
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLignin derived activated carbon particulates as an electric supercapacitor: carbonization and activation on porous structures and microstructures†
Sixiao Hu and
You-Lo Hsieh *
*
Fiber and Polymer Science, University of California, Davis, CA 95616, USA. E-mail: ylhsieh@ucdavis.edu; Fax: +1 530 752 7584; Tel: +1 530 752 0843
First published on 13th June 2017
Abstract
Microporous and mesoporous particulate activated carbons (PACs) with unique hierarchical microstructures were facilely synthesized from alkali lignin via simultaneous carbonization and alkali hydroxide activation. Both NaOH and KOH activated PACs contained slit-like micropores and carbon microstructures with circular nanoplates of graphite-like basal planes and amorphous carbon clusters. The micropores broadened with increasing temperatures, holding time and alkaline hydroxides. The basal planes sizes and order were enhanced with increasing temperatures and holding time but lowering impregnation ratios, while amorphous carbon showed no particular patterns due to its much higher reactivity toward alkali hydroxides. The PACs with the highest micropore surface area and pore volume (1100 m2 g−1, 0.43 cm3 g−1) were obtained at 900 °C, 30 min and an impregnation ratio of 1 and fabricated into electrical supercapacitors to exhibit excellent 226 F g−1 specific capacitance, 7.8 W h kg−1 energy density and 47 kW kg−1 power density as well as over 92% capacitance retention after 5000 cycles.
Introduction
Activated carbons are high surface porous materials, typically in the forms of powders, granulates or fibers, that are widely used in water filtration, air purification, oil spill clean-up, energy and fuel storage, etc.1 More recently, activated carbons have also found successful use in electrical supercapacitors, which are intermediate energy storage devices with properties falling between dielectric capacitors and electrochemical batteries, that could store high power (up to a few hundred farads per gram) in milliseconds and electrical charges at the electrode/electrolyte interfaces.2,3 The high specific capacitance, power and energy density of such supercapacitors have been associated with good electrical conductivity, large surface area and microporosity of activated carbons.4,5 The pore characteristics are in particular critical to achieve excellent electrochemical properties. The presence of mesopores was found crucial to the transfer of electrolytes to the micropore regions.6 The relative pore size in relationship to the electrolyte dimensions, rather than the absolute pore size, influenced the capacitance.7 Pore sizes close to that of monovalent electrolyte ion diameter could help to strip its hydration shell to squeeze into the narrow pore to improve electrolyte adsorption onto the electrodes.8,9Various highly porous activated carbon based supercapacitors have been prepared from commercially available particulate activated carbons (PACs),10 graphene,11 anthracite,12 aromatic hydrocarbon resin,13 bituminous coal14 as well as olive pits,15 cottons16 and tea-leaves.17 The optimal specific capacitance of these activated carbons mainly ranged from 80 to 330 F g−1 for aqueous electrolytes and from 50 to 150 F g−1 for organic electrolytes. Such variances have been attributed to their different carbon microstructures and pore structures which were challenging to fine-tune due to the chemical heterogeneity of the precursors and the not well understood effects of pyrolysis parameters.
Lignocellulosics have been among the major sources of activated carbons whose properties depend on the precursors' lignin, cellulose and hemicelluloses compositions and respective structures.18,19 Lignin contributes the most to the carbon mass due to its high carbon content and crosslinked polyphenolic structure whereas holocelluloses decompose extensively and gasified during pyrolysis.20 At over 1.1 million metric tons per year, isolated lignin, the major by-product of chemical pulping and biofuel production,21 is readily available and under-utilized. We have previously prepared microporous activated carbon fibers (ACFs) by electrospinning aqueous low sulfonate content alkali lignin (ALls) with ca. 10% fiber-forming polyethylene oxide, followed by simultaneous carbonization and activation at 850 °C.22 These super-fine (submicron wide), high specific surface (up to 1400 m2 g−1) ACFs were fabricated into supercapacitor cells to exhibit outstanding energy storage performance in aqueous KOH electrolyte with specific capacitance as high as 344 F g−1,23 higher than those prepared from commercially available PACs and other activated carbons previously mentioned.10–17 Producing PACs from lignin is attractive as the abundant and readily available feedstock is conducive to industrial scale manufacturing. In fact, highly porous PACs with up to 3000 m2 g−1 specific surface have been derived from kraft lignin via zinc chloride24 and alkali hydroxide25 activation. However, their microstructures have not been characterized and their applications unexplored.
With the demonstrated success of microporous ACFs as supercapacitors,23 this work further streamlined the simultaneous carbonization and activation approach to convert lignin directly to PACs, bypassing both electrospinning and incorporating fiber-forming synthetic polymer. The effects of in situ impregnation of alkali hydroxides as well as pyrolytic conditions on their carbon and pore structures were characterized by Raman spectroscopy, scanning and transmission electron microscopy, atomic force microscopy and gas adsorption. The PACs with the highest specific surface were further fabricated into supercapacitors and their detailed electrochemical properties including specific capacitance, equivalent series resistance, cycling stability, energy and power density were evaluated.
