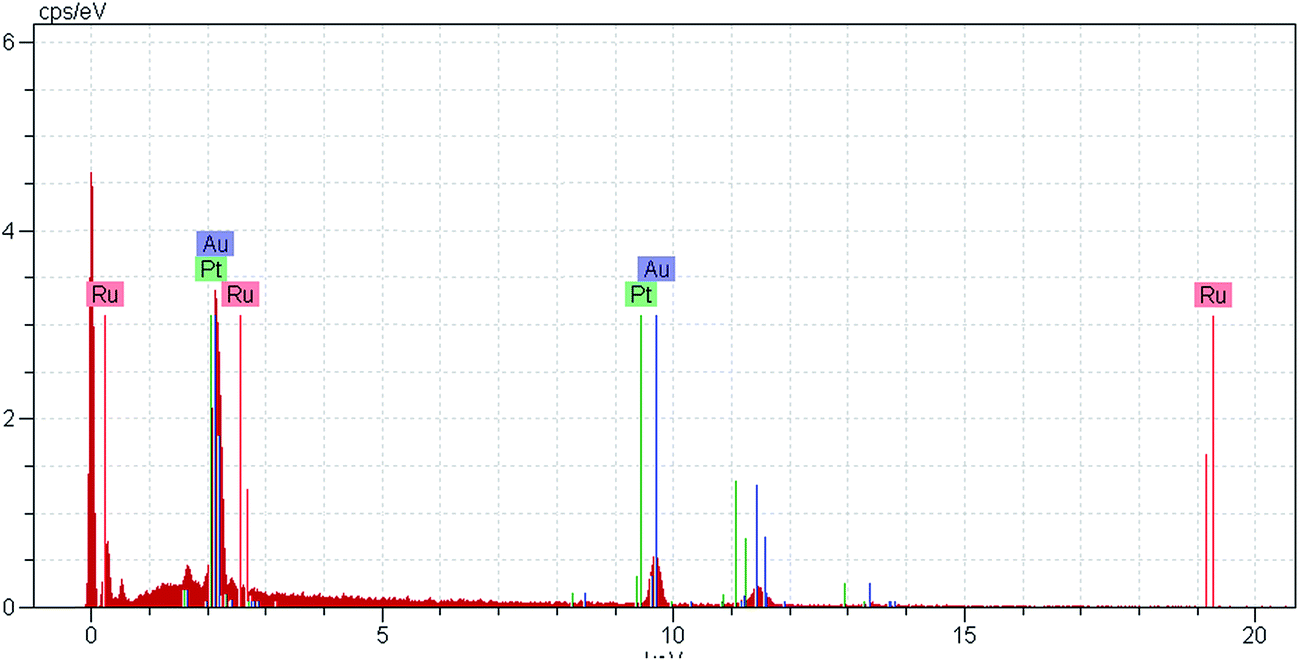 | ||
| Fig. 4 EDX pattern of RuPtNPGF electrode. | ||
| Element | Line | Int | W% | A% |
|---|---|---|---|---|
| Ru | K | 12.35 | 11.46 | 20.14 |
| Pt | L | 3.56 | 3.30 | 3.01 |
| Au | L | 91.81 | 85.23 | 76.85 |
To investigate the electrochemical behavior of NPGF, PtNPGF, RuNPGF, and RuPtNPGF electrodes, the CV method was used. Representative CVs obtained at a potential range from 0 V to 1.5 V at scan rate 10 mV s−1 for the above electrodes in 0.5 M H2SO4 solution are shown in Fig. 5. For the CV of the NPGF electrode a broad, double-layer region below ca. 0.6 V, the anodic oxide formation peak above 1.2 V in a positive scan direction, and the cathodic removal of oxide at 0.86 V in a negative scan direction are well-known features of gold electrochemistry.33 The CV of the PtNPGF electrode was obtained after deposition of an ultrathin Pt layer on the NPGF electrode. In the positive scan direction of CV, Pt oxidation is apparent from ∼0.61 V and shows an ascending aspect, which leads to a significant enhancement in current at the anodic potential limit. In the negative scan direction, the reduction peaks at 0.9 V and 0.3 V are related to gold oxide and platinum oxide, respectively. A significant decrease and shift to more positive potential in the negative scan direction of gold oxide indicate the deposition of an ultrathin platinum layer on an NPGF electrode such that only a few gold sites remain exposed. Such an electrochemical action has been presented previously for Pt.38–40 A rapid increase in oxidation current on the surface of the RuNPGF electrode is observed in the range of 1.0 and 1.2 V that conforms with the electrochemical formation of surface RuO2, considered a reversible oxide at E < 1.3 V (vs. Ag/AgCl), as noted elsewhere.41 Moreover, a significant observation is the enhancement in the double-layer charging current, due to a common feature of polycrystalline Ru bulk electrodes;38 also the Au oxide reduction profile becomes weak for the RuNPGF electrode. This observation confirms the presence of ruthenium on the NPGF electrode surface. The CV of RuPtNPGF shows a reduced peak in the potential region between 0.4 V and 0.8 V, which is attributed to reduction in surface oxide formed during the anodic scan between 1 and 1.4 V.