Experimental
Chemicals
Alkali lignin (low sulfonate) (ALls) (Mw = 60 kDa, spruce origin) were acquired from Sigma-Aldrich (USA), sodium hydroxide (anhydrous pallets, A.C.S. grade, 85% minimum purity) and potassium hydroxide (anhydrous pallets, A.C.S. grade 97% minimum purity) were from Fisher Scientific (USA). All the chemicals were used as received.Preparation of PACs
Aqueous mixtures were prepared at 0.5, 1 and 2 alkali hydroxide/ALls w/w ratios at total concentrations of 15, 20 and 30 wt% respectively, then dried (60 °C, 72 h) to precursor granulates to be placed in a quartz tube (2 cm inner diameter) of an electric furnace (Mini-Mite, Lindberg/Blue). Simultaneous carbonization and activation was performed by heating at 10 °C min to 105 °C and held for 30 min to drive off moisture, then to 850 °C (held for 30 min), 900 °C (held for 5, 30 and 60 min), or 950 °C (held for 30 min), all under flowing N2 at 100 mL min−1. The heat processed granulates were cooled to ambient temperature within 12 h, also under flowing N2 at 100 mL min−1, then washed with deionized water to remove residual alkali metals and other small hydrocarbon impurities and dried at 105 °C for 30 min to particulates activated carbons (PACs).Analytical methods
The thermal properties and yields of the ALls were determined by thermogravimetric analysis (TGA) (TGA-50, Shimadzu) by heating at 10 °C min−1 under flowing N2 at 50 mL min−1 to 850, 900 and 950 °C, then holding for 30 min. The atomic compositions of PACs were analyzed by energy-dispersive X-ray spectroscopy (EDX) adjunct to a scanning electron microscope (SEM) (FEI-XL 30, FEI). The sp2 and sp3 carbon configurations and proportions in PACs were investigated by Raman spectroscopy (RM1000, Renishaw) with excitation source of a 514.5 nm Ar line. The G and D peaks in Raman spectrums were resolved into respective G1 & G2 and D1 & D2 peaks using four-Gaussian deconvolution for non-graphitic carbons23,26,27 using OriginPro (version 8.1.34.90) and with coefficient of determination (COD) calculated. According to the three-stage graphene amorphization trajectory, the grain sizes of basal planes and sp2 carbon were estimated by eqn (1) (ref. 28) and (2),29 respectively. Their sums were used to approximate the total PAC grain size to correlate with morphological features observed by electron microscopy. The defects of the basal planes and sp2 carbon clusters were examined via the full width at half maximum (FWHM) of D1 and G2 peak, respectively.| I(D)/I(G) = C(λ)/La; C(515 nm) = 4.4 nm | (1) |
| I(D)/I(G) = C′(λ)/La2; C(515 nm) = 0.005 | (2) |
For surface area and pore characterization, PACs were dried at 50 °C for 48 h then measured at 77 K by a nitrogen adsorption–desorption analyzer (ASAP 2020, Micrometrics). The single point total pore volume was estimated from the adsorption branch of the isotherm at a relative pressure P/Po close to 1. The Brunauer–Emmett–Teller (BET) surface area was calculated from the isotherm in the BET linear region where relative pressure P/Po ranged from 0.01 to 0.1, with the exception of PACs from NaOH at impregnation ratio of 2 where the BET were calculated from relative pressure P/Po ranged from 0.01 to 0.3. The mesopore surface area was derived from the adsorption branch whereas pore and neck size distributions were derived from both adsorption and desorption branches of the isotherm using Barret–Joyner–Halenda (BJH) method. Micropore surface area and pore hydraulic diameter distribution from 0.7 to 1.6 nm were derived from t-plot using Mikhail, Brunauer and Bodor MP method30 and Harkins & Jura equation.31 Micropore volume (Vmp) was derived from the tangent line of a contiguous range of t-plot using the surface area of the filled pores via eqn (3),
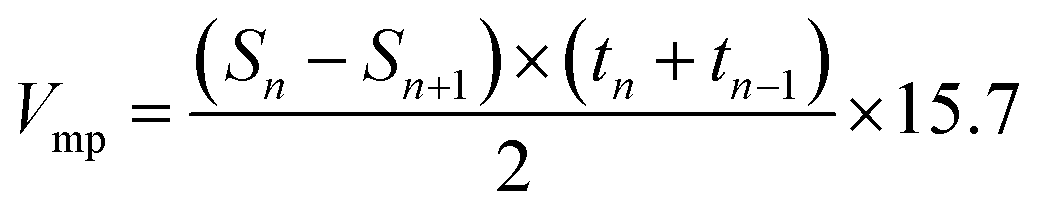 | (3) |
The morphology and structure of PACs were observed by SEM, atomic force microscopy (AFM) (MFP 3D, Asylum Research) and transmission electron microscopy (TEM) (JEOL 3000, JEOL). SEM samples were sputter coated with gold for 1 min and observed under a working voltage of 5 kV. TEM samples were prepared by dispersing a small amount of PACs in water (∼0.01 g L−1) and sonicate (2510, Branson) for 30 min first, then placing a drop of the sonicated suspension onto a carbon grid and dry in air. AFM samples were prepared by grinding PACs with mortar and pestle first, then disperse a small amount of grinded powder in water (∼0.05 g L−1). The suspension was then centrifuged, and a drop of the supernatant was placed on a glass slide to air dry. The sample was then analyzed with a silicon tip (AC160 TS, Olympus) in the tapping mode.
Supercapacitor cells
PACs activated by NaOH and KOH at 1 alkali hydroxide/ALls impregnation ratio at 900 °C for 30 min were made into supercapacitor cells. 3.2 mg NaOH and 4.0 mg KOH-activated PACs were deposited on nickel foams, respectively, followed by roller pressing and mold punching into the spherical electrodes in 1.5 cm diameter. The electrochemical properties of these cells were evaluated through cyclic voltammetry (CV) on a potentiostat/galvanostat (EG&G Princeton Applied Research, Model 263A), galvanostatic charge/discharge on an eight channels battery analyzer (MTI Corporation, mode BST8-MA), and electrochemical impedance spectroscopy (EIS) on a frequency response detector coupled with the EG&G 263A with the frequency from 100 kHz to 10 mHz at room temperature. The specific capacitance was estimated from CV curve (Cc–v) and galvanostatic charge/discharge curve (Ci–t) via eqn (4) and (5) respectively, the energy and power density from eqn (6) and (7) respectively, where V is the operating voltage, ΔV is the voltage change (1 V), Im is the discharge current, Δt is the discharge time, m is the average mass of single electrode, R is the equivalent series resistance (ESR).