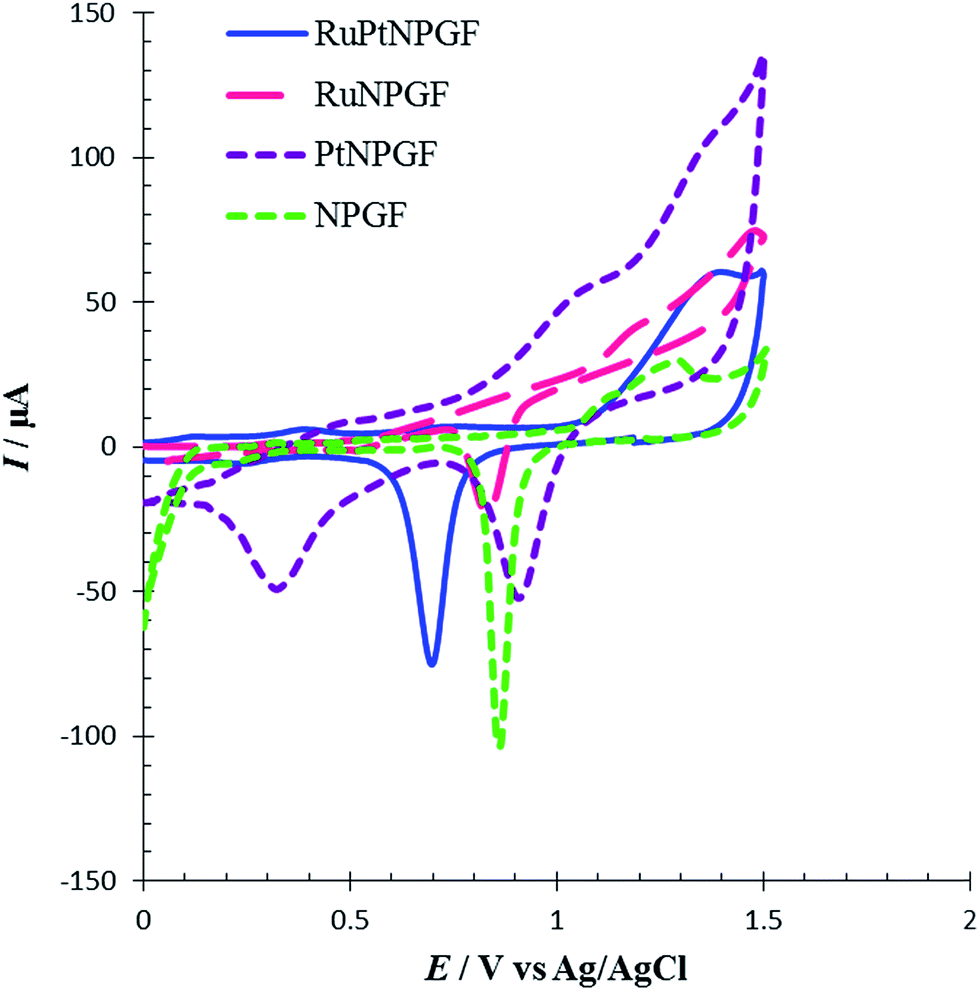 | ||
| Fig. 5 Cyclic voltammograms for NPGF, PtNPGF, RuNPGF and RuPtNPGF electrodes in 0.5 mol L−1 H2SO4 solution. Scan rate 10 mV s−1. | ||
The electrochemical performance of SG, NPGF, and RuPtNPGF electrodes was investigated by CV using the Fe (CN)63−/4− system as a benchmark redox reaction for various modified electrodes. CVs recorded at a scan rate of 10 mV s−1 for a 5 mM Fe (CN)63− and 5 mM Fe (CN)64− mixture contained in 0.1 M NaNO3 solution in the potential range between −0.4 V to 0.8 V are displayed in Fig. 6. The SG, NPGF, and RuPtNPGF electrodes show ΔEP as ∼345, 291, and 245 mV, respectively. In the RuPtNPGF electrode both the anodic and cathodic peak current of Fe (CN)63−/4− increased considerably, as compared with that of the NPGF and SG electrodes. Thus, the RuPtNPGF electrode improved the electron and mass transfer due to the increased surface area and lower electric resistance of the surface that are desirable for an electrochemical sensor.
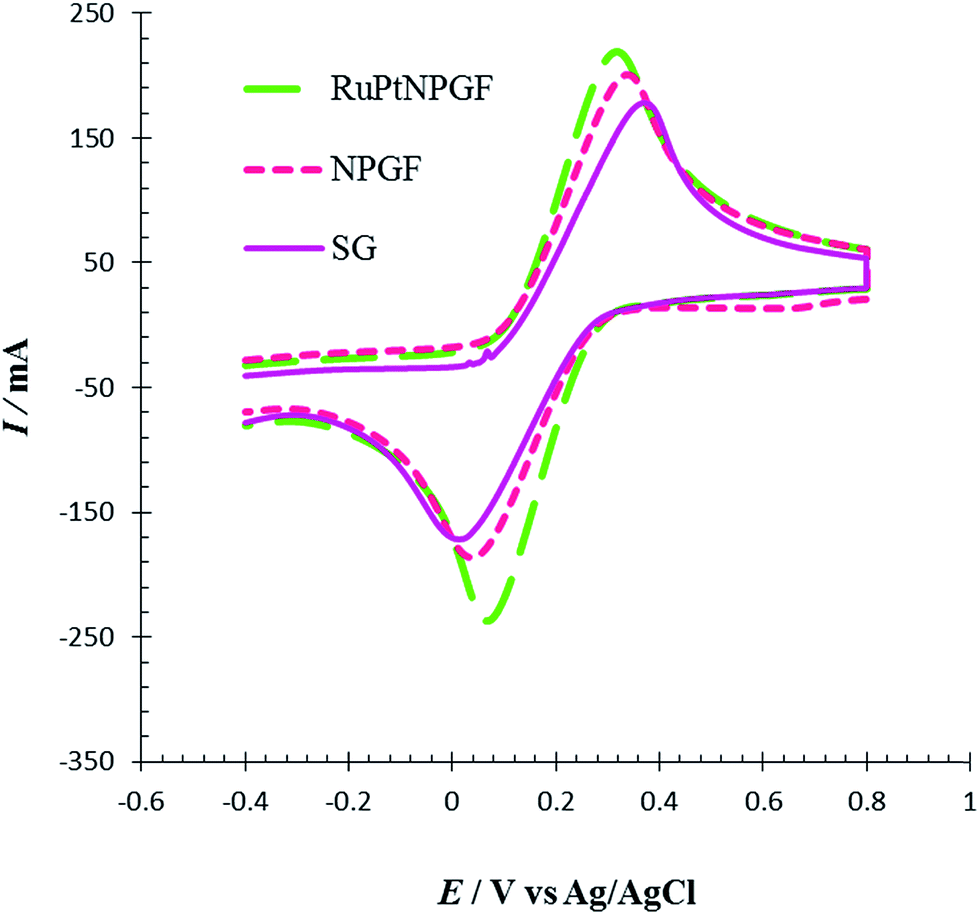 | ||
| Fig. 6 Cyclic voltammograms for SG, NPGF and RuPtNPGF electrodes in 5 mM K3[Fe(CN6)] + 5 mmol L−1 K4[Fe(CN6)] contained in 0.1 mol L−1 NaNO3 solution. Scan rate 10 mV s−1. | ||
The electrochemical behavior of Met in the surface of the RuPtNPGF electrode was investigated in 0.1 M PBS pH = 7 by cyclic voltammetry in the potential ranges −0.5 V to 1 V at a scan rate 10 mV s−1. Fig. 7 compares the cyclic voltammogram of the RuPtNPGF electrode in PBS without (curve a) and with (curve b) 100 μM Met. In the absence of Met, the voltammogram of the RuPtNPGF electrode shows the characteristic features of Ru and Pt. Upon addition of Met, the voltammetric characteristic of the RuPtNPGF electrode changes significantly and shows a sharp anodic peak for oxidation of Met in the range from 0.5 V to 0.85 V vs. Ag/AgCl. This anodic peak starts at 0.5 V and reaches a maximum height at a potential of 0.73 V. The RuPtNPGF electrode does not shows any redox peak in the presence of Met in the negative scan. Such a behavior implies an irreversible chemical reaction during the electro-oxidation of Met.
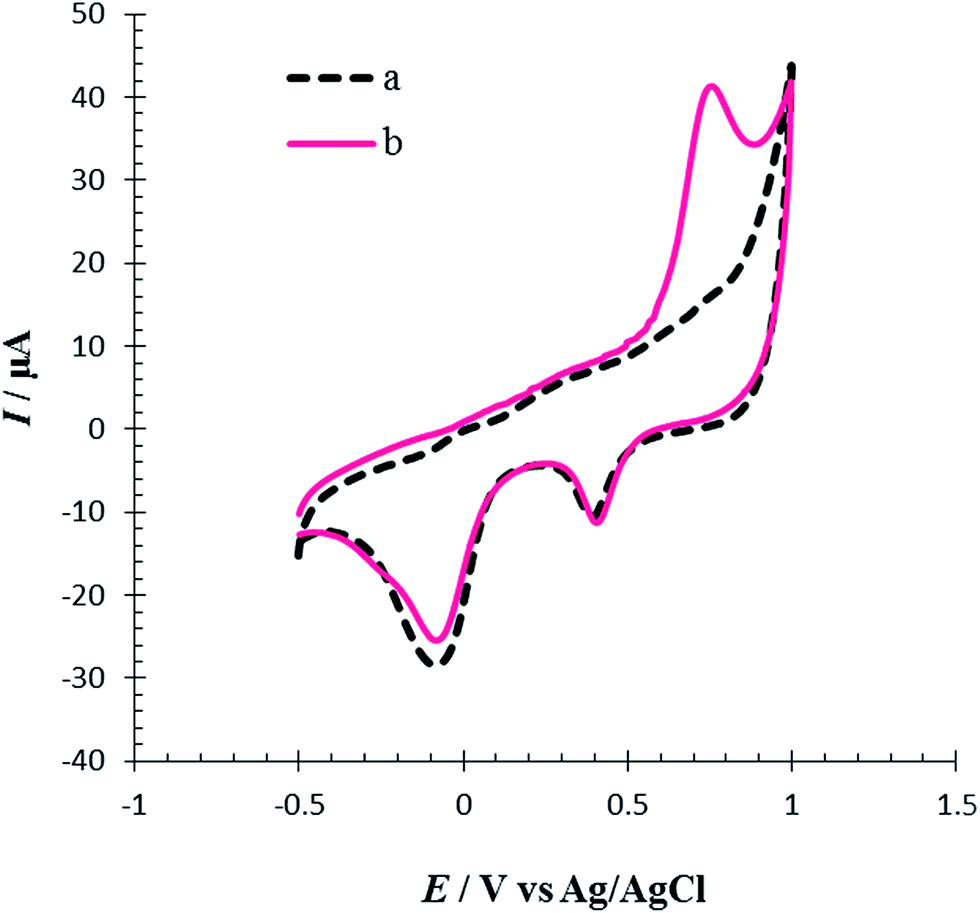 | ||
| Fig. 7 Cyclic voltammograms of RuPtNPGF electrode (a) without and (b) with 100 μmol L−1 Met solution in 0.1 M phosphate buffer (pH = 7) solution. Scan rate 10 mV s−1. | ||
The solution pH was varied from 5.0 to 9.0 to determine its effect on the catalytic oxidation of Met at the RuPtNPGF electrode. Fig. 8a shows the electrochemical behavior of Met in 0.1 M PBS with various pH, in the pH range 5.0–9.0, at the RuPdNPGF electrode using cyclic voltammetry. It was observed that the anodic peak current of Met reaches a maximum value at pH = 7.0, and decreases gradually as the pH increases (Fig. 8b). Also, the anodic peak potential of Met at the surface of the RuPtNPGF electrode shifts to a less positive value with increasing pH of the buffer solution (Fig. 8c). Therefore, pH = 7.0 was selected as the optimum pH for the following electrochemical detection of Met.
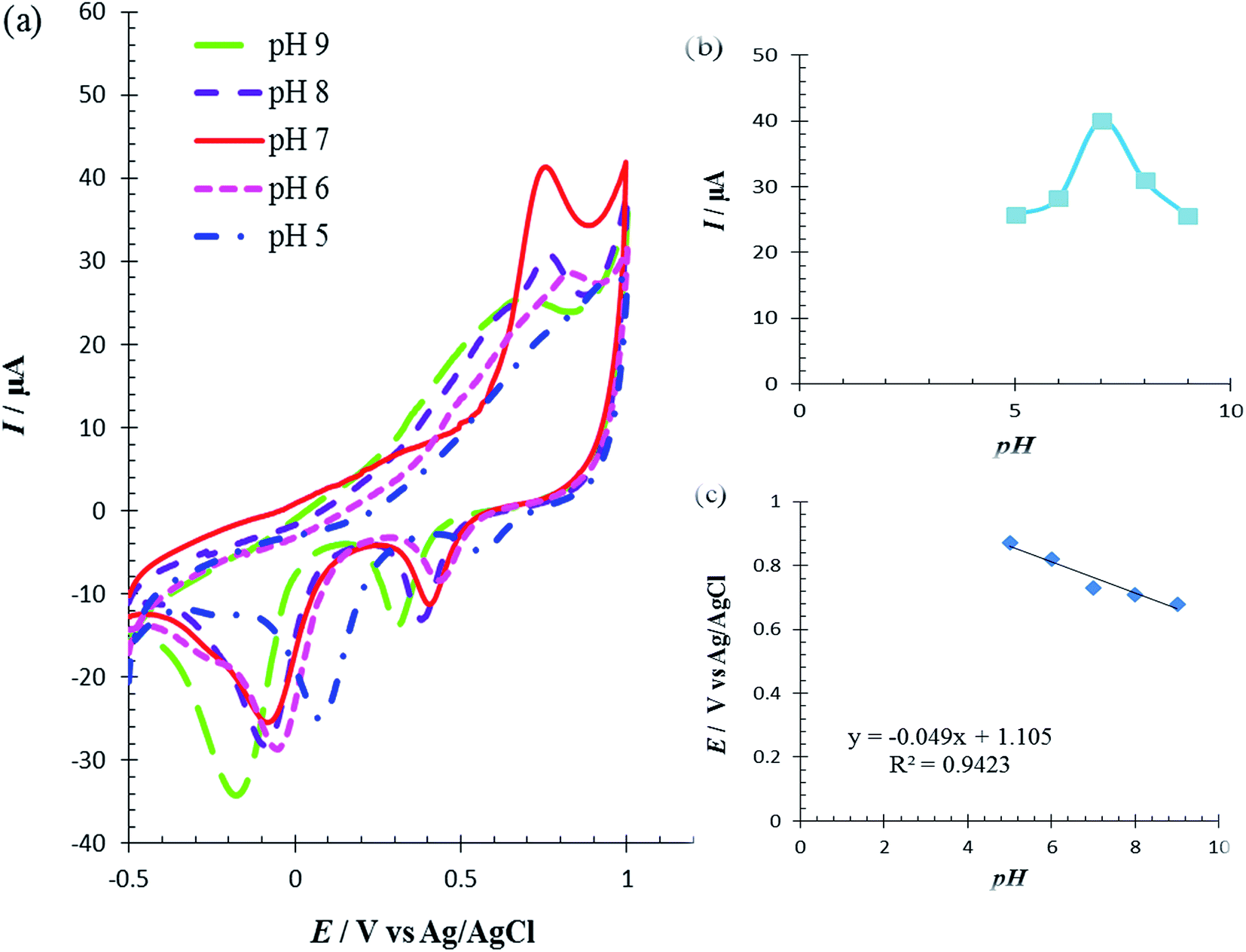 | ||
| Fig. 8 (a) Cyclic voltammograms (b) the effect of pH on anodic peak current (c) the influence of pH on anodic peak potential for oxidation of 100 μmol L−1 Met at the surface of the RuPtNPGF electrode at various pH (5–9) in 0.1 mol L−1 phosphate buffer solution. Scan rate 10 mV s−1. | ||
The irreversible oxidation process of Met which is pH dependent could be due to the oxidation of Met which has a sulfur atom in an intermediate position to a sulfone as the final product, as described as in eqn (1).32
| R–S–CH3 + 2H2O → R–SO2–CH3 + 4H+ + 4e− | (1) |
Met has four electrons involved in an irreversible process.42,43 The slope 0.049 V/pH of curve E/pH (Fig. 8c) indicated that electron transfer was accompanied by an equal number of protons in the electrode reaction of Met. This result confirms that the above-proposed mechanism is correct for Met oxidation at the surface of the RuPtNPGF electrode.