 | (4) |
 | (5) |
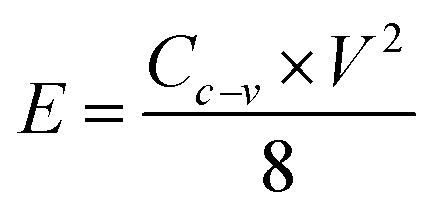 | (6) |
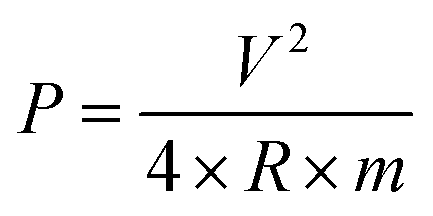 | (7) |
Results and discussion
Pyrolytic behaviors and PAC yields
The mass profile of precursor ALls during pyrolysis in TGA showed 9% loss at 150 °C from moisture evaporation, another 32% at 600 °C and additional 13 to 22% at 850 °C, 900 °C and 950 °C, leaving substantial chars of 45.9%, 37.1% and 35.8%, respectively (Fig. 1). The major mass loss between 150 and 600 °C is consistent with the loss of benzene methoxyl groups, phenylpropanoid side chains and phenol derivatives reported by others.32,33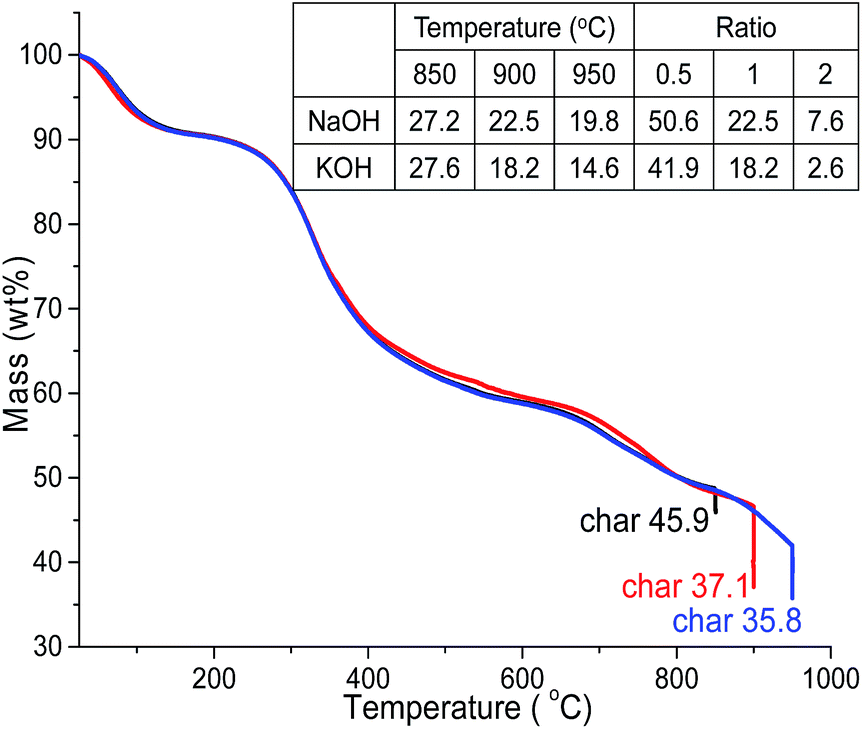 | ||
| Fig. 1 TGA of ALls heat to 850, 900 and 950 °C and held for 30 min (N2, 10 °C min). Inserted table: yields (wt%) of PACs heated to 850, 900 and 950 °C and held for 30 min (1 impregnation ratio) and activated at 0.5, 1 and 2 alkali hydroxide/ALls impregnation ratios (900 °C, 30 min). | ||
At 1 alkali hydroxide/Alls impregnation ratio, NaOH activation generated 27.2%, 22.5% and 19.8% PACs whereas those activated by KOH produced 27.6%, 18.2% and 14.6% PACs at 850 °C, 900 °C and 950 °C, respectively, both yielding less with increasing temperatures (Fig. 1). PAC yields were 15 to 20% less than lignin chars at the same temperatures, as expected from alkali hydroxide induced oxidation of chars into CO and CO2,34 and slightly lower with KOH than NaOH especially at higher temperatures, likely due to the lower ionization energy of potassium. Both PAC series contained mainly carbon, with 97.3–97.7% C in those from NaOH and 94.2–97.0% C in PACs from KOH (Fig. S1†), showing highly effective carbonization.
At 900 °C, PACs activated by KOH and NaOH at 0.5 impregnation ratio yielded 50.6% and 41.9%, respectively, higher than the 37.1% char of ALls precursor, but significantly decreased to 22.5 and 18.2% then even further to 7.6 and 2.6% at 1 and 2 impregnation ratios, respectively. This yield-impregnation relationship may be attributed to opposing effects of two concurrent processes. Alkali metals have shown to promote significant demethylation and slight demethoxylation when pyrolyzed from 300 to 500 °C (ref. 35) to form alkali metal salts by substituting hydrogen from the tarry phenolic substances, thus increasing mass. At the same time, alkali hydroxide oxidation consumes chars,36 lowering mass. Hydrogen substitution is thought to be more prevalent at low alkali impregnation ratio, retaining mass, whereas oxidation of carbon chars dominates with excess alkali metals at higher impregnation ratios, losing major mass. It was clear that the yields of PACs were dependent on the alkali hydroxides types, i.e., higher with NaOH than KOH, and significantly decreased with increasing loadings as well as pyrolytic temperatures.
Carbon microstructures by Raman spectroscopy
The Raman spectrum of ALls char and all PACs showed prominent G peaks in 1500–1600 cm−1 and D peaks in 1300–1400 cm−1 ranges, indicating fairly heterogeneous carbon micro-structures (Fig. 2 & S2†). The broad G and D peaks were resolved into G1 & G2 and D1 & D2 peaks with high correlations (COD > 0.997). The similar and sharper G1 and D1 peaks were attributed to the winding short basal plane of graphite with bond angle order, while the broader G2 and D2 peaks were associated with the amorphous sp2 carbon clusters with bond disorder.27 The basal planes were likely consisted of lignin benzene ring and conjugated polycyclic hydrocarbons formed during carbonization while the amorphous carbon clusters could be the pyrolysis products of non-aromatic side chains. The G1 peaks of all PACs were located in the 1580–1600 cm−1 region, indicating the graphite-like basal plane to be nanocrystalline according to the three-stage model of graphene to diamond amorphization.29 The G1 peak positions also varied only slightly, indicating little change in the structure of the basal planes under various pyrolysis conditions. The FWHM of D1 peak, a more sensitive measure of structural defects for graphitic materials,37–41 was used to evaluate basal plane defects. While G2 FWHM, a measure of structural disorder related to bond angle and strength distortion in various amorphous carbons41–43 was used to indicate amorphous carbon defects. The D peak positions offered little insight on the PACs' microstructures since D peak could be the lower frequency shoulder of the G peak and the fitted values were the least accurate.29 | ||
| Fig. 2 Raman spectrum of (a) ALls char (b) NaOH and (c) KOH-activated PAC (900 °C, 30 min, N2, 10 °C min, ratio 1). | ||
ALls char exhibited G1 peak with 57 cm−1 FWHM, much larger than the 13 cm−1 FWHM of pure crystal graphite,44 consistent with the moderately graphitized lignin with imperfect conjugated polycyclic hydrocarbons. For both alkali hydroxide activated PACs, their higher I(D1)/I(G1) and lower I(D2)/I(G2) ratios than those in ALls char indicate reduced sizes of both basal planes and amorphous carbon clusters. As a result, the overall PACs grain sizes were smaller in the 18–28 nm range than ca. 30 nm of ALls char. This was attributed to the redox reactions between alkali hydroxides and the chars, lowering carbon yields as discussed previously as well as inducing defects in both basal plane and amorphous carbon. The higher structural disorder in PACs was also evident by the higher G1 and G2 FWHMs than those of the un-activated ALls char. However, the G1 FWHMs of all PACs varied little, suggesting the basal plane was not affected significantly by the impregnation ratios, temperatures and holding times due to its relative recalcitrant nature. The G2 FWHMs showed much stronger dependence on the pyrolysis parameters, suggesting more extensive reaction occurred between the alkali hydroxides and amorphous carbon.
With increasing carbonization temperatures from 850 to 950 °C, both I(D1)/I(G1) ratios and D1 FWHMs reduced, indicating the formation of larger basal planes with fewer defects. Carbonization at 900 °C generated the most structural disorder and defects in the amorphous carbon as shown by the lowest frequencies of the G2 peaks and the highest FWHMs. Noted, the G2 FWHM of NaOH-activated PACs decreased significantly to 20 cm−1 at 950 °C, possibly due to the formation of small aromatic hydrocarbons with little bond disorder. Such structural change could result from the extensive bond cleavage caused by the radically increased reactivity of sodium in the gas phase. With increasing temperatures, the I(D2)/I(G2) ratios increased slightly, indicating somewhat larger amorphous carbon domains from possibly two opposing effects on carbon microstructures. While enhanced graphitization via elimination of existed defects could increase the structural orderness,45 more defects could be generated as the reactivity of alkali hydroxides and reduced alkali metal increased. The former effects would dominate on the lignin basal plane due to its rigid structures, lowering reactivity with alkali hydroxides.
Similarly, increasing holding time from 5 to 60 min slightly decreased I(D1)/I(G1) for both PACs, suggesting slightly enhanced graphitization and enlarged basal planes, possibly due to decomposition of the existed defects. However, higher I(D2)/I(G2) ratio also indicated increasing amorphous carbon dimensions. As the amorphous carbon were more reactive, more extensive reaction with alkali hydroxides would occur with longer time, generating new defects.
With increasing impregnation ratios, the I(D1)/I(G1) ratio and D1 FWHM of both PACs increased, indicating smaller and more defective basal planes possibly due to the more extensive redox reaction. However, neither G2 FWHMs nor I(D2)/I(G2)s showed particular trends, indicating non-specific impregnation ratio effects on amorphous carbon clusters as simultaneous formation of new defects and decomposition of existing defects occurred.
Both alkali hydroxide activated PACs consisted of two carbon microstructures: nanographite-like basal planes consisting mainly conjugated polycyclic hydrocarbons and amorphous sp2 carbon clusters. Carbonization and activation posed competing effects on carbon microstructures, while increasing temperature and holding time would induce enhanced graphitization, it would also lead to more extensive activation to generate defects. The basal planes were structurally more rigid than amorphous carbons, thus less susceptible than the amorphous carbon. Increasing heating temperature and time mainly induced the decomposition of defects and led to enlarged basal planes with fewer defects, but increasing alkali hydroxides loadings expectedly resulted in smaller basal planes with more defects. However, the amorphous carbons were much more reactive toward alkali hydroxides, and thus the combined effects of carbonization and activation could not be generalized.
Porous structures by BET
All PACs exhibited type IV nitrogen adsorption–desorption isotherms with H4 hysteresis loops of different sizes (Fig. S3–S5†), typically associated with microporous and mesoporous structures with slit-like micropores arrays interconnecting with mesopores.46 All PACs showed step-downs at 0.45 P/Po in their isotherms. The BJH neck size distribution derived from the desorption branch exhibited distinct artificial peaks at 4 nm from the thermal physical state of N2 fluid during cavitation (Fig. S3c and d†). Such presence suggested ink-bottle structure with necks size less than the 5 nm.46,47With increasing temperatures from 850 to 950 °C, larger and more distinct hysteresis loops were observed with all PACs (1 impregnation ratio), suggestive of increasing extent of mesopores. The BJH mesopore surface area and pore volume as well as the single point total pore volume maximized at 950 °C for PACs activated by both NaOH (500 m2 g−1, 0.49 cm3 g−1 and 0.75 cm3 g−1) and KOH (371 m2 g−1, 0.30 cm3 g−1 and 0.71 cm3 g−1). However, total BET surface area, t-plot derived micropore surface area and micropore volume peaked at 900 °C for PACs activated by NaOH (1321, 1108 m2 g−1 and 0.43 cm3 g−1) and KOH (1540, 1093 m2 g−1 and 0.43 cm3 g−1). The transitions from microporous to mesoporous structures were more evident in NaOH activated PACs where micropores widened from 0.55–1.0 nm to 0.6–1.4 nm (Fig. S3e†), and mesopores enlarged from <2 nm to <4 nm (Fig. S3g†), with increasing temperatures. KOH-activated PACs had relatively consistent micropores that peaked near 0.7 nm (Fig. S3f†), and mesopores mostly <4 nm (Fig. S3h†). This is consistent with the higher kinetic energy and liquid to gas phase change of sodium (Tb = 882 °C) with increasing temperatures while micropores may collapse into larger and more stable mesopores. These two concurrent processes may explain the initial increase and then decrease in micropores but a steady increase in mesopores with increasing temperatures from 850 to 950 °C.