The effect of scan rate on the oxidation of Met was also studied (Fig. 9). The effect of scan rate was explored over the range 5.0 to 60.0 mV s−1 using a fixed concentration of Met. We have checked the relationship between the peak current and the scan rate. A linear correlation between the scan rate and the peak currents was gained with the equation I (μA) = 1.6023ν (mV s−1) + 20.194 and R2 = 0.9908. This indicated that the redox process was adsorption controlled. As the potential scan rate increased, the oxidation peak potential changed to more positive values, confirming the limitation of the kinetics of the electrochemical reaction.
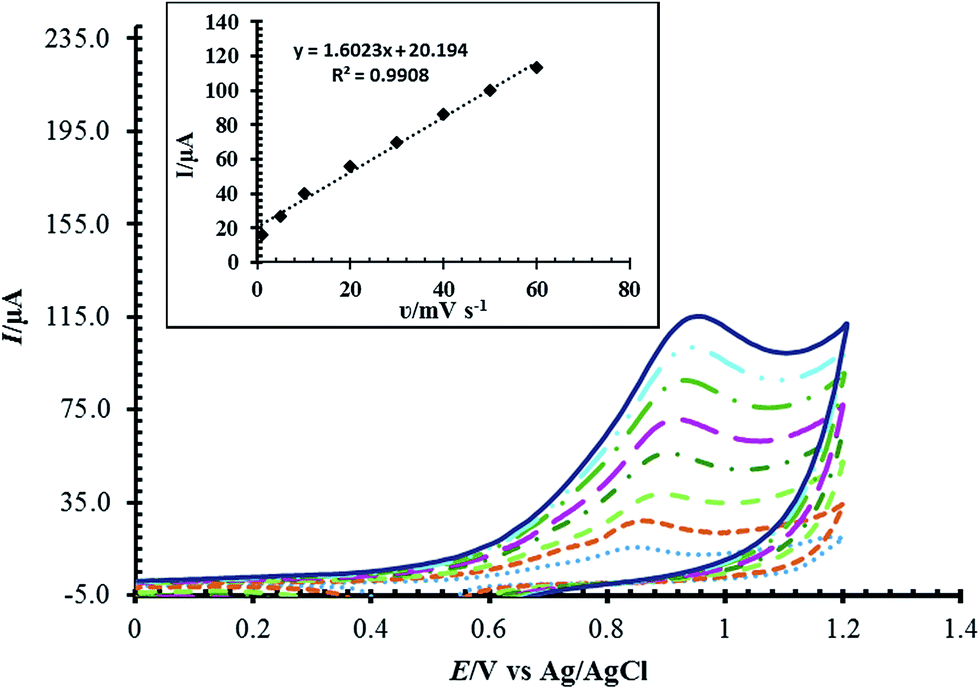 | ||
| Fig. 9 Cyclic voltammograms of RuPtNPGF electrode in the presence of 100 μmol L−1 Met in 0.1 mol L−1 phosphate buffer (pH = 7) solution at various scan rates 5–60 mV s−1. | ||
To demonstrate the special effect of the bimetallic RuPt catalyst in the RuPtNPGF electrode for the oxidation of Met, the CV characteristics of the oxidation of 100 μM Met at the SG, NPGF, PtNPGF, and RuNPGF electrodes were also studied and compared with those obtained at the RuPtNPGF electrode in PBS (0.1 M, pH 7.0) with 100 μM Met (Fig. 10). As can be seen in the CV of the SG electrode, at the surface of this electrode no electrochemical activity for methionine oxidation can be observed.