With increasing holding time from 5 to 60 min (900 °C, impregnation ratio 1), total BET surface area, micropore surface area and pore volume peaked at 30 min for both PACs (Fig. S4†), while mesopore surface area and pore volume maximized at 60 min for NaOH (275 m2 g−1 and 0.33 cm3 g−1) and 30 min for KOH (321 m2 g−1 and 0.24 cm3 g−1) min, respectively. The mesopores in both PACs were in the 2 to 4 nm range (Fig. S4g and h†), while the micropore size distributions were more dependent on the holding time. Both PACs contained micropores between 0.55 and 1 nm (Fig. S4e and f†), but larger micropores up to 1.4 nm was observed for NaOH and KOH-activated PACs at 60 and 30 min, respectively. NaOH-activated PACs had more mesopores with longer holding time likely from formation of new mesopores and broadening of existing micropores. In contrast, KOH activation led to both maximal mesopore and micropores at 30 min. The loss of potassium atoms/clusters via evaporation during longer holding time attributed to it's lower boiling point (758 °C) led to less activation and fewer new micro/mesopores, while existing ones collapsed to less porous structures.
With increasing impregnation ratios (900 °C, 30 min holding time), the step-downs decreased to 0.4 P/Po and the artificial peak disappeared at 2 ratio, indicating absence of ink-bottle structure. The hysteresis also became very small, indicating the mesopores either becoming less accessible, or no longer interconnected to the microporous arrays. The structural changes were reflected on the pore size distributions as well. NaOH-activated PACs had essentially no pores at 0.5 impregnation ratio, prominent micropores peaked ca. 0.7 nm at 1, and significant micro/mesopores peaked ca. 2 nm at 2 (Fig. S5e and g†). The micropore broadening also occurred in KOH-activated PACs with peaks shifting from 0.6 to 0.75 nm with increasing KOH (Fig. S5f and h†). For both NaOH and KOH, micropore surface area and pore volume as well as total pore volume maximized at the ratio of 1 whereas mesopore surface area and pore volume peaked at impregnation ratio of 2, reaching 1847 m2 g−1 and 1.10 cm3 g−1 for NaOH and 483 m2 g−1 and 0.31 cm3 g−1 for KOH. Higher alkali loading clearly enlarged pore sizes and altered porous structures. For NaOH-activated PACs in particular, the micropores completely widened to rather uniform mesopores as the ratio increased from 1 to 2.
In conclusion, both PACs also showed very similar adsorption–desorption isotherm and hysteresis indicating almost identical porous structure (Fig. 3). Both alkali hydroxide activated PACs reached similar maximal micropore surface area and pore volume under the same condition, i.e. 1 impregnation ratio, 900 °C, 30 min, and were fabricated into supercapacitor cells. PAC activated by NaOH exhibited narrower micropore distribution from 0.6 to 1 nm, while that by KOH had wider micropore range from 0.6 to 1.4 nm. Increasing the impregnation ratio, temperature, holding time all led to increased extent of mesopores, with the impregnation ratio being the most significant. At 0.5 and 1 alkali hydroxide/lignin ratios, both NaOH and KOH activation produced mainly slit-like micropores interconnected with limited mesopore that were only accessible through small necks. At a higher 2 impregnation ratio, pores widened, leading to the dramatic increase of accessible mesopores with KOH activation and the complete shift from micropores to small mesopores with NaOH activation.
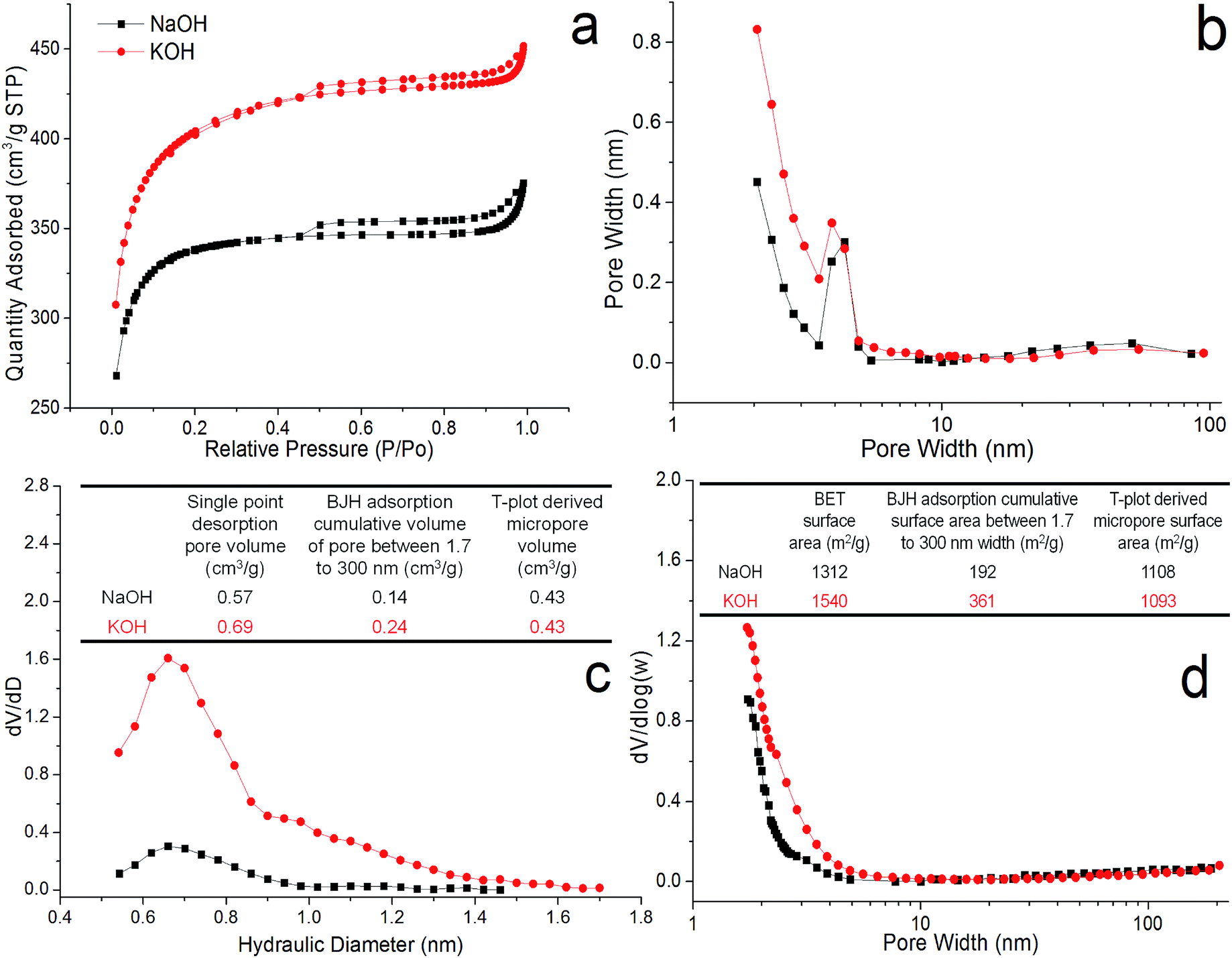 | ||
| Fig. 3 PACs porous structures (900 °C, ratio 1, 30 min, N2): (a) nitrogen adsorption–desorption isotherm; (b) BJH neck size distribution; (c) micropore hydraulic diameter distribution; (d) BJH mesopore/cavity width distribution. Inset tables: (c) pore volume; (d) surface area. | ||
Morphology by SEM, TEM and AFM
All PACs were irregularly shaped in sizes ranging from a few hundred nm to a few μm (Fig. S6a–d,† insets) and with nano-scale features (Fig. S6e–h†). All PACs exhibited distinct surface hairline cracks (Fig. S6a–d†), agreeing well with the slit-like pore geometry by the BET analysis. The internal micropores and mesopores discerned by BET seemed to originate from the spacing between these nanostructures (Fig. S6e–h†). As the impregnation ratio increased to 2, the surface porosities became more evident (Fig. 4a and c), while the hierarchical structures consisting of circular shaped particles were clearly observed in both PACs (Fig. 4b and d). These circular particles were estimated to be ca. 14–25 nm and ca. 10–16 nm in diameters from NaOH and KOH activation, respectively (Fig. S6†), consistent with the 18–28 nm grain sizes estimated from earlier Raman analysis. | ||
| Fig. 4 SEM (a and c) and TEM (b and d) of PACs activated by (a and b) NaOH and (c and d) KOH (900 °C, 30 min, impregnation ratio of 2, N2, 10 °C min). | ||
To further examine these nano-scale features, PACs were ground, dispersed in water, sonicated and centrifuged to collect the supernatants for TEM and AFM observations. The supernatant from KOH-activated PACs showed no disintegrated PACs from sonication, indicating tightly bound graphene layers from strong pi–pi interactions. The supernatant from NaOH-activated PACs, on the other hand, showed circular particles with 1 to 3 nm thickness or in the form of nanoplates (Fig. 5). These circular nanoplates are amounted to ca. 3 to 9 graphene-like layers, given the 0.335 nm interplanar spacing in graphene.48 The formation of these multi-layer carbon nanoplates might be closely associated with stacking of lignin phenylpropanoids via the strong pi–pi inter and intra molecular interactions during drying prior to carbonization.
 | ||
| Fig. 5 TEM and AFM with high profile of ground, aq. dispersed and sonicated NaOH-activated PAC (900 °C, 30 min, impregnation ratio 2). | ||
The morphology analysis illustrated the PAC hierarchal structures assembled from circular nanoplates, the spacings among which gave rise to internal porosities. While the porous structures have always been a crucial factor in determining any interfacial absorption phenomena in PACs, the size and compositions of these nanoplates building block could be equally important, in particular during electrolyte absorption when PACs were used as supercapacitor electrodes.
Electrochemical properties
Both alkali activated PACs (1 impregnation ratio, 900 °C, 30 min) that exhibited the highest micropore surface and pore volume, i.e., c.a. 1100 m2 g−1 and 0.43 cm3 g−1, were fabricated into supercapacitors. Both capacitor cells exhibited the rectangular CV curves (Fig. 6a), indicative of double layer capacitive behaviours. At a 20 mV s−1 scanning rate, the specific capacitance and energy density estimated from the CV curve were significantly higher for NaOH-activated PACs, i.e., 226 F g−1 and 7.85 W h kg−1, respectively, than those activated by KOH (132 F g−1 and 4.58 W h kg−1 for, respectively). A higher specific capacitance of 248 F g−1 was also derived from galvanostatic charge/discharge curve for NaOH-activated PACs than the 149 F g−1 value for KOH-activated PACs (Fig. 6b). Despite their similar total surface, micropore area and pore volume, NaOH-activated PACs exhibited much higher capacitance. Such differences may be attributed to the narrower size distribution (0.6–1.0 nm vs. 0.6–1.4 nm) in the micropores region as reported in previous studies.49–51 The much larger nanoplates in NaOH-activated PACs (14–25 nm vs. 10–16 nm) could be another contributing factor, since the capacitance parallel to the basal plane is much larger than that in the perpendicular direction, and larger nanographite would contribute more to the overall capacitance (Fig. 4).52 Both supercapacitors from NaOH and KOH-activated PACs also exhibited excellent cycling stability after 5000 cycles, retaining over 95% and 92% capacitance, respectively (Fig. 6c). The Nyquist plots of both supercapacitors exhibited typical small semicircle at the high frequency region followed by a nearly vertical line, indicative of the double layer capacitive behaviours of the cells at fast ion diffusion rate, also consistent with the CV analysis (Fig. 6d). The ESRs derived from the Nyquist plots were 1.7 and 1.9 ohms and 46.9 and 32.9 kW kg−1 power densities for NaOH and KOH-activated PACs, respectively. The supercapacitor with NaOH-activated PACs, in particular, showed comparable specific capacitance and energy density than those with PACs previously reported.10,11,13–17 Furthermore, both specific capacitance and energy density of NaOH-activated PACs were 80% higher than the similarly NaOH-activated carbon fibers derived from the same lignin from our own previous work, and only 30% less than the best KOH-activated carbon fibers.23 Most significantly, PACs could be fabricated directly, without electrospinning nor any polymer carrier, thus much simpler and efficient, making PACs more appealing for industrial production.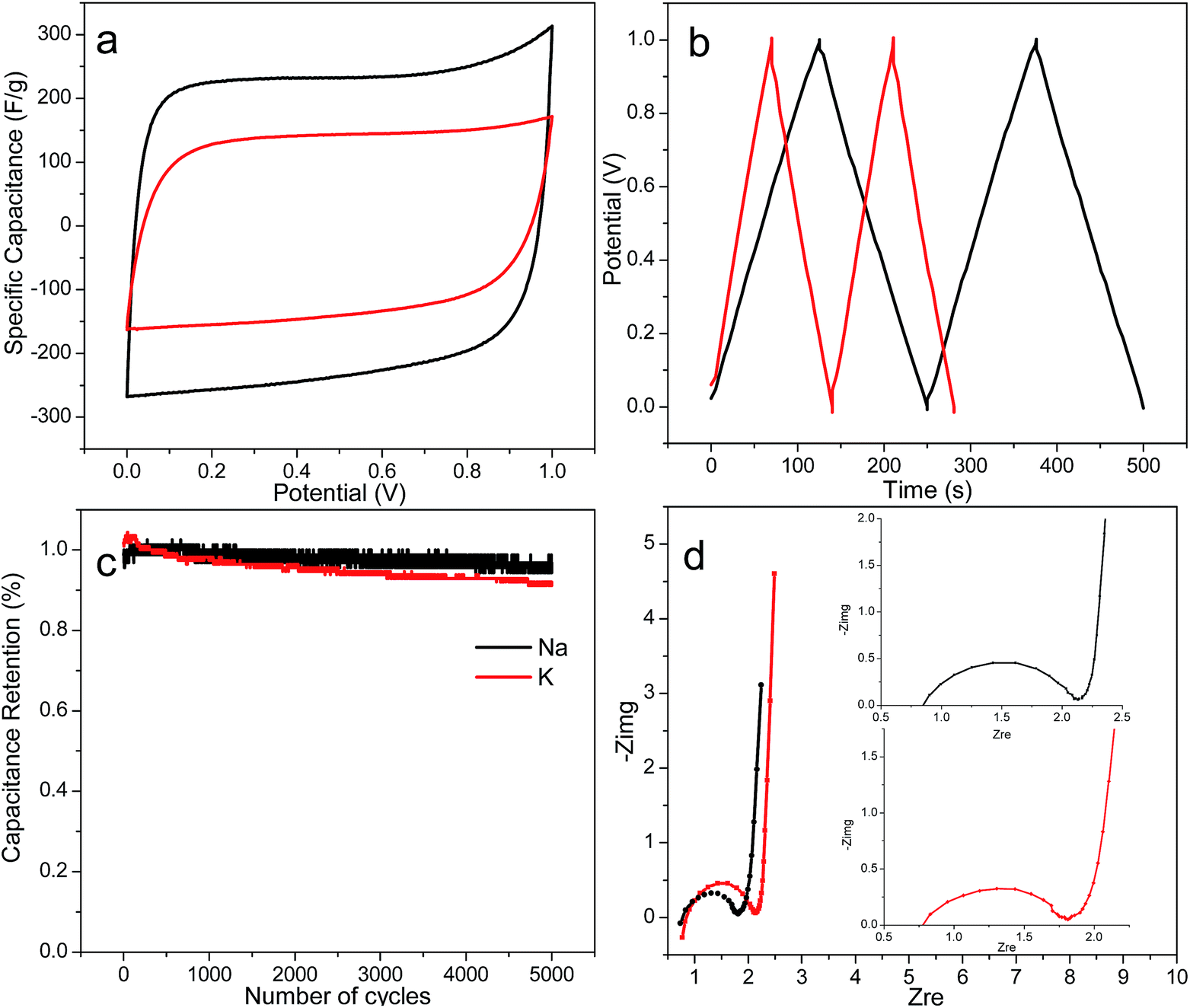 | ||
| Fig. 6 (a) Cyclic voltagramms at 20 mV s−1, (b) galvanostatic charge/discharge curves at 1 A g−1, (c) capacitance retention over 5000 cycles under 1 A g−1, and (d) Nyquist plots of NaOH and KOH-activated PACs based supercapacitors. | ||
Conclusions
Highly porous particulate activated carbons (PACs) were facilely synthesized from low sulfonate content alkaline lignin (ALls) via simultaneous carbonization and activation with NaOH and KOH. The PACs exhibited hierarchical structures consisting of circular nanoplates with diameter ranging from 10 to 25 nm, and thickness from 1 to 3 nm. The PACs' carbon microstructures contained graphite-like basal planes and amorphous sp2 carbon clusters. The recalcitrant basal planes were much less susceptible to temperature, holding time and impregnation ratios than the amorphous carbon clusters. Therefore, the size and structural orderness of basal planes increased with temperature and holding time but decreased with impregnation ratio, while that of amorphous carbons showed no particular pattern. The spacing between the nanoplates led to internal porosities, with optimal 1100 m2 g−1 micropore surface area and 0.43 cm3 g−1 pore volume were generated from both alkali hydroxides at 1 impregnation ratio, 900 °C and 30 min holding time. Increasing temperature, holding time and impregnation ratio all led to broadening of the micropores into mesopores, with impregnation ratio being most significant. Electric supercapacitors fabricated from PACs with optimal microporosities exhibited excellent respective specific capacitance, power and energy density of 226 F g−1, 46.9 kW kg−1 and 7.85 W h kg−1 for NaOH-activated PACs, significantly higher than the respective 132 F g−1, 32.9 and 4.58 kW kg−1 for KOH-activated PACs, while both exhibited exceptional cycling stability, retaining 95% and 92% respective capacitance after 5000 cycles. This work has illustrated the relations between heat parameters on the morphology, microstructures and porous structures of PACs from lignin, an abundant under-utilized pulping and biofuel by-product, as well as demonstrated their high performance as electric supercapacitors.Acknowledgements
The authors acknowledge Dr Sanliang Zhang and Professor Ning Pan for access to their facility to characterize electrochemical properties of supercapacitors.References
- T. D. Burchell, Carbon Materials for Advanced Technologies, Elsevier Science, Pergamon, 1st edn, 1999 Search PubMed
.
- Y. Wang, Z. Q. Shi, Y. Huang, Y. F. Ma, C. Y. Wang, M. M. Chen and Y. S. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CAS
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC
- A. Ghosh and Y. H. Lee, Chemsuschem, 2012, 5, 480–499 CrossRef CAS PubMed
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765–772 CrossRef CAS
- G. Gryglewicz, J. Machnikowski, E. Lorenc-Grabowska, G. Lota and E. Frackowiak, Electrochim. Acta, 2005, 50, 1197–1206 CrossRef CAS
- M. Endo, T. Maeda, T. Takeda, Y. J. Kim, K. Koshiba, H. Hara and M. S. Dresselhaus, J. Electrochem. Soc., 2001, 148, A910–A914 CrossRef CAS
- L. Eliad, G. Salitra, A. Soffer and D. Aurbach, J. Phys. Chem. B, 2002, 106, 10128–10134 CrossRef CAS
- J. M. Di Leo and J. Maranon, J. Mol. Struct.: THEOCHEM, 2005, 729, 53–57 CrossRef
- J. Gamby, P. Taberna, P. Simon, J. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef CAS
- Y. Zhu, S. Murali, M. D. Stoller, K. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed
- D. Lozano-Castello, D. Cazorla-Amoros, A. Linares-Solano, S. Shiraishi, H. Kurihara and A. Oya, Carbon, 2003, 41, 1765–1775 CrossRef CAS
- M. Endo, Y. J. Kim, H. Ohta, K. Ishii, T. Inoue, T. Hayashi, Y. Nishimura, T. Maeda and M. S. Dresselhaus, Carbon, 2002, 40, 2613–2626 CrossRef CAS
- E. Raymundo-Pinero, K. Kierzek, J. Machnikowski and F. Beguin, Carbon, 2006, 44, 2498–2507 CrossRef CAS
- E. Redondo, J. Carretero-González, E. Goikolea, J. Ségalini and R. Mysyk, Electrochim. Acta, 2015, 160, 178–184 CrossRef CAS
- G. Ma, D. Guo, K. Sun, H. Peng, Q. Yang, X. Zhou, X. Zhao and Z. Lei, RSC Adv., 2015, 5, 64704–64710 RSC
- C. Peng, X.-b. Yan, R.-t. Wang, J.-w. Lang, Y.-j. Ou and Q.-j. Xue, Electrochim. Acta, 2013, 87, 401–408 CrossRef CAS
- W. M. A. W. Daud and W. S. W. Ali, Bioresour. Technol., 2004, 93, 63–69 CrossRef CAS PubMed
- L. Khezami, A. Chetouani, B. Taouk and R. Capart, Powder Technol., 2005, 157, 48–56 CrossRef CAS
- J. J. M. Orfao, F. J. A. Antunes and J. L. Figueiredo, Fuel, 1999, 78, 349–358 CrossRef CAS
- C. S. A. Higson, NNFCC Renewable Chemicals Factsheet: Lignin, 2011 Search PubMed
- S. Hu and Y. L. Hsieh, J. Mater. Chem. A, 2013, 1, 11279–11288 CAS
- S. Hu, S. Zhang, N. Pan and Y.-L. Hsieh, J. Power Sources, 2014, 270, 106–112 CrossRef CAS
- J. Hayashi, A. Kazehaya, K. Muroyama and A. P. Watkinson, Carbon, 2000, 38, 1873–1878 CrossRef CAS
- V. Fierro, V. Torne-Fernandez and A. Celzard, Microporous Mesoporous Mater., 2007, 101, 419–431 CrossRef CAS
- M. Schmirler, F. Glenk and B. J. M. Etzold, Carbon, 2011, 49, 3679–3686 CrossRef CAS
- N. Shimodaira and A. Masui, J. Appl. Phys., 2002, 92, 902–909 CrossRef CAS
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS
- R. S. Mikhail, S. Brunauer and E. E. Bodor, J. Colloid Interface Sci., 1968, 26, 45–53 CrossRef CAS
- W. D. Harkins and G. Jura, J. Chem. Phys., 1943, 11, 431–432 CrossRef CAS
- M. A. Scheijen, J. J. Boon, W. Hass and V. Heemann, J. Anal. Appl. Pyrolysis, 1989, 15, 97–120 CrossRef
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. H. Lin, W. G. Chan and M. R. Hajaligol, Fuel, 2004, 83, 1469–1482 CrossRef CAS
- P. Ehrburger, A. Addoun, F. Addoun and J. B. Donnet, Fuel, 1986, 65, 1447–1449 CrossRef CAS
- M. Kleen and G. Gellerstedt, J. Anal. Appl. Pyrolysis, 1995, 35, 15–41 CrossRef CAS
- Y. Yamashita and K. Ouchi, Carbon, 1982, 20, 41–45 CrossRef CAS
- A. Jorio, C. Fantini, M. S. S. Dantas, M. A. Pimenta, A. G. Souza, G. G. Samsonidze, V. W. Brar, G. Dresselhaus, M. S. Dresselhaus, A. K. Swan, M. S. Unlu, B. B. Goldberg and R. Saito, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66(11), 115411–8 CrossRef
- V. M. Irurzun, M. P. Ruiz and D. E. Resasco, Carbon, 2010, 48, 2873–2881 CrossRef CAS
- S. Lee, J. W. Peng and C. H. Liu, Carbon, 2009, 47, 3488–3497 CrossRef CAS
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martinez-Alonso and J. M. D. Tascon, J. Mater. Chem., 1998, 8, 2875–2879 RSC
- A. C. Ferrari, S. E. Rodil and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67(15), 155306-20 CrossRef
- C. Casiraghi, A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(8), 085401-14 CrossRef
- A. C. Ferrari and J. Robertson, Philos. Trans. R. Soc., A, 2004, 362, 2477–2512 CrossRef CAS PubMed
- S. Piscanec, M. Lazzeri, J. Robertson, A. C. Ferrari and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75(3), 035427-22 CrossRef
- J. RodriguezMirasol, T. Cordero and J. J. Rodriguez, Carbon, 1996, 34, 43–52 CrossRef CAS
- P. I. Ravikovitch and A. V. Neimark, Langmuir, 2002, 18, 9830–9837 CrossRef CAS
- M. Thommes, B. Smarsly, M. Groenewolt, P. I. Ravikovitch and A. V. Neimark, Langmuir, 2006, 22, 756–764 CrossRef CAS PubMed
- L. A. Girifalco and R. A. Lad, J. Chem. Phys., 1956, 25, 693–697 CrossRef CAS
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P.-L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed
- S. Kondrat, C. Perez, V. Presser, Y. Gogotsi and A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 CAS
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed
- J.-P. Randin and E. Yeager, J. Electroanal. Chem. Interfacial Electrochem., 1975, 58, 313–322 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: EDX, Raman spectrum, BET, SEM and TEM of PACs activated with alkali hydroxides at varied ratios and under different pyrolytic conditions. See DOI: 10.1039/c7ra00103g |
| This journal is © The Royal Society of Chemistry 2017 |