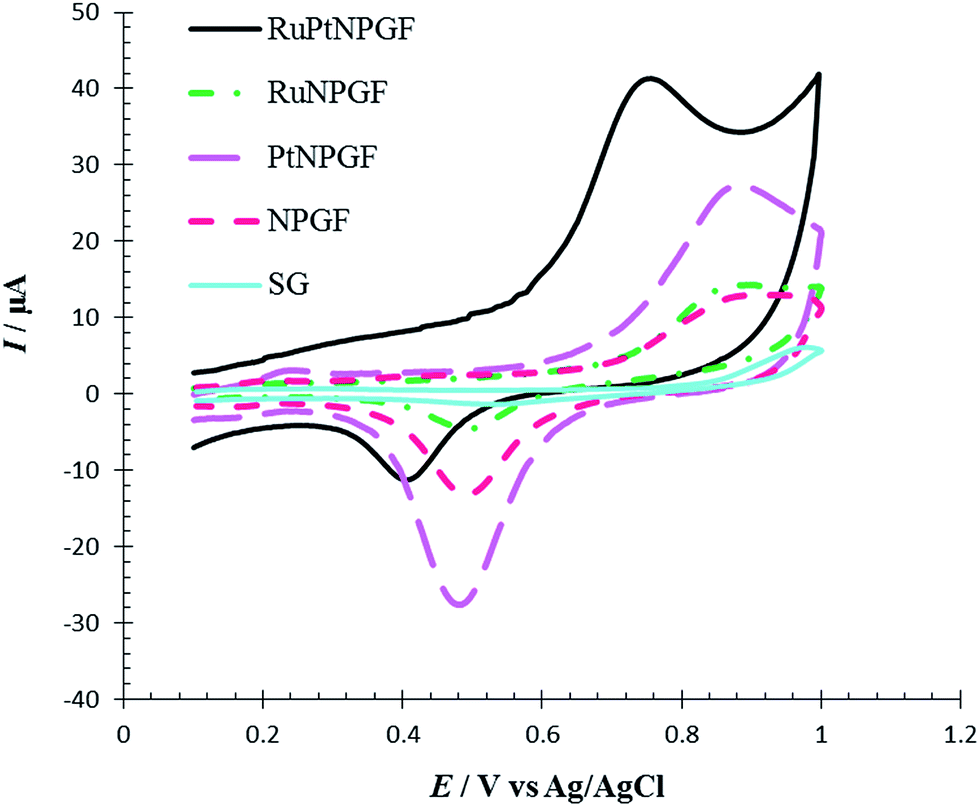 | ||
| Fig. 10 Cyclic voltammograms for oxidation of 100 μmol L−1 Met at the surface of SG, NPGF, PtNPGF, RuNPGF and RuPtNPGF electrodes in 0.1 mol L−1 phosphate buffer (pH = 7) solution. Scan rate 10 mV s−1. | ||
At the surface of the RuPtNPGF electrode, the onset potential of Met oxidation was 0.5 V, which is the lowest value compared with that on NPGF, PtNPGF, and RuNPGF electrodes (0.7 V). While the anodic peak potential for Met oxidation at the NPGF, RuNPGF and PtNPGF electrodes were 889, 882 and 868 mV, respectively, the potential at RuPtNPGF was 730 mV. Also, it is obvious that the electrocatalytic current at the RuPtNPGF electrode is ca. 3.13, 2.79 and 1.49 times those obtained for NPGF, RuNPGF and PtNPGF electrodes, respectively. It is observed that the combination of RuPt bimetallic catalyst and NPGF significantly improved the performance of the electrode for Met oxidation and, also, the electrocatalytic activity of bimetallic RuPt catalyst electrodes for Met oxidation was higher than for either monometallic Ru or Pt catalysts or the NPGF electrode. Obviously, the synergistic effect of the RuPt and NPGF cumulatively decreased the overvoltage as well as promoted the electron transfer of the Met, which is significant for the detection of Met.
The influence of different ratios of Ru/Pt on the Met electro-oxidation reaction was investigated by cyclic voltammetry. So, in the presence of different molar ratios of Pt and Ru ions, the replacement reaction was conducted in order to control the surface Ru/Pt composition. Fig. 11 shows CVs of oxidation of 100 μM of Met in PBS (pH = 7, 0.1 M) at the surface of RuPtNPGF electrodes with different ratios of Ru:Pt of 1:1, 1:4 and 4:1 at a scan rate of 10 mV s−1. This figure shows that the ratio Ru:Pt has a marked influence on the electrochemical activity of the RuPtNPGF electrode for oxidation of Met, with a clear maximum for Ru:Pt = 4:1. The current values of the anodic peak of Met oxidation decrease in the following order: Ru:Pt = 4:1 (18.80 μA) > Ru:Pt = 1:1 (8.32 μA) > Ru:Pt = 1:4 (6.10 μA). Moreover, the oxidation of Met at Ru:Pt = 4:1 occurred at a lower potential (730 mV) compared to Ru:Pt = 1:1 (800 mV) and Ru:Pt = 1:4 (0.826 mV). Thus, the RuPtNPGF electrode with a higher amount of Ru showed a superior performance toward Met oxidation.
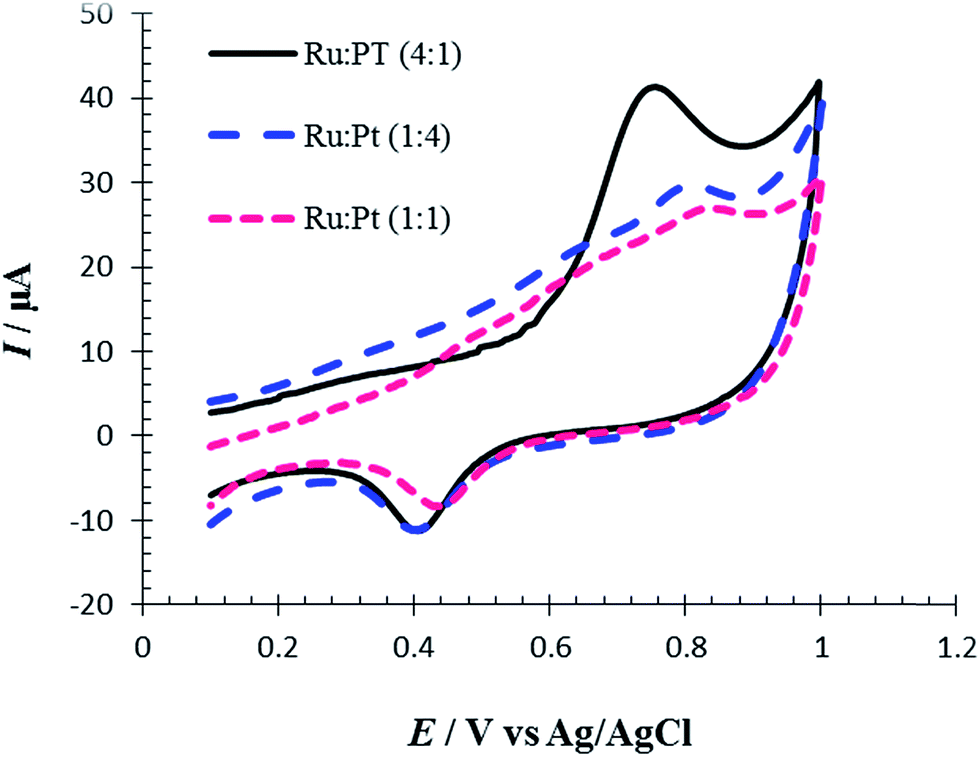 | ||
| Fig. 11 Cyclic voltammograms for electro-oxidation of 100 μmol L−1 Met at the surface of RuPtNPGF electrodes with different ratio of Ru:Pt. Scan rate 10 mV s−1. | ||
According to the literature,44,45 RuPt materials with different ratios showed about 50% Ru alloyed with Pt when the materials have higher amounts of Ru, while for materials with lower amounts of Ru only 10% Ru was alloyed with Pt. Thus, the increase in electrocatalyst activity of the RuPtNPGF electrode with a ratio of Ru:Pt = 4:1 for Met oxidation may be attributed to an increase in the degree of Ru alloyed with Pt. Similar behavior has been reported for RuPt catalysts toward methanol and co electro-oxidation.46,47
To compare the electrocatalytic properties of codeposition or layer by layer deposition of the Pt/Ru layer that formed on the NPGF electrode for Met oxidation, the cyclic voltammetry method was used. For this purpose, we fabricated an Ru-PtNPGF electrode (by repeated replacements, first forming a Pt monolayer-coating, followed by a Ru monolayer-coating), a Pt–RuNPGF electrode (by repeated replacements, first forming a Ru monolayer-coating, followed by a Pt monolayer-coating) and an RuPtNPGF electrode (codeposition of Ru and Pt). Fig. 12 shows CVs of Met oxidation in PBS (0.1 M, pH = 7.0) on the surface of the Ru–PtNPGF, Pt–RuNPGF and RuPtNPGF electrodes. It can be seen from Fig. 12 that the three catalysts have very different electrochemical behaviors. This observation indicated that the electrocatalytic activity of the RuPtNPGF electrode is much higher than the electrocatalytic activity of the Pt–RuNPGF and Ru–PtNPGF electrodes for Met oxidation.
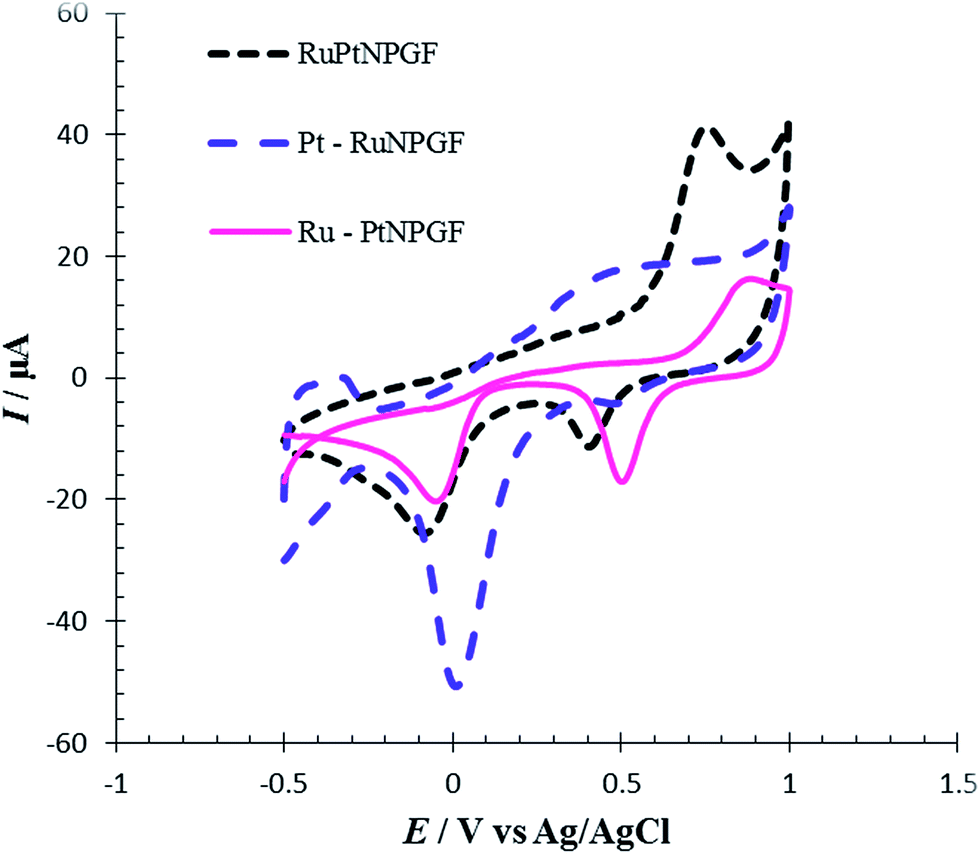 | ||
| Fig. 12 Cyclic voltammograms for electro-oxidation of 100 μmol L−1 Met at the surface of RuPtNPGF, Ru–PtNPGF and Pt–RuNPGF electrodes at scan rate 10 mV s−1. | ||
The differential pulse voltammetry (DPV) method, a very sensitive method, was used to estimate the linear calibration range of Met and the lower range of detection. DPV waves for Met were obtained in 0.1 M PBS (pH = 7.0) in the surface of the RuPtNPGF electrode (Fig. 13a). The calibration curves for these determinations, which are displayed in Fig. 13b and c, exhibit two linear range segments with different slopes for Met concentration: for 6 to 105 nmol L−1 Met and 3 to 102 μmol L−1 Met. The detection limit for determination of Met using this method was 2 nmol L−1.
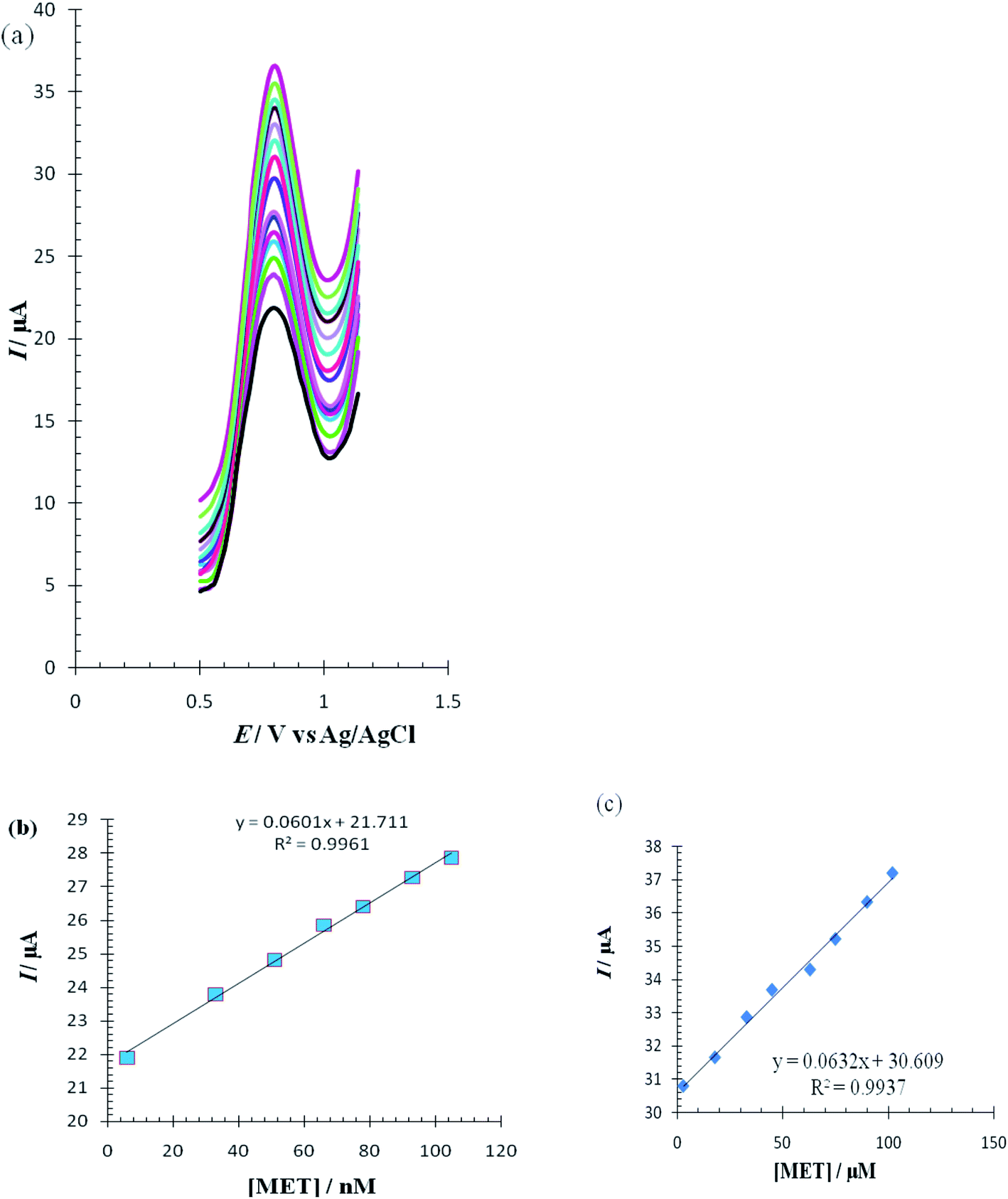 | ||
| Fig. 13 (a) Differential pulse voltammograms of Met under optimum conditions; (b) shows plot of 6, 33, 51, 66, 78, 93, 105 nmol L−1 of Met and (c) shows plot of 3, 18, 33, 45, 63, 75, 90, 102 μmol L−1 of Met in phosphate buffer solution (pH = 7, 0.1 mol L−1). | ||
The effect of various interfering foreign species such as glucose, captopril, ascorbic acid, uric acid, cysteine and sodium chloride, on the oxidation peak of the Met was studied using the DPV method at a range of potential from 0.0 V to 1.2 V. It is observed that there was no response to any of these compounds on the surface of the RuPtNPGF electrode. Moreover, no change in the anodic peak of Met oxidation was observed for 100 μmol L−1 Met in the presence of these species (400 times), indicating that the RuPtNPGF electrode possessed high selectivity for Met.
The practical application of the RuPtNPGF electrode for electro-oxidation of Met was demonstrated by measuring the concentration of Met from human urine samples. The samples of human urine were obtained from volunteers against an informed consent and used without any dilution. Determination of Met was performed using the standard addition method. We added a known concentration of Met to the urine sample. The recovery results of different additions of Met in human urine samples are summarized in Table 2. As can be seen from Table 2, the proposed method showed good recoveries for the spiked Met in human urine samples, indicating that the present method could be applied to determine Met in real samples. Therefore, this sensor can be used as a very highly sensitive detection device for Met in the clinical laboratory.
| Sample | Methionine added (μmol L−1) | Methionine founded (μmol L−1) | Recovery (%) |
|---|---|---|---|
| 1 | — | — | — |
| 10 | 10.20 ± 0.3 | 102.00 | |
| 20 | 29.4 ± 0.7 | 98.01 | |
| 2 | — | — | — |
| 30 | 29.6 ± 0.9 | 98.66 | |
| 50 | 78.3 ± 1.4 | 97.88 |
The long-term stability of the RuPtNPGF electrode was tested over a three week period. After this, DPV recorded for RuPtNPGF showed a peak potential for Met oxidation that was unchanged and the current signal showed a less than 2.7% decrease relative to the initial response. The oxidation peak current remained the same with a relative standard deviation of 1.9% for 15 repetitive DPV measurements in BPS (0.1 mol L−1, pH = 7), indicating that this electrode has better repeatability.
The reproducibility of the Met sensor was evaluated by a comparison of the currents of different electrodes constructed under the same conditions. The DPV response of the 5 RuPtNPGF electrodes to 100 μmol L−1 Met was examined, the results providing an RSD of 2.3%. Good stability and reproducibility showed that the catalyst layer on the surface of the electrode was structurally stable, free from morphological change, and not significantly affected by repeated measurement under rigorous conditions.
To estimate the performance of the proposed electrode as a sensor for the determination of Met, the results of this study were compared with results reported by other research groups for the electrocatalytic oxidation of Met at the surface of chemically modified electrodes (Table 3).
| Electrode | Linear range (μmol L−1) | Limit of detection (μmol L−1) | Potential of anodic peak (V) vs. Ag/AgCl | Reference |
|---|---|---|---|---|
| a GCE: glassy carbon electrode, CPE: carbon paste electrode. | ||||
| Ru(II)Den CP | 0.0064–10 | 0.002 | +1.0 | 29 |
| Pt–TiO2/CNT/GCE | 3–100 | 1 | +1.1 | 30 |
| p-4α-CuIITAPc–GC | 50–500 | 0.027 | +1.26 | 31 |
| nAu-Cyst-CPE | 1–100 | 0.59 | +0.95 | 32 |
| RuPtNPGF | 0.006–0.105 and 3–102 | 0.003 | +0.789 | This work |
Conclusion
The preparation of an electrocatalyst consisting of an RuPt monolayer coating on a nonporous gold film (RuPtNPGF) electrode has been demonstrated. The RuPtNPGF electrode was applied to the electro-oxidation of Met in aqueous media. Outstanding features, such as a low onset potential for oxidation, low detection limit, wide linear range, and high reproducibility and repeatability, demonstrate the successful application of the RuPtNPGF electrode for the determination of Met. Also, we have shown that the electrocatalytic activity of the RuPtNPGF electrode is much higher than the electrocatalytic activity of PtNPGF and RuNPGF electrodes for Met oxidation. Therefore, the electronic interactions between the Ru and Pt atoms play a main role in promoting Met electro-oxidation. The results suggest that the method developed in this study provides a comfortable, cheap and fast way to form a bimetallic sensor with excellent performance for electrocatalytic oxidation. Finally, the RuPtNPGF electrode indicated good recovery for Met in human urine samples, which showed that the present method may be used for practical applications.Acknowledgements
The authors gratefully acknowledge financial support of the Research Council of Payame Noor University.References
- G. Doria, J. Conde, B. Veigas, L. Giestas, C. Almeida, M. Assunção, J. Rosa and P. V. Baptista, Sensors, 2012, 12, 1657–1687 CrossRef CAS PubMed.
- S. Tokonami, Y. Yamamoto, H. Shiigi and T. Nagaoka, Anal. Chim. Acta, 2012, 716, 76–91 CrossRef CAS PubMed.
- N. Toshima and Y. Wang, Adv. Mater., 1994, 6, 245–247 CrossRef CAS.
- N. Toshima and P. Lu, Chem. Lett., 1996, 729–730 CrossRef CAS.
- L.-u. Rahman, R. Qureshi, M. M. Yasinzai and A. Shah, C. R. Chim., 2012, 15, 533–538 CrossRef CAS.
- G. A. Somorjai and Y. Li, Introduction to surface chemistry and catalysis, John Wiley & Sons, 2010 Search PubMed.
- K.-J. Chen, C.-F. Lee, J. Rick, S.-H. Wang, C.-C. Liu and B.-J. Hwang, Biosens. Bioelectron., 2012, 33, 75–81 CrossRef CAS PubMed.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- S. H. Chang, W. N. Su, M. H. Yeh, C. J. Pan, K. L. Yu, D. G. Liu, J. F. Lee and B. J. Hwang, Chem.–Eur. J., 2010, 16, 11064–11071 CrossRef CAS PubMed.
- Y. Kim, J. W. Hong, Y. W. Lee, M. Kim, D. Kim, W. S. Yun and S. W. Han, Angew. Chem., 2010, 122, 10395–10399 CrossRef.
- W. Li, X. Wang, Z. Chen, M. Waje and Y. Yan, J. Phys. Chem. B, 2006, 110, 15353–15358 CrossRef CAS PubMed.
- W. Xhou, W. Li, Z. Zhou, S. Song, Z. Wei, G. Sun, P. Tsiakaras and Q. Xin, Chem. Res. Chin. Univ., 2002, 24, 858–862 Search PubMed.
- K.-J. Chen, K. C. Pillai, J. Rick, C.-J. Pan, S.-H. Wang, C.-C. Liu and B.-J. Hwang, Biosens. Bioelectron., 2012, 33, 120–127 CrossRef CAS PubMed.
- J. H. Mueller, Exp. Biol. Med., 1922, 19, 161–163 CrossRef.
- A. Salimi and M. Roushani, Electroanalysis, 2006, 18, 2129–2136 CrossRef CAS.
- A. DiRocco, M. Tagliati, D. Dorfman, J. Moise and D. Simpson, Neurology, 1996, 46(2), 6124 Search PubMed.
- S. Toyokuni, K. Okamoto, J. Yodoi and H. Hiai, FEBS Lett., 1995, 358, 1–3 CrossRef CAS PubMed.
- P. Jandik, J. Cheng, J. Evrovski and N. Avdalovic, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 759, 145–151 CrossRef CAS.
- A. Zinellu, S. Sotgia, M. F. Usai, E. Zinellu, A. M. Posadino, L. Gaspa, R. Chessa, A. Pinna, F. Carta and L. Deiana, Anal. Biochem., 2007, 363, 91–96 CrossRef CAS PubMed.
- S. P. Stabler, P. D. Marcell, E. R. Podell and R. H. Allen, Anal. Biochem., 1987, 162, 185–196 CrossRef CAS PubMed.
- D. R. Murphy-Chutorian, M. P. Wexman, A. J. Grieco, J. A. Heininger, E. Glassman, G. E. Gaull, S. K. Ng, F. Feit, K. Wexman and A. C. Fox, J. Am. Coll. Cardiol., 1985, 6, 725–730 CrossRef CAS PubMed.
- S. H. Mudd, N. Braverman, M. Pomper, K. Tezcan, J. Kronick, P. Jayakar, C. Garganta, M. G. Ampola, H. L. Levy and S. E. McCandless, Mol. Genet. Metab., 2003, 79, 6–16 CrossRef.
- M. López-Torres and G. Barja, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 1337–1347 CrossRef PubMed.
- S. C. Lu, H. Tsukamoto and J. M. Mato, Alcohol, 2002, 27, 155–162 CrossRef CAS PubMed.
- S. Melnyk, M. Pogribna, I. Pogribny, R. J. Hine and S. J. James, J. Nutr. Biochem., 1999, 10, 490–497 CrossRef CAS PubMed.
- F. Blasco, M. Medina-Hernandez and S. Sagrado, Anal. Chim. Acta, 1997, 348, 151–159 CrossRef CAS.
- A. Samadi-Maybodi and K. Abolfazli, Int. J. Electrochem. Sci., 2009, 4, 684–693 CAS.
- L. Cheng, G. E. Pacey and J. A. Cox, Anal. Chem., 2001, 73, 5607–5610 CrossRef CAS PubMed.
- F. Chekin, S. Bagheri and S. B. AbdHamid, Sens. Actuators, B, 2013, 177, 898–903 CrossRef CAS.
- A. J. Jeevagan and S. A. John, Bioelectrochemistry, 2012, 85, 50–55 CrossRef CAS PubMed.
- L. Agüı, J. Manso, P. Yáñez-Sedeño and J. Pingarrón, Talanta, 2004, 64, 1041–1047 CrossRef PubMed.
- N. Tavakkoli, N. Soltani and E. Khorshidi, Electrochim. Acta, 2015, 164, 1–11 CrossRef CAS.
- N. Tavakkoli, S. Nasrollahi and G. Vatankhah, Electroanalysis, 2012, 24, 368–375 CrossRef CAS.
- S. Trasatti and O. Petrii, Pure Appl. Chem., 1991, 63, 711–734 CrossRef CAS.
- K. Bonroy, J.-M. Friedt, F. Frederix, W. Laureyn, S. Langerock, A. Campitelli, M. Sára, G. Borghs, B. Goddeeris and P. Declerck, Anal. Chem., 2004, 76, 4299–4306 CrossRef CAS PubMed.
- R. Szamocki, A. Velichko, C. Holzapfel, F. Mücklich, S. Ravaine, P. Garrigue, N. Sojic, R. Hempelmann and A. Kuhn, Anal. Chem., 2007, 79, 533–539 CrossRef CAS PubMed.
- T. S. Mkwizu, M. K. Mathe and I. Cukrowski, Langmuir, 2009, 26, 570–580 CrossRef PubMed.
- M. Khosravi and M. K. Amini, Int. J. Hydrogen Energy, 2010, 35, 10527–10538 CrossRef CAS.
- S. H. Yoo and S. Park, Adv. Mater., 2007, 19, 1612–1615 CrossRef CAS.
- S. Hadz, H. Angerstein-Kozlowska, M. Vukovič and B. Conway, J. Electrochem. Soc., 1978, 125, 1471–1480 CrossRef.
- W. e. T. Tan and J. e. K. Goh, Electroanalysis, 2008, 20, 2447–2453 CrossRef CAS.
- C. E. Banks and R. G. Compton, Analyst, 2006, 131, 15–21 RSC.
- V. Radmilovic, H. Gasteiger and P. Ross, J. Catal., 1995, 154, 98–106 CrossRef CAS.
- J. Guo, T. Zhao, J. Prabhuram, R. Chen and C. Wong, Electrochim. Acta, 2005, 51, 754–763 CrossRef CAS.
- M. M. Tusi, N. S. de Oliveira Polanco, M. Brandalise, O. V. Correa, J. C. Villalba, F. J. Anaissi, A. O. Neto and E. Spinacé, Int. J. Electrochem. Sci., 2011, 6, 484–491 CAS.
- Y. Jin-hua, W. Feng-bin and X. Xing-huaC, J. Electrochem., 2014, 20, 416–425 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |