
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diazepinium perchlorate: a neutral catalyst for mild, solvent-free acetylation of carbohydrates and other substances†
Santosh Kumar Giri,
Rajesh Gour and
K. P. Ravindranathan Kartha *
*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research, SAS Nagar, Punjab-160062, India. E-mail: rkartha@niper.ac.in
First published on 28th February 2017
Abstract
Diazepinium perchlorate, an essentially neutral organic salt possessing excellent stability, has been found to be well suited for the acetylation of free as well as partially protected sugars, phenols, thiophenols, thiols and other alcohols as well as amines. The diazepinium perchlorate-catalyzed acetylation is mild, organic and solvent-free and leaves acid sensitive protecting groups such as TBDMS/TBDPS/Tr ethers and isopropylidene/benzylidene acetals present on a substrate unaffected. Regioselective hydroxyl protection in partially protected carbohydrate derivatives/polyhydroxylic compounds was possible and was proved to be a convenient time-saving alternative to the conventional synthesis of such compounds. Easy preparation of the catalyst, mild reaction conditions and an environmentally benign protocol are some of the notable features of this reaction. The results obtained on the acetylation of phenols and thiophenols could be rationalized through their local nucleophilicity index obtained from DFT calculations.
1. Introduction
Acyl transfer reaction is one of the most commonly used reactions in synthetic organic chemistry, in particular, synthetic carbohydrate and natural products chemistry, for the protection of hydroxyl groups as esters and therefore application of new catalysts for this category of reactions is in continuous demand. It is usually carried out by reacting the substrate with the desired acyl donor in the presence of a catalyst. Both acidic as well as basic catalysts are equally effective and have used for this purpose. Examples of substances that are neutral in nature and used as acyl transfer agents can also be seen in the literature. An exhaustive list of the various classes of catalysts used in the peracetylation of carbohydrates can be seen from a recently published review.1 Additionally, for non-carbohydrate alcohols a variety of other catalysts such as Bu3P,2 CoCl2,3 TaCl2,4 TMSOTf,5 iminophosphorane bases (with enol esters)6 and distannoxane catalysts, etc. have also been used in the presence of excess amounts of acetic anhydride in various solvents.7 The relatively strong basicity (pKa = 13.4)8 of the 2,3-dihydro-5,7-dimethyl-1,4-diazepine (1a) and the excellent stability of its neutral perchlorate salt 2 (ref. 9) (Fig. 1) attracted our attention in this context and prompted us to explore the suitability of the latter as a possible acylation catalyst with potential benefits over many of the hitherto known acylation catalysts. In particular, it was felt that the mild nature of 2 may offer good possibilities for the selective formation/hydrolysis of isopropylidene acetals besides possible selective acetolysis of one or more benzyl ether groups in sugars carrying multiple benzyl ether protecting groups. Thus, herein we describe the use of 2 as an efficient acyl transfer catalyst for the acetylation of various classes of organic compounds containing –OH, –SH and NH2 groups in stoichiometric acetic anhydride (see Scheme 1 to follow). | ||
| Fig. 1 Diazepinium perchlorate catalysts. | ||
 | ||
| Scheme 1 Diazepinium perchlorate-catalyzed acyl transfer reaction using Ac2O as the acyl donor. | ||
2. Results and discussion
The diazepinium salt 2 (ref. 10) was prepared by the method reported by Lloyd et al.9 and was used for the acetylation reaction as described below. Considering the more challenging demand of a carbohydrate molecule structurally, as compared to many of the other simple hydroxylic compounds, a common monosaccharide, D-glucose (5), was chosen as the substrate for the initial experiments on acetylation using perchlorate 2 as the promoter. Thus when 5 was treated with acetic anhydride in the presence of 2 (10 and 20 mol% in two separate parallel experiments; see Table 1, entries 1 and 2), no acetylated product was found to be formed during a reaction time of up to 24 h at 25 °C. Compound 5 remained insoluble. This was not very surprising from the neutral nature of 2. Under the same conditions at 30 mol% of 2, however, 5 reacted to a notable degree to give a 40% yield of the desired 6 (entry 3, Table 1). A partial dissolution of 5 had been observed in this case. And, when the reaction was performed at a slightly higher temperature of nearly 40 °C (entry 4, Table 1) the sugar started to go into solution in a few minutes and a complete dissolution of 5 was observed in 10 min (entry 4, Table 1). A clear colourless solution had been resulted which had also become hot, typical of an exothermic reaction. Analysis of the reaction mixture by TLC (eluent, EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-heptane, 2:3) showed the presence of only one product (spot), that corresponded to an authentic sample of the expected penta-O-acetate 6 (ref. 11) in Rf value; and importantly, no partially acetylated glucose derivative was present in the mixture (as evidenced by the absence of any slower moving products on TLC). Compound 6 isolated in near-quantitative yield (2.15 g, 99%) following a simple aqueous workup and crystallization was practically pure and in a form suitable for employing in reactions further. In a subsequent reaction wherein 2 was used at 25 mol% concentration (entry 5, Table 1), the same reaction was found to be complete in about 20 min of reaction time with 6 again being obtained in near-quantitative yield. Acetylation of 5 using Ac2O as the acyl donor agent in the presence of 2 (25–30 mol%) carried out in common solvents such as MeCN, CH2Cl2, etc. for comparative purposes was found to be inferior indeed in achieving the objective (results not shown in the table); and was therefore not pursued further. Thus, solvent-free reaction with a slight excess over the stoichiometric requirement of Ac2O in the presence of 25–30 mol% 2 at 35–40 °C was found to be optimum for the complete acetylation of a compound such as 5.
:n-heptane, 2:3) showed the presence of only one product (spot), that corresponded to an authentic sample of the expected penta-O-acetate 6 (ref. 11) in Rf value; and importantly, no partially acetylated glucose derivative was present in the mixture (as evidenced by the absence of any slower moving products on TLC). Compound 6 isolated in near-quantitative yield (2.15 g, 99%) following a simple aqueous workup and crystallization was practically pure and in a form suitable for employing in reactions further. In a subsequent reaction wherein 2 was used at 25 mol% concentration (entry 5, Table 1), the same reaction was found to be complete in about 20 min of reaction time with 6 again being obtained in near-quantitative yield. Acetylation of 5 using Ac2O as the acyl donor agent in the presence of 2 (25–30 mol%) carried out in common solvents such as MeCN, CH2Cl2, etc. for comparative purposes was found to be inferior indeed in achieving the objective (results not shown in the table); and was therefore not pursued further. Thus, solvent-free reaction with a slight excess over the stoichiometric requirement of Ac2O in the presence of 25–30 mol% 2 at 35–40 °C was found to be optimum for the complete acetylation of a compound such as 5.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R-OHa | Acyl donorb | Catalyst (mol%) | Temp. (°C) | Time | R-OAc | Yieldc (%) |
| a 1 mmol substrate was taken for the reaction.b The acyl donor was taken in excess.c Isolated yields are reported.d Anomeric ratio of α:β = 98:2 was obtained.e Yield (conversion) was determined by gas chromatography (GC). |
|||||||
| 1 | D-Glucose (5) | Ac2O | 10 | 25 | 24 h | D-Glucose pentaacetate (6) | — |
| 2 | 5 | Ac2O | 20 | 25 | 24 h | 6 | — |
| 3 | 5 | Ac2O | 30 | 25 | 24 h | 6 | 40 |
| 4 | 5 | Ac2O | 30 | 35–40 | 10 min | 6 | 99d |
| 5 | 5 | Ac2O | 25 | 35–40 | 20 min | 6 | 99 |
| 6 | 5 | AcOH | 30 | 35–40 | 24 h | — | — |
| 7 | n-BuOH | AcOH | 30 | 35–40 | 30 min | n-BuOAc | 75e |
| 8 | 5 | AcCl | 30 | 35–40 | 24 h | 6 | 20 |

Further, although a reaction of 5 carried out with AcOH as the acetyl donor agent in the presence of 2 (30 mol%) was proved incapable of serving the purpose (entry 6, Table 1), was found somewhat effective for the acetylation of simple alcohols such as n-BuOH (entry 7, Table 1). Thus, under these conditions (entry 7, Table 1) conversion of the alcohol to an extent of 75% (by GC) had been observed. On the other hand, when AcCl was used as the acyl donor in the reaction of 5, the acetylated product 6 (entry 8, Table 1) was obtained in 20% yield. It may be noted that reaction of polyhydroxylic compounds such as 5 with AcCl (conducted in the absence of any acid scavenger) is usually accompanied by release of stoichiometric quantities of HCl (gas). Taking into account the solubility of hydrogen halides in Ac2O/AcOH, this opportunity can be utilized in effectively trapping the HCl generated in situ and utilizing the same for the conversion of the glycosyl acetate formed in the reaction to the corresponding glycosyl chloride in the same pot.13 This possibility in the instance above shall be investigated in the future.
The diazepinium perchlorate (2)-catalysed acetylation can be depicted as shown in Scheme 1. When a solution of perchlorate 2 in Ac2O (0.28 M) was prepared and the mass spectrum (LCMS-LTQ ESI) was recorded after 30 min of standing, peaks corresponding to the following m/z values were obtained: (i) 125.02 of [2 − ClO4]+ and (ii) 349.12 of [2 + Ac2O + Na]+, the latter clearly suggesting the possible 2-Ac2O adduct in the solution. On the other hand under the same conditions n-Bu4N(ClO4) gave only one peak, of m/z 242.19 that corresponded to [Bu4N]+ as also CsClO4 but at an m/z value of 125.01 that corresponded to [Ac2O + Na]+. LiClO4, however, did not give any notable mass spectral signals at all under these conditions. It may be emphasized that among the four perchlorates mentioned above (that is, 2, n-Bu4N(ClO4), CsClO4, and LiClO4), LiClO4 was also insoluble in Ac2O.14 Again, in reactions in Ac2O using these catalysts (25 mol% with respect to the sugar, D-glucose) for acetylation at 35–40 °C for 30 min, only the case of 2 led to the formation of the acetylated product 6. However, in the case of LiClO4, the reaction employing the catalyst at 50 mol% level for 12 h led to a successful acetylation as described by Huang and coworkers.15 Further, in the case of acetylation of the monosaccharide 5 promoted by 2, mass spectroscopic analysis of the reaction mixture revealed that the intensity of the peak corresponding to m/z = 349.12 ([2 + Ac2O + Na]+) was significantly weaker towards the completion of the reaction as compared to the initial stages of the reaction.
In view the above observations made in the course of the mass spectral analysis of the acetylation mixture, a DFT study on the system was also carried out and the results of this have been shown in Tables 2 and 3 (respectively, the energy-minimized 2-Ac2O complex 2b showing the possible H-bond distances and listing of the energy values observed). The energy-minimized structure clearly shows that the H-6 of the diazepinium moiety is very much within the range of hydrogen bond distances considered reasonable for C–H hydrogens.16,17 A value of +0.282 NBO charge for the H-6 as against the values of −0.563 and −0.555 for the acetic anhydride–oxygen atoms is also consistent with H-6 acting as a Lewis acid as depicted in Scheme 1. It was then thought worthwhile to compare the complex 2b with another formed between a conventional acid/Lewis acid catalyst and Ac2O. Thus a recently reported MeSO3H–Ac2O that has been found to be highly efficient in acetylating various hydroxylic substrates was chosen for this purpose.18 The minimized structure of the complex 2d along with the hydrogen bond distances and the energy of stabilization are also shown in Tables 2 and 3. Thus, while the stronger hydrogen bond in complex 2d along with a higher energy of stabilization (compared to those of 2b) are consistent with this (latter) powerful acetylation system,18 it also indeed lends support to the mechanism of Lewis acid-like catalysis as proposed in Scheme 1 for the activation of Ac2O in the acetylation reaction promoted by 2.
| Entry | Structure of the complex | Atom | NBO charge |
|---|---|---|---|
| 1 | 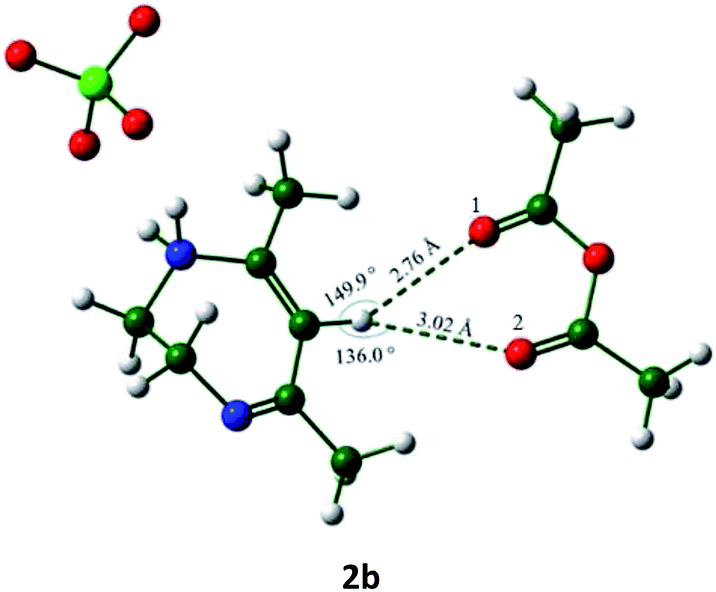 |
H | 0.282 |
| O-1 | −0.563 | ||
| O-2 | −0.555 | ||
| 2 | 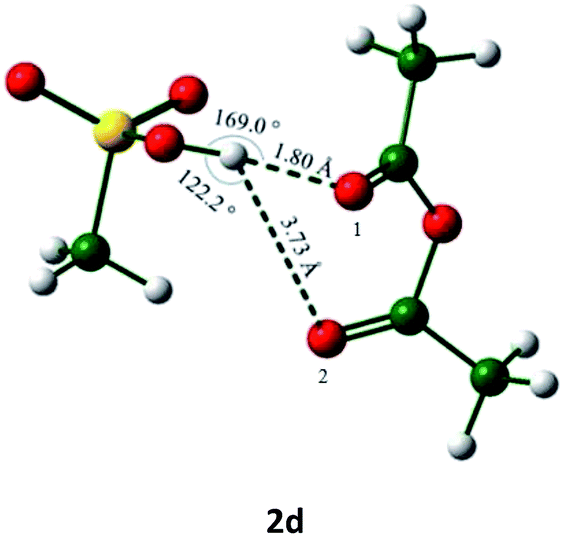 |
H | 0.541 |
| O-1 | −0.611 | ||
| O-2 | −0.548 |
| Entry | Catalyst/complexing agent | Calculated absolute energy (kcal mol−1) | |||
|---|---|---|---|---|---|
| E1 | E2 | E3 | E3 − E2 | ||
| a E1 = absolute energy of substrate; E2 = sum of (catalyst + Ac2O) energy; E3 = energy of complex with catalyst and Ac2O; E3 − E2 = energy difference. | |||||
| 1 | Diazepinium perchlorate (2) | −718239.306 | −957727.049 | −957731.374 | −4.325 |
| 2 | MeSO3H | −416830.609 | −656318.352 | −656327.777 | −9.424 |
| 3 | Ac2O | −239487.742 | — | — | — |
The bond angle α in C–H⋯O interaction is in between 90° and 180° in complex 2b which are necessary for the H-bonding (entry 1, Table 2) to be facilitated with ease. The data summarized in Table 3 on the calculated energies for 2, Ac2O and their complex 2b, for which the net energy of stabilization was found to be lower than the value for MeSO3H–Ac2O complex (2d) as expected, also is in agreement with the activation of Ac2O proposed for acetylation in Scheme 1.
Arguably, analogous to the situation in metal triflate-catalyzed acetylations where an in situ-formed TfOH could be attributed to partially account for the catalysis of the acylation reaction,19 an in situ-formation of HClO4 in 2-Ac2O system can also be considered reasonable and to similarly account for the observed catalysis. In view of this possibility, we tried to examine the acidity of In(OTf)3–Ac2O system,12 a typical metal triflate–Ac2O system, as well as 2-Ac2O system using a standard pH meter. Thus, while a “pH” value of 2.73–3.2 was obtained for neat Ac2O, the corresponding reading obtained for a solution of In(OTf)3 (catalytic) in Ac2O was well below zero (and outside the limits of the pH meter). However, the corresponding reading for the 2-Ac2O system was 3.44. Similarly, for the 2-Ac2O–glucose system (after completion of the acetylation of the sugar) the “pH” reading obtained was 2.71. Thus, in contrast to Ac2O turning highly acidic upon dissolution of a catalytic amount of the metal triflate in it, the dissolution of 2 only rendered the resultant solution turn less acidic. This is in accord with the reaction sequence outlined in Scheme 1. In order to verify the possibility for any base-catalyzed activation of the alcohol residue by the perchlorate anion, work is underway for synthesizing diazepinium salts with other counter anions such as those of triflic acid, hexafluorophosphoric acid, tetrafluoroboric acid, etc. This will also enable us to conclude on the possibilities for finding low melting diazepinium salts that we hope could also possibly serve as ionic liquids.
In the acetylation of D-glucose described above, it may also be pointed out that the by-product (AcOH) of the reaction served as an excellent solvent for the per-O-acetylated product but could also be removed easily in the aqueous alkali workup involved in the acetylation. Needless to mention, in small scale reactions it can also be conveniently removed by co-distillation with toluene under a reduced pressure.
When the reaction under identical conditions was carried out using D-galactose (7, entry 1, Table 4) as the starting material, the corresponding pentaacetate 8 was also obtained in 98% yield. Likewise another common reducing sugar (D-mannose, 9) was also converted at ease to its per-O-acetylated derivative 10 in 96% yield (entry 2, Table 4). In the same manner, disaccharides [lactose (11), cellobiose (13) and maltose (15)] could also be converted to their corresponding octaacetates (12, 14 and 16, respectively) with equal efficiency (entries 3–5, Table 4).
| Entry | Substrate | Product | Reaction time (min) | % yieldb (α:β) |
Ref. |
|---|---|---|---|---|---|
| a The substrate was treated with Ac2O (1.2 equiv. per OH/SH group except for entry 7, 11, 12, 13) in the presence of diazepinium perchlorate (2, 25 mol%) under neat reaction conditions at room temperature (40 °C).b Isolated yield has been reported.c 3.0 equiv. of 2 was used at room temperature (40 °C).d 2.5 equiv. of Ac2O was used at room temperature (40 °C).e 50 mol% of 2 needed for the reaction. | |||||
| 1 |  |
 |
30 | 98 (2:3) |
11 |
| 2 |  |
 |
20 | 96 (3:1) |
11 |
| 3 |  |
 |
45 | 96 (2:1) |
11 |
| 4 | 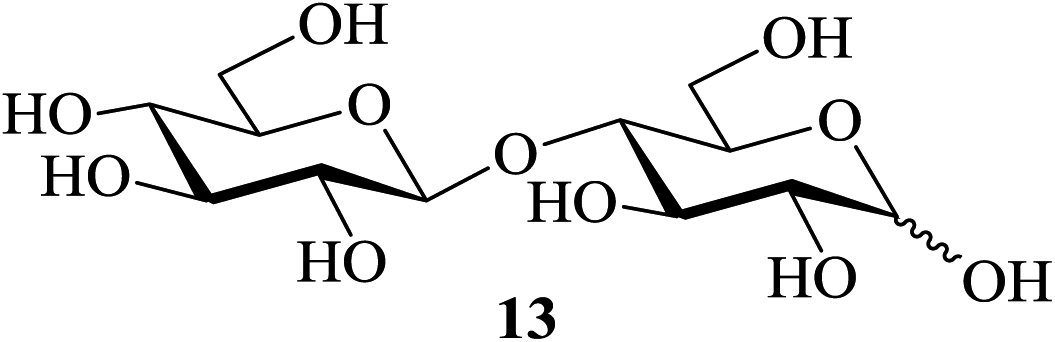 |
 |
180 | 99 (1:2) |
20 |
| 5 |  |
 |
45 | 96 (1:5) |
21 |
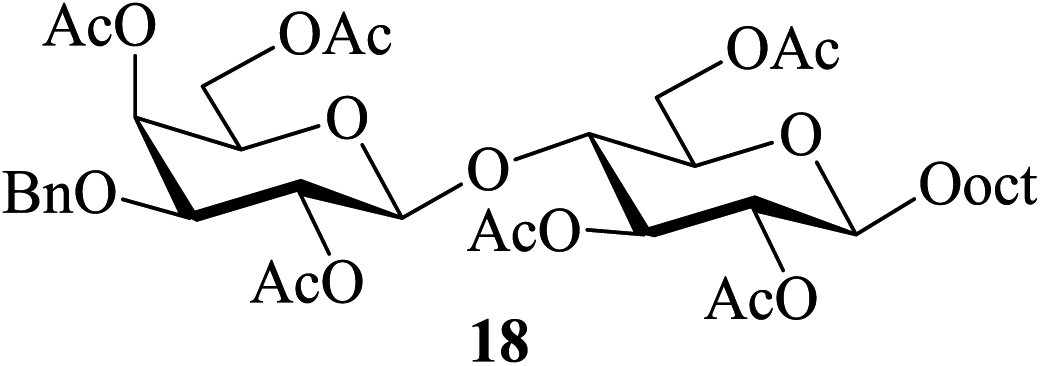
| 6c | 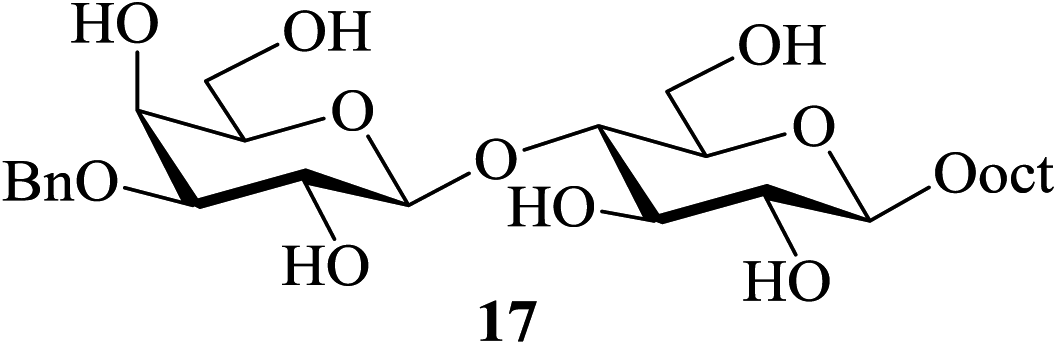 |
 |
24 h | 95 | — |
| 7 | 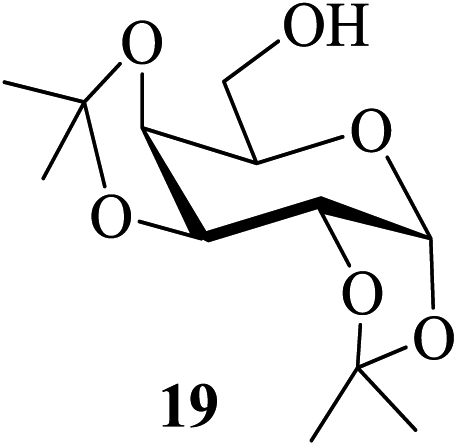 |
 |
8 h | 98d,e | 18 |
| 8 |  |
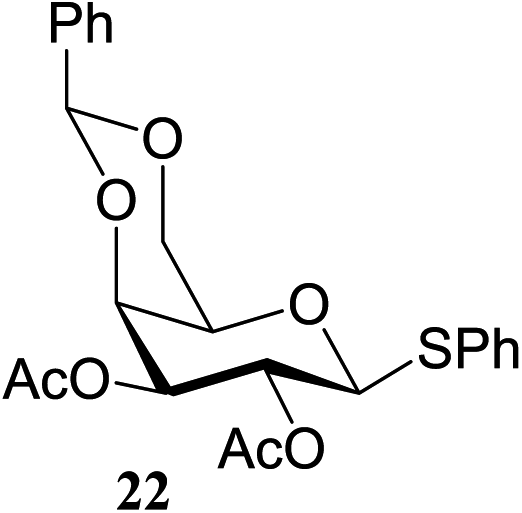 |
60 | 97 | 22 |
| 9 |  |
 |
15 | 95 | — |
| 10 |  |
 |
120 | 96 | 23 |
| 11 | 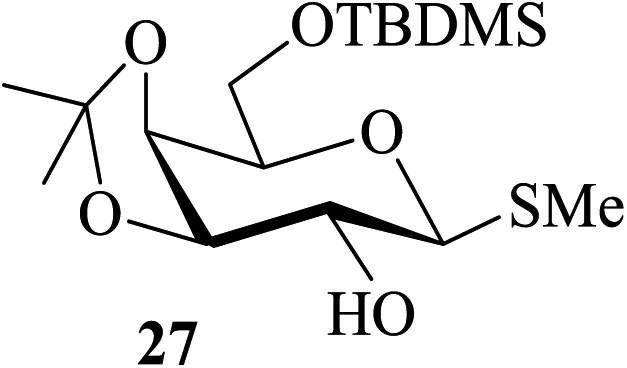 |
 |
5 h | 96d | 24 |
| 12 |  |
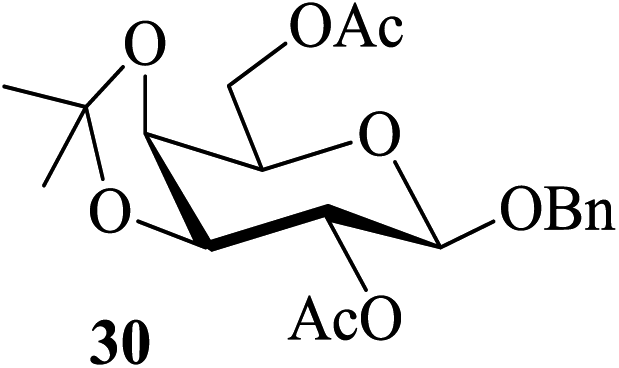 |
12 h | 98d | 25 |
| 13 |  |
 |
120 | 96d | 26 |
| 14 | 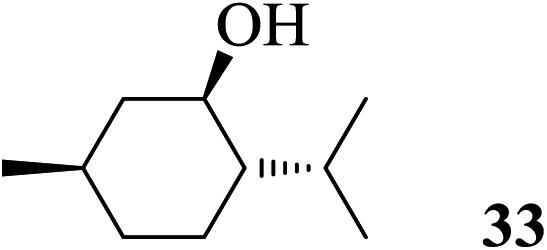 |
 |
30 | 99 | 26 |
| 15 | 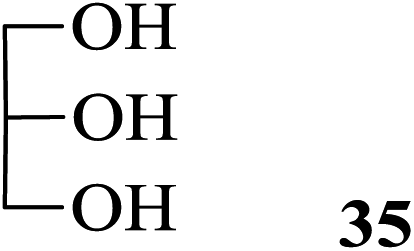 |
 |
30 | 97 | 27 |
| 16 |  |
 |
30 | 98 | 18 |
 |
 |
||||
| 17 | 39 R1 = CH2OH, R2 = H | 40 R1 = CH2OAc, R2 = H | 30 | 99 | 26 |
| 18 | 41 R1 = OH, R2 = H | 42 R1 = OAc, R2 = H | 3 h | 96 | 28 |
| 19 | 43 R1 = OH, R2 = CH3 | 44 R1 = OAc, R2 = CH3 | 2.5 h | 95 | 29a |
| 20 | 45 R1 = OH, R2 = NO2 | 46 R1 = OAc, R2 = NO2 | 24 h | No reaction | — |
| 21 | 47 R1 = SH, R2 = CH3 | 48 R1 = SAc, R2 = CH3 | 15 | 96 | 29a |
| 22 | 49 R1 = SH, R2 = OH | 50 R1 = SAc, R2 = OAc | 2 h | 96 | 29a |
| 23 | 41 + 45 | 42 + unchanged 45 | 3 h | 95 | 28 |
| 24 | 51 R1 = OH, R2 = NH2 | 52 R1 = OAc, R2 = NHAc | 1 h | 99d | 30 |
| 25 |  |
 |
10 | 96d | 31 |
| 26 |  |
 |
15 | 97 | 30 |
| 27 |  |
 |
60 | 95 | 32 |
In view of the above successful results obtained with 2, its analogues 3 and 4 were also investigated for its possible application in the above class of acylation reactions using 5 as the substrate. It was found that under the reaction conditions described above, the perchlorates 3 and 4 were in fact ineffective as a promoter for the acetylation of 5. These salts were therefore not investigated further.
In most of the acid-catalyzed acetylation reactions of sugars difficulties arise when acetylation of their derivatives bearing acid-labile substituents such as acetals and trityl/TBDMS/TBDPS ethers are to be carried out. One of the approaches towards overcoming such problems has been to perform the reaction under milder and controlled conditions (for example, at lower temperatures, reduced catalyst-concentration, etc.). Therefore care-free methods requiring no such special conditions for the success of this reaction would be greatly beneficial. It was in this context that the following sets of partially protected sugar derivatives were examined as substrates for acetylation in the presence of 2. Thus, the partially benzylated lactoside 17, the isopropylidene acetal of galactopyranose 19, the hexopyranosides-derived benzylidene acetals 21 and 23, the trityl ether 25, the silyl ether 27 and the benzyl glycoside 29 also gave their respective acetylated derivatives 18, 20, 22, 24, 26, 28 and 30 respectively in excellent (near-quantitative) yields in reactions carried out at 35–40 °C (entries 6–12, Table 4). The NMR spectra of these compounds obtained as crude products after the aqueous workup of the reaction mixture (please see the ESI†) stand as testimony to their excellent purity.
In order to reconfirm the stability of the acid-sensitive protecting groups on the substrates in the acetylating solution, the reaction mixture from a parallel reaction carried out on the acetal-protected 29 was left for four weeks after the formation of 30 was complete.
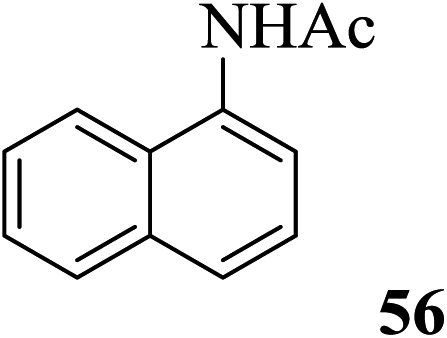
The product isolated subsequently by aqueous work up was indeed found to be the unchanged 30. The importance of this observation lies in the fact that all these compounds are extremely vulnerable to acetolytic conditions (Ac2O-acidic catalyst systems) usually employed for the reaction. Further, alcohols such as β-cholesterol (31), (−)-menthol (33), glycerol (35), dithiothreitol (37) and benzyl alcohol (39) also gave their acetylated derivatives 32, 34, 36, 38 and 40 (entries 13–17, Table 4), respectively, in excellent yields on treatment with Ac2O in the presence of 2. Likewise, examples of phenols, thiophenols and their derivatives were also tested as substrates for acetylation. It was found that phenol (41), 4-methylphenol (43), 4-methylthiophenol (47) and 4-hydroxythiophenol (49) gave their respective acetylated derivatives 42, 44, 48 and 50 (entries 18, 19, 21 and 22, Table 4) in excellent yields. Thus, no chemoselectivity towards acetylation under the above condition was observed in the case of the phenolic hydroxyl and sulfhydryl groups. On the other hand, 4-nitrophenol (45) in the presence of 2 in Ac2O did not give the acetylated product 46 even after 24 h of treatment under conditions identical for phenol (entry 20, Table 4). As theoretical data of 4-methylphenol (43) and 4-nitrophenol (45) like pKa and σ (Hammett constants)29b are 10.174, −0.17 and 7.161, 0.78 indicated that latter case is more acidic and rather electrophilic (entries 19 and 20, Table 4). Thus, the deactivating effect of the para-substituted electron withdrawing nitro group was clearly evident. Moreover, the reported values29b for the pKa and the σ (Hammett constants) for these compounds are also supportive of this observation. As a result thus, when a mixture of 41 and 45 was subjected to treatment with Ac2O in the presence of 2, only the former underwent the reaction enabling the isolation of 42 by aqueous workup in excellent yield along with the near quantitative recovery of 45 (entry 23, Table 4). Therefore this could serve as a simple protocol for the effective separation of such compounds in a mixture. In continuation of this study, a set of typical examples of amino compounds were also subjected to acetylation under the conditions noted above. Thus, aminophenols/alcohols such as 51 and 53 as well as amines such as 55 and 57 when treated with Ac2O in the presence of 2 also underwent the reaction smoothly to give the corresponding acetylated derivatives 52, 54, 56 and 58, respectively (Table 4, entries 24–27) in excellent yields. Whereas in case of 51 and 53 (Table 4, entries 24–25), no chemoselectivity in the N-/O-acetylation was observed.
2.1 Rationalization of the reactivity differences observed in the phenolic/thiophenolic compounds
In the acylation reactions such as those described above, the reactivity differences observed in the case of substituted phenols/thiophenols, etc. are generally justified (and as described herein) by the activating/deactivating effect of the substituent groups present on the aromatic ring. However, no quantitative estimates in respect of the above, in terms of the calculated nucleophilicity of phenol/thiophenol substrates have been available. In view of this an attempt was made to perform a computational study by using Gaussian09 package employing a density functional B3LYP/6-31+G(d) method to calculate the nucleophilicity parameters as local nucleophilicity index (Nk−), global nucleophilicity index (N), and Fukui function for electrophilic attack (fk−). The Fukui function allows to predict the most electrophilic and nucleophilic sites in a molecule which can be further used to determine the reactivity of the molecules under study. When 39, 41, 43, 47, 49, 59, 61, 63, 67, 69, 71 and 73 were treated with acetic anhydride in the presence of 2 at 40 °C yielded respectively 40, 42, 44, 48, 50, 60, 62, 64, 68, 70, 72 and 74 in excellent yields (Table 5, entries 1–16). On the other hand, the fluoro- (65) and the phenol, thiophenol and the substituted phenols/thiols used in this study have been listed in Table 5 in the decreasing order of their nucleophilicity, based on the calculated local nucleophilicity index, Nk− (see in ESI†). It can be seen from the table that for the substrates used in this study, the nucleophilicity index in general followed the order, thiophenols > substituted thiophenols > phenols > substituted phenols, with the exception of p-nitrothiophenol occupying a place among that of the substituted phenols, not surprisingly though considering the strong electron withdrawing (inductively as well as, significantly more strongly, by resonance) nature of this substituent group. The experimental results obtained for these compounds are also in general in accordance with the above, as can be seen from the reaction time noted in Table 5 for the respective compounds.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Phenol/thiophenol | Calculated values | Experimental observations/results | |||
| Substrate | Product | Nk− | fk− | Reaction time (h) | Yieldc (%) | |
| a Nk− = local nucleophilicity, fk− = Fukui function.b All reactions were carried out using 1 mmol substrate in 1 mL of acetic anhydride in the presence of 25 mol% 2.c Isolated yields are reported.d Not isolated. | ||||||
| 1 | Ph-SH (59) | Ph-SAc (60) | −0.35 | −0.36 | 0.5 | 99 |
| 2 | p-Me-Ph-SH (47) | p-Me-Ph-SAc (48) | −0.32 | −0.32 | 0.25 | 96 |
| 3 | o-Br-Ph-SH (61) | o-Br-Ph-SAc (62) | −0.29 | −0.35 | 1.5 | 99 |
| 4 | p-Cl-Ph-SH (63) | p-Cl-Ph-SAc (64) | −0.25 | −0.31 | 1.5 | 99 |
| 5 | p-HO-Ph-SH (49) | p-AcO-Ph-SAc (50) | −0.17 | −0.20 | 2 | 96 |
| 6 | p-Me-Ph-OH (43) | p-Me-Ph-OAc (44) | −0.16 | −0.15 | 2.5 | 95 |
| 7 | Ph-OH (41) | Ph-OAc (42) | −0.16 | −0.16 | 3 | 96 |
| 8 | p-F-Ph-OH (65) | p-F-Ph-OAc (66) | −0.14 | −0.16 | 24 | Tracesd |
| 9 | o-Cl-Ph-OH (67) | o-Cl-Ph-OAc (68) | −0.14 | −0.16 | 3.5 | 98 |
| 10 | p-O2N-Ph-SH (69) | p-O2N-Ph-SAc (70) | −0.12 | −0.35 | 4 | 98 |
| 11 | p-Cl-Ph-OH (71) | p-Cl-Ph-OAc (72) | −0.12 | −0.14 | 3.5 | 98 |
| 12 | p-Br-Ph-OH (73) | p-Br-Ph-OAc (74) | −0.11 | −0.13 | 4 | 90 |
| 13 | Bn-OH (39) | Bn-OAc (40) | −0.10 | −0.12 | 0.5 | 99 |
| 14 | m-O2N-Ph-OH (75) | m-O2N-Ph-OAc (76) | −0.06 | −0.17 | 24 | 73 |
| 15 | p-O2N-Ph-OH (45) | p-O2N-Ph-OAc (46) | −0.06 | −0.16 | 24 | No reaction |
| 16 | o-O2N-Ph-OH (77) | o-O2N-Ph-OAc (78) | −0.05 | −0.18 | 24 | No reaction |

However, the experimental results obtained in the case of p-fluorophenol (entry 8, Table 5) showing it to be virtually unreactive under the conditions, has been an exception in this case. The reactivity differences between 4-fluorophenol (65) and 2-chlorophenol (67) have also been noted previously by others.46a This certainly points to the important additional factors, such as the intermolecular hydrogen bonding, in operation in the case of this halogenophenol. Likewise, although p-thiocresol (47) was proved experimentally to be completing the reaction faster as expected from the activating effect of the p-Me substituent in it, was shown to be marginally lower in the order of nucleophilicity index (entries 1 and 2, Table 5). Similarly, p-cresol (43) though was more reactive (reacting completely in 2.5 h) than phenol (41, that required 3 h in comparison), had identical nucleophilicity indices (entries 6 and 7, Table 5). In the case of the three (o-, m- and p-) nitrophenols (entries 14–16, Table 5), the m-nitrophenol (75) was proved more reactive than the other two (77 and 45) as expected. The strong electron withdrawing nature of the nitro group is still evident (in spite of the absence of any resonance effect) from the relatively low yielding reaction observed in it (entry 14 versus 15 and 16, as well as other entries, Table 5).
Additional information emerged from the experiments carried out on various sugars and their partially protected analogues was that, in general, the partially protected sugars required longer time for the completion of the reaction. Although it is consistent with the exothermic nature of the reaction, it also served to suggest that selective acetylation leading to partially protected sugars could perhaps be achievable in the case of partially protected sugar derivatives with more than one hydroxyl functionality, perhaps placed strategically, on them.33,34
Thus, the 6-O-silylated mannosides 79 and 81 were employed as substrates for acetylation with Ac2O in the presence of 2 (entries 1 and 2, Table 6). Indeed, the respective di-O-acetylated products 80 and 82, with the OH-4 remaining free, were obtained as the sole respective products in excellent yields (entries 1 and 2, Table 6). The partially protected compounds 80 and 82 are extremely valuable glycosyl acceptor substrates in oligosaccharide synthesis and their preparation from their respective precursor-glycosides would have involved significantly more steps. In contrast, the outcome of the acetylation of the p-nitrophenyl galactoside 83 carried out subsequently was comparatively less dramatic in its effectiveness (entry 3, Table 6) in that the selectively protected 84 could be obtained in only 60% yield, the remaining being the tetra-O-acetate 85 although no difficulty in the separation of the two (84 and 85) by column chromatography on silica gel was experienced. Thus, under the conditions of acetylation studied the HO-2 of the galactoside 83 was found less reactive than the other secondary hydroxyls present in the molecule. Among the secondary hydroxyl groups of galactopyranosides such as 83 the HO-3 is known for its relative high reactivity compared to HO-2 and HO-4. However, the lower reactivity of HO-2 compared to that of HO-4 observed in the above case was somewhat surprising and could have been due to the deactivating nature of the p-nitrophenyl group present on O-1 of the sugar moiety leading to a possibly reduced, despite perhaps only marginally, nucleophilicity of its O-2. In an attempt to rationalize this observation further the acetylation of m-nitrophenyl β-D-galactoside 86 was carried out under the same conditions as for the analogous galactoside 83. Here again, the selectively 2,3,6-tri-O-acetylated product 87 was obtained along with the tetra-O-acetylated galactoside derivative 88. However, compounds 87 and 88 were obtained in 80% and 20% yields (entry 4, Table 6), respectively, after separation by chromatography on a column of silica gel. Thus, although the m-nitrophenyl group at the anomeric centre of 86 is expected to influence the nucleophilicity of its O-4 to a lesser extent, as compared to the p-nitrophenyl substituent present in 83, a significantly enhanced yield of 87 (bearing a free hydroxyl on C-4) was indeed obtained in the reaction of 86.
| Entry | Substrate | Catalyst (mol%) | Time (h) | Ratio of productsb | Yieldc (%) |
|---|---|---|---|---|---|
| a The substrate was treated with Ac2O (2 equiv.) in the presence of diazepinium perchlorate 2 at room temperature (35–40 °C).b On the basis of isolated yield and 1H NMR, ratios of the products were reported.c Isolated yield of the product was reported.d 90 and 91 are inseparable in the ratio of 1:0.6 and 92 and 93 are inseparable in the ratio of 1:0.7 where overall ratio of 90:91:92:93 = 1:0.6:2:1.4. |
|||||
| 1 |  |
30 | 2 |  |
96 |
| 2 |  |
30 | 4 |  |
95 |
| 3 |  |
100 | 3 |  |
95 |
| 4 | 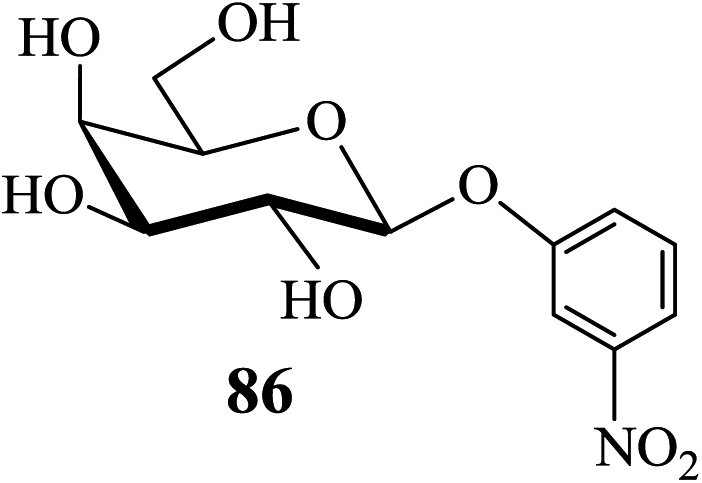 |
100 | 3 |  |
96 |
| 5 |  |
100 | 3 |  |
92d |
| 6 |  |
100 | 4 |  |
90 |
| 7 |  |
100 | 4 |  |
96 |
| 8 |  |
50 | 5 |  |
97 |
| 9 | 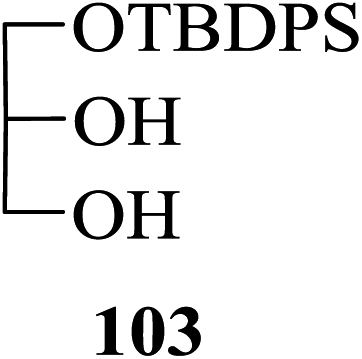 |
30 | 4 | 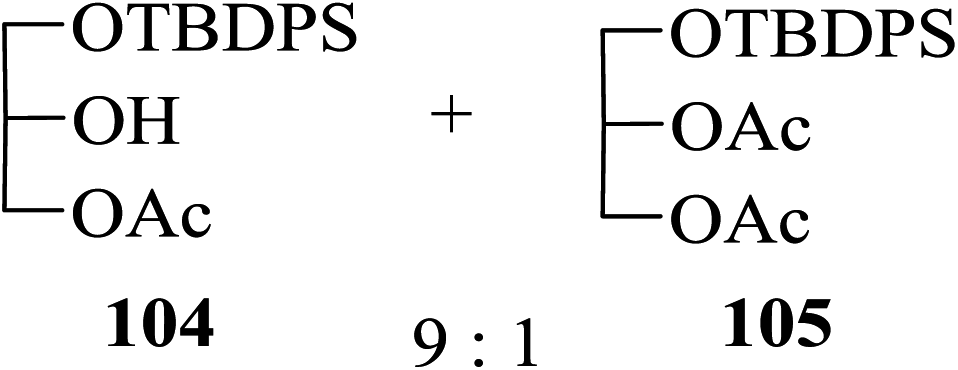 |
96 |
It therefore appeared that the influence of the m-nitrophenyl group present at the anomeric centre on 86 may have been more steric (than electronic) in nature. It may be noted that as compounds 87 and 88 had significantly different Rf values their separation was achieved efficiently and therefore the extremely valuable partially protected galactoside 87 could be obtained in pure form without any practical difficulty. Its traditional synthesis would have indeed been multi-step in nature. At this point a study on the acetylation of the o-nitrophenyl β-D-galactoside 89 was thought beneficial.
Thus, when 89 was treated with Ac2O in the presence of 2 and the product emerged was analyzed by TLC, it was revealed that the outcome was of a more complex nature. A set of three spots with Rf values 0.45 (A), 0.43 (B) and 0.23 (C) was obtained on TLC (eluent, EtOAc:n-heptane, 2:1) of the product. Attempts to separate them by chromatography on a column of silica, resulted in the elution of the first two (A and B) closely-moving components as a mixture. Analysis of this mixture by NMR spectroscopy, however, enabled the identification of these products as the 2,4,6-tri-O-acetylated compound 90 and the 3,6-di-O-acetylated compound 91 (A and B, respectively) in the ratio 1:0.6 (entry 5, Table 6). Following the above, component C was eluted and was identified by NMR spectroscopy as a mixture of two regioisomeric diacetates, namely, the 4,6-di-O-acetylated compound 92 and the 2,6-di-O-acetylated compound 93 in a ratio of 1:0.7. The proportion in which these eluates (A + B and C) were present in the mixture was 1:2 [(90 + 91):(92 + 93)], thus the overall ratio of 90:91:92:93 formed being 1:0.6:2:1.4. A surprising observation on the outcome of the foregoing reaction was the unusually low proportions of the product-fraction 91 bearing a 3-O-acetyl moiety, in spite of the known relatively high reactivity of the HO-3 of unprotected D-galactopyranosides. Moreover, compound 91 was the sole 3-O-acetyl group-containing product formed in the acetylation of 89. In contrast, the 2-O-acetyl group-containing products formed in the acetylation of 89 were significantly more, indicating a relatively more facile acetylation at HO-2. This was suggestive of a possible steric interaction (1,3-cis-) exerted by the anomeric O-o-nitrophenyl substituent in 89 on its HO-3.
Moreover, in the case of the 4,6-O-benzylidene-protected diol 94 the mono- and di-O-acetates 95 and 96, obtained in 2:1 ratio, were the only products of acetylation, with no selectively 2-O-acetylated product being obtained in the reaction. Our attempts to gather theoretical support for the observations noted in the regioselective acetylations of galactosides 83, 86, 89 and 90 described above, by subjecting these structures to studies by Gaussian09 package unfortunately were turned out inconclusive (and therefore are not discussed here).
p-Acetyl-o-methoxyphenyl β-D-glucoside 97 when treated with acetic anhydride in the presence of 2 the selectively 2,3,6-tri-O-acetylated product 98 was obtained in 80% yield along with the tetra-O-acetylated glucoside derivative 99 obtained in 20% yield (entry 7, Table 6) in consistence with the known order of reactivity of a glucopyranoside-hydroxyl groups. Likewise, in the case of the reaction of methyl 6-O-trityl α-D-glucoside 100 with Ac2O in the presence of 2 also, the selectively 2,3-di-O-acetylated product 101 was indeed the major partially acetylated product formed (65% yield along with tri-O-acetylated product 102 in 32% yield, entry 8, Table 6). Moreover, preference for the acetylation of a primary hydroxyl group over a secondary was also evident from the results obtained from the reaction of the partially protected glycerol derivative 103 yielding selectively mono-O-acetylated product 104 as the major product (along with di-O-acetate 105 in a ratio of 9:1 in 96% combined yield, entry 9, Table 6).
Further, a comparison of different reported catalysts with the diazepinium salt 2 for acetylation reaction of 5 and 19 has been illustrated in Table 7. Thus, it becomes clear that while the current method is comparable to the best literature methods in terms of the reaction yield, it is solvent-free and requires only close-to-stoichiometric quantities of the acyl donor agent (like some of the known entries in the table) and, unlike many of the others, is greener (organic, non-corrosive, non-hazardous and not sensitive to air/moisture).
|
Catalyst | Ac2O (equiv.) | Solvent | Time (min) | Reported yield (%) | Ratio (α/β) | |
|---|---|---|---|---|---|---|---|
| Name | Mol% | ||||||
| a The reaction was performed in CH3CN at 80 °C.b The reaction was performed under microwave irradiation.c The reaction was performed at 40 °C.d The reaction was carried out at ice-bath temperature.e Reaction did not go to completion.f Degraded product was obtained. | |||||||
 |
In(OTf)3 (ref. 12) | 0.16 | 8.8 | None | 3 | 100 | 14/1 |
| Ce(OTf)3 (ref. 35) | 1 | 7.5 | Ac2O | 150 | >98 | — | |
| Sc(OTf)3 (ref. 36) | 0.5 | 6 | None | 90 | 96 | — | |
| CoCl2 (ref. 3) | 5 | 10 | CH3CN | 6 h | 75a | — | |
| InCl3 (ref. 37) | 0.1 | 7.5 | CH3CN | 1.5 | 94b | 4/1 | |
| I2 (ref. 11) | 3.5 | 9.5 | None | 30 | 90 | 25/1 | |
| [bmim][HSO4]38 | 100 | 5.5 | Ac2O | No reaction | — | ||
| [bmim][OAc]38 | 100 | 5.5 | Ac2O | 15 | 90 | — | |
| LiClO4 (ref. 15a) | 50 | 5.8 | None | 12 h | 94c | — | |
| CuClO4 (ref. 39) | 1 | 5.05 | None | 30 | 97 | 1/0 | |
| Montmorilonite-K40 | 1 g | 13.8 | None | 6 h | 94 | — | |
| Imidazole41 | 60 | 6 | CH3CN | 5 h | 98 | 4/1 | |
| Pyridine42a | Excess | 7.5 | CH3CN | 18 h | 33 | 3.7/1 | |
| —do—42b | Excess | Excess | Ac2O | 18 h | 95 | — | |
| Diazepinium perchlorate (2) | 25 | 6.6 | None | 30 | 99c | 49/1 | |
 |
I2 (ref. 11) | 2.5 | 13 | None | 5 | 98d | — |
| LiClO4 (ref. 15b) | 10 | 1.1 | None | 48 h | 96c | — | |
| Al2O3 (ref. 43) | 200 | 1.5 | None | 12 h | 30e | — | |
| InCl3 (ref. 37) | 0.1 | 7.5 | CH3CN | 2 | 35b,f | — | |
| Pyridine42 | Excess | 2.5 | Pyridine | 45 | 95 | — | |
| Imidazole41 | 60 | 1.2 | CH3CN | 2 h | 98 | — | |
| DABCO44 | 100 | 1.2 | None | 20 | 98 | — | |
| Diazepinium perchlorate (2) | 50 | 2.5 | None | 8 h | 98c | — | |

3. Conclusions
Diazepinium perchlorate (2) has been found to be an attractive alternative neutral catalyst for acyl transfer reactions of mono- and disaccharides and their various partially protected derivatives as well as various alcohols, phenols, thiols, thiophenols and amines using acetic anhydride as the acyl donor agent. Regioselective acetylations of various partially protected monosaccharides were also successful and were proved effective time-saving alternatives to their conventional multi-step synthesis. The results obtained on the acetylation of phenols and thiophenols were rationalized through their local nucleophilicity index from DFT calculations.4. Experimental
4.1 General
All reagents (Aldrich India Ltd) used were of analytical grade and were used as purchased without further purification. Reactions were monitored by TLC, which was performed using 0.2 mm pre-coated silica gel 60 F254 aluminium sheets. Compounds were detected by dipping the TLC plates in an ethanolic solution of sulphuric acid (5%, v/v) and heating or using a reagent prepared by dissolving a mixture of ammonium molybdate (4.2 g), cerium sulfate tetrahydrate (0.2 g) and sulphuric acid (6.2 mL) in water (94 mL). Column chromatography was performed using silica gel (100–200 mesh, ASTM). 1H/13C NMR spectra were recorded in CDCl3 at 400/100 MHz on a Bruker AvanceIII spectrometer. The chemical shifts have been reported in ppm using TMS as the internal standard. The multiplicity of the signals have been shown as, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet) with the coupling constant (J value) expressed in Hz. Dept 135 and 2D NMR like COSY, HSQC and HMBC were also used for characterizing synthesized molecules. The anomeric ratios of products reported in Table 1 were determined from their NMR spectra. Mass spectra were obtained on Ultraflex TOF/TOF MALDI or HRMS equipment. Specific rotations were determined on an AUTOPOL IV polarimeter at 20–24 °C. IR spectra were recorded on a Nicolet FT-IR Impact 410 instrument either as neat or as KBr pellets. Melting points were determined on a melting point apparatus.4.2 Typical procedure for the preparation of 2
A mixture of acetylacetone (10 mL, 97.8 mmol) and ethylenediamine (10 g, 166.3 mmol) was heated at 120 °C for 2 min. Acetic acid (20 mL) was then added to it and the reaction mixture was stirred for 10 min at 120 °C. It was then brought to room temperature, and after adding water (200 mL) followed by aqueous HClO4 (20%, 60 mL) and stirring, the mixture was allowed to stand overnight to form crystals. The crystals were then separated by filtration through a fritted glass funnel and were washed with cold water followed by n-hexane/n-heptane on the funnel. They were then dried under reduced pressure (48 h) to yield 2 as a colourless crystalline product (36 g, 96%). The product was characterized by comparison of its melting point and spectral data (NMR and MS) with literature values which were in agreement with each other. Colourless needle shaped crystal; mp 140–141 °C; 1H NMR (CD3OD): δ (ppm) 5.03 (s, 1H, CH), 4.88 (bs, 4H, 2 × CH2), 3.65 (bs, 2H, NH2), 2.22 (s, 6H, 2 × CH3); 13C NMR: (CD3OD): δ (ppm) 167.3, 90.9 (CH), 47.9 (CH2), 47.5 (CH2), 22.4 (2 × CH3). IR: (KBr) νmax 3335, 2477, 1620, 1552, 1328, 1100, 819, 703, 619 cm−1; MALDI-TOF/TOF: m/z calcd for C7H13NaClN2O4 247.04. Found: 247.69 [M + Na]+.4.3 General procedure for the acetylation reaction in the presence of 2
The substance to be acetylated was added portionwise to a mixture of 2 (25–30 mol%) and acetic anhydride (1.2–2 mol equiv./OH/SH/NH2 group) and the mixture was stirred at 35–40 °C. On completion of the reaction (as confirmed by TLC), the mixture was directly poured onto crushed ice, neutralized with NaHCO3 and was extracted with EtOAc in a separating funnel. The organic extract was then washed with brine and was dried over anhydrous Na2SO4. After separation by filtration it was concentrated under reduced pressure and vacuum-dried to yield the respective peracetylated product in near-quantitative yields. The crude product thus obtained was practically pure (see the NMR spectra) and were suitable for further use directly. Analytically pure samples were obtained by either recrystallization or by chromatography on a column of silica. Compounds 6,11 8,11 10,11 12,11 14,20 16,21 20,18 22,22 26,23 28,24 30,25 32,26 34,26 36,27 38,18 40,26 42,28 44,29a 46,29a 48,29a 50,29a 52,30 54,31 56,30 58,32 60,28 62,45 64,28 66,28 68,46 70,18 72,28 74,29 76,18 78,28 85,47 88,47 96,48 99,49 101,50 102 (ref. 18) and 105 (ref. 51) (Tables 1–6) have been reported in the literature. Compounds 18, 24, 80, 82, 84, 87, 90, 91, 92, 93, 95, 98 and 104 are new and their characterization data have been described below. For the NMR spectra of these latter compounds, please see the ESI† provided.4.4 Typical procedure for a practical-scale preparation of 6
D-Glucose (20 g, 111 mmol) was added portionwise (over a period of 15 min) to a solution of 2, (6 g, 26.7 mmol) in acetic anhydride (80 mL) during stirring at room temperature (35–40 °C) for 15 min by which time TLC (EtOAc:n-heptane, 2:3, v/v) showed complete consumption of the sugar. The solution had become hot due to the exothermic nature of the reaction. It was then poured, while being stirred, onto crushed ice in a beaker containing sodium bicarbonate and was left in a refrigerator for the solids to separate. The solids were then collected over a glass filter by filtration at the pump and were washed with cold water followed by cold n-hexane/n-heptane. Drying under reduced pressure yielded the desired product, 1,2,3,4,6-penta-O-acetyl-D-glucopyranose (6, 42.8 g, 99%) as a colourless solid homogenous in all respects with an authentic sample of 6 and as confirmed by physical constants and spectral data.
4.5 Typical procedure for the regioselective acetylation of the triol 79: preparation of 80
Octyl 6-O-tert-butyldimethylsilyl-α-D-mannopyranoside (79, 1 g, 2.31 mmol) was added to a solution of diazepinium perchlorate (2, 165 mg, 30 mol%) in acetic anhydride (5 mL) and was stirred for 2 h at 35–40 °C when TLC (EtOAc:n-heptane, 2:3, v/v) showed complete consumption of the triol 79. The reaction mixture was diluted with EtOAc (20 mL) and was washed successively with cold aqueous NaHCO3 solution and water (to neutrality) and dried (Na2SO4). Removal of the solvent under reduced pressure and purification of the residue on a short column of silica (eluent, EtOAc:Hex, 30:70) afforded octyl 2,3-di-O-acetyl-6-O-tert-butyldimethylsilyl α-D-mannopyranoside (80, 1.16 g, 96%) as a colourless liquid as was confirmed by spectral analysis (see below).
4.6 Characterization of compounds
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 137.2, 128.4, 127.9, 127.7, 101.2, 100.5, 76.3, 72.9, 72.6, 71.7, 71.4, 70.8, 70.6, 70.2, 65.3, 62.3, 61.4, 31.9, 31.7, 29.7, 29.3, 29.2, 25.7, 22.6, 20.8, 20.7, 20.6 (3 × CH3 of COCH3), 14.2, 14.1. IR: (neat) νmax 3475, 2926, 2855, 2255, 1751, 1458, 1369, 1226, 1170, 1054, 913, 743, 648 cm−1; ESI-HRMS: m/z calcd for C39H60NO17 814.3861. Found: 814.3861 [M + NH4]+.O), 97.4 (C-1), 72.8, 71.8, 71.3, 69.8, 68.1, 64.2 (C-6), 31.8, 29.3, 26.0, 25.8, 22.6, 20.9, 18.2, 14.0, −5.4 {2 × CH3 of Si(CH3)2}; IR: (neat) νmax 3436, 2925, 2854, 1751 (CO), 1634, 1372, 1249, 1084, 836 cm−1; ESI-HRMS: m/z calcd for C24H46NaO8Si 513.2864. Found: 513.2854 [M + Na]+.
O), 137.2, 128.4, 127.9, 127.7, 101.2, 100.5, 76.3, 72.9, 72.6, 71.7, 71.4, 70.8, 70.6, 70.2, 65.3, 62.3, 61.4, 31.9, 31.7, 29.7, 29.3, 29.2, 25.7, 22.6, 20.8, 20.7, 20.6 (3 × CH3 of COCH3), 14.2, 14.1. IR: (neat) νmax 3475, 2926, 2855, 2255, 1751, 1458, 1369, 1226, 1170, 1054, 913, 743, 648 cm−1; ESI-HRMS: m/z calcd for C39H60NO17 814.3861. Found: 814.3861 [M + NH4]+.O), 97.4 (C-1), 72.8, 71.8, 71.3, 69.8, 68.1, 64.2 (C-6), 31.8, 29.3, 26.0, 25.8, 22.6, 20.9, 18.2, 14.0, −5.4 {2 × CH3 of Si(CH3)2}; IR: (neat) νmax 3436, 2925, 2854, 1751 (CO), 1634, 1372, 1249, 1084, 836 cm−1; ESI-HRMS: m/z calcd for C24H46NaO8Si 513.2864. Found: 513.2854 [M + Na]+.![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2Ph), 5.10 (dd, J1 = 10.0 Hz, J2 = 3.4 Hz, 1H, H-3), 4.80 (s, 1H, H-1), 4.54 (t, J = 4.0 Hz, 1H, CNH), 3.99 (dd, J1 = 10.4 Hz, J2 = 4.0 Hz, 1H, CH of CH2O of serine), 3.92 (t, J = 4.2 Hz, 1H, H-4), 3.89 (dd, J1 = 7.2 Hz, J2 = 3.0 Hz, 3H, H-6 & CH of CH2O of serine), 3.77 (s, 3H, OCH3), 3.68–3.63 (m, 1H, H-5), 2.09 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 0.90 (s, 9H, C(CH3)3), 0.08 (s, 6H, Si(CH3)2); 13C NMR: (CDCl3): δ (ppm) 170.7, 170.2, 170.1, 170.0 (CO), 156, 136, 128.5, 128.3, 98.2 (C-1), 72.2, 71.5, 69.3, 68.9, 67.1, 63.5, 54.2, 52.8, 29.6 (2 × CH3 of COCH3), 25.8 (CH3 of C(CH3)3), 20.9 (2 × CH3 of COCH3), 18.2 (CH3 of COCH3), −5.4 (CH3 of Si(CH3)2); IR: (neat) νmax 3361, 3034, 2927, 2855, 1748 (CO), 1530, 1462, 1438, 1371, 1248, 1141, 1048, 984, 836 cm−1; ESI-HRMS: m/z calcd for C28H44NO12Si 614.2633. Found: 614.2632 [M + H]+.O), 161, 143.0, 125.7, 116.6, 100.4 (C-1), 72.5, 71.3, 68.5, 66.9, 61.9, 20.7 (CH3 of COCH3), 20.5 (2 × CH3 of COCH3); IR: (neat) νmax 3439, 2924, 2927, 1747 (CO), 1611, 1593, 1518, 1372, 1345, 1240, 1074, 862, 751 cm−1; ESI-HRMS: m/z calcd for C18H21NaNO11 450.1007. Found: 450.1003 [M + Na]+.O), 157.0, 149.1, 130.17, 123.7, 118.1, 111.3, 99.2 (C-1), 71.6, 70.6, 68.3, 66.9, 61.7, 20.6 (CH3 of COCH3), 20.5 (3 × CH3 of COCH3); IR: (neat) νmax 3459, 2933, 1746 (CO), 1634, 1532, 1352, 1230, 1080, 1141, 1048, 749 cm−1; ESI-HRMS: m/z calcd for C21H26O11Na 477.1373. Found: 477.1370 [M + Na]+.O), 149.4, 141.4, 134.5, 125.6, 123.5, 119.4, 103.6 (C-1), 74.0, 72.8, 71.5, 69.2, 66.7, 61.9, 20.7 (3 × CH3 of COCH3); IR: (Neat) νmax 3445, 2924, 2854, 1743 (CO), 1607, 1529, 1487, 1370, 1237, 1073, 748 cm−1. ESI-HRMS: m/z calcd for C18H21NNaO11 450.1007. Found: 450.1006 [M + Na]+.O), 150.3, 141.4, 133.6, 125.1, 123.4, 118.9, 100.4 (C-1), 74.0, 71.1, 68.7, 66.7, 62.2, 20.7 (2 × CH3 of COCH3); IR: (neat) νmax 3445, 2924, 2854, 1743 (CO), 1607, 1529, 1487, 1370, 1237, 1073, 748 cm−1. ESI-HRMS: m/z calcd for C16H19NNaO10 408.0901. Found: 408.0900 [M + Na]+.O), 150.3, 141.3, 134.4, 125.6, 123.3, 119.4, 102.8 (C-1), 72.9, 71.8, 71.3, 68.7, 62.7, 20.8 (2 × CH3 of COCH3); IR: (Neat) νmax 3419, 2924, 1742 (CO), 1607, 1528, 1370, 1238, 1070, 749 cm−1. ESI-HRMS: m/z calcd for C16H19NNaO10 408.0907. Found: 408.0903 [M + Na]+.O), 149.5, 140.5, 133.6, 125.0, 123.2, 118.6, 100.4 (C-1), 72.2, 71.8, 71.5, 68.4, 61.9, 20.7 (2 × CH3 of COCH3); IR: (neat) νmax 3419, 2924, 1742 (CO), 1607, 1528, 1370, 1238, 1070, 749 cm−1. ESI-HRMS: m/z calcd for C16H19NNaO10 408.0901. Found: 408.0902 [M + Na]+.O), 161.6, 142.9, 137.3, 129.1, 128.2, 126.1, 125.7, 116.7, 101.0, 100.5, 73.4, 73.0, 68.7, 68.0, 66.8, 21.0 (CH3 of COCH3); IR: (neat) νmax 3482, 2992, 2936, 2864, 1755 (CO), 1593, 1519, 1347, 1230, 1089, 994, 910, 850, 750 cm−1; anal. calcd for C21H21NO9: C, 58.47; H, 4.91; N, 3.25. Found: C, 58.12; H, 5.02; N, 2.87. ESI-MS: m/z calcd for C21H21NO9 431.12. Found: 431.21 [M]+.O), 171.5, 171.4, 169.5 (CO), 150.4, 150.2, 133.3, 122.4, 117.1, 111.6, 99.9 (C-1), 75.1, 74.4, 70.9, 68.7, 62.6, 56.1, 26.4 (CH3 of ArCOCH3), 20.8 (3 × CH3 of COCH3), 14.1; IR: (neat) νmax 3459, 2924, 2853, 2855, 1747 (CO), 1674, 1593, 1416, 1367, 1233, 1080, 1035, 908, 805, 744 cm−1; ESI-HRMS: m/z calcd for C21H26O11Na 477.1373. Found: 477.1370 [M + Na]+.O), 135.5, 133.0, 129.9, 127.8, 71.7, 65.2, 64.4, 26.7 {C(CH3)3}, 21.0 (CH3 of COCH3); IR: (neat) νmax 3435, 2963, 1770 (CO), 1428, 1371, 1260, 1136, 1012, 799 cm−1. ESI-HRMS: m/z calcd for C21H28NaO4Si 395.1649. Found: 395.1647 [M + Na]+.
2Ph), 5.10 (dd, J1 = 10.0 Hz, J2 = 3.4 Hz, 1H, H-3), 4.80 (s, 1H, H-1), 4.54 (t, J = 4.0 Hz, 1H, CNH), 3.99 (dd, J1 = 10.4 Hz, J2 = 4.0 Hz, 1H, CH of CH2O of serine), 3.92 (t, J = 4.2 Hz, 1H, H-4), 3.89 (dd, J1 = 7.2 Hz, J2 = 3.0 Hz, 3H, H-6 & CH of CH2O of serine), 3.77 (s, 3H, OCH3), 3.68–3.63 (m, 1H, H-5), 2.09 (s, 3H, COCH3), 2.03 (s, 3H, COCH3), 0.90 (s, 9H, C(CH3)3), 0.08 (s, 6H, Si(CH3)2); 13C NMR: (CDCl3): δ (ppm) 170.7, 170.2, 170.1, 170.0 (CO), 156, 136, 128.5, 128.3, 98.2 (C-1), 72.2, 71.5, 69.3, 68.9, 67.1, 63.5, 54.2, 52.8, 29.6 (2 × CH3 of COCH3), 25.8 (CH3 of C(CH3)3), 20.9 (2 × CH3 of COCH3), 18.2 (CH3 of COCH3), −5.4 (CH3 of Si(CH3)2); IR: (neat) νmax 3361, 3034, 2927, 2855, 1748 (CO), 1530, 1462, 1438, 1371, 1248, 1141, 1048, 984, 836 cm−1; ESI-HRMS: m/z calcd for C28H44NO12Si 614.2633. Found: 614.2632 [M + H]+.O), 161, 143.0, 125.7, 116.6, 100.4 (C-1), 72.5, 71.3, 68.5, 66.9, 61.9, 20.7 (CH3 of COCH3), 20.5 (2 × CH3 of COCH3); IR: (neat) νmax 3439, 2924, 2927, 1747 (CO), 1611, 1593, 1518, 1372, 1345, 1240, 1074, 862, 751 cm−1; ESI-HRMS: m/z calcd for C18H21NaNO11 450.1007. Found: 450.1003 [M + Na]+.O), 157.0, 149.1, 130.17, 123.7, 118.1, 111.3, 99.2 (C-1), 71.6, 70.6, 68.3, 66.9, 61.7, 20.6 (CH3 of COCH3), 20.5 (3 × CH3 of COCH3); IR: (neat) νmax 3459, 2933, 1746 (CO), 1634, 1532, 1352, 1230, 1080, 1141, 1048, 749 cm−1; ESI-HRMS: m/z calcd for C21H26O11Na 477.1373. Found: 477.1370 [M + Na]+.O), 149.4, 141.4, 134.5, 125.6, 123.5, 119.4, 103.6 (C-1), 74.0, 72.8, 71.5, 69.2, 66.7, 61.9, 20.7 (3 × CH3 of COCH3); IR: (Neat) νmax 3445, 2924, 2854, 1743 (CO), 1607, 1529, 1487, 1370, 1237, 1073, 748 cm−1. ESI-HRMS: m/z calcd for C18H21NNaO11 450.1007. Found: 450.1006 [M + Na]+.O), 150.3, 141.4, 133.6, 125.1, 123.4, 118.9, 100.4 (C-1), 74.0, 71.1, 68.7, 66.7, 62.2, 20.7 (2 × CH3 of COCH3); IR: (neat) νmax 3445, 2924, 2854, 1743 (CO), 1607, 1529, 1487, 1370, 1237, 1073, 748 cm−1. ESI-HRMS: m/z calcd for C16H19NNaO10 408.0901. Found: 408.0900 [M + Na]+.O), 150.3, 141.3, 134.4, 125.6, 123.3, 119.4, 102.8 (C-1), 72.9, 71.8, 71.3, 68.7, 62.7, 20.8 (2 × CH3 of COCH3); IR: (Neat) νmax 3419, 2924, 1742 (CO), 1607, 1528, 1370, 1238, 1070, 749 cm−1. ESI-HRMS: m/z calcd for C16H19NNaO10 408.0907. Found: 408.0903 [M + Na]+.O), 149.5, 140.5, 133.6, 125.0, 123.2, 118.6, 100.4 (C-1), 72.2, 71.8, 71.5, 68.4, 61.9, 20.7 (2 × CH3 of COCH3); IR: (neat) νmax 3419, 2924, 1742 (CO), 1607, 1528, 1370, 1238, 1070, 749 cm−1. ESI-HRMS: m/z calcd for C16H19NNaO10 408.0901. Found: 408.0902 [M + Na]+.O), 161.6, 142.9, 137.3, 129.1, 128.2, 126.1, 125.7, 116.7, 101.0, 100.5, 73.4, 73.0, 68.7, 68.0, 66.8, 21.0 (CH3 of COCH3); IR: (neat) νmax 3482, 2992, 2936, 2864, 1755 (CO), 1593, 1519, 1347, 1230, 1089, 994, 910, 850, 750 cm−1; anal. calcd for C21H21NO9: C, 58.47; H, 4.91; N, 3.25. Found: C, 58.12; H, 5.02; N, 2.87. ESI-MS: m/z calcd for C21H21NO9 431.12. Found: 431.21 [M]+.O), 171.5, 171.4, 169.5 (CO), 150.4, 150.2, 133.3, 122.4, 117.1, 111.6, 99.9 (C-1), 75.1, 74.4, 70.9, 68.7, 62.6, 56.1, 26.4 (CH3 of ArCOCH3), 20.8 (3 × CH3 of COCH3), 14.1; IR: (neat) νmax 3459, 2924, 2853, 2855, 1747 (CO), 1674, 1593, 1416, 1367, 1233, 1080, 1035, 908, 805, 744 cm−1; ESI-HRMS: m/z calcd for C21H26O11Na 477.1373. Found: 477.1370 [M + Na]+.O), 135.5, 133.0, 129.9, 127.8, 71.7, 65.2, 64.4, 26.7 {C(CH3)3}, 21.0 (CH3 of COCH3); IR: (neat) νmax 3435, 2963, 1770 (CO), 1428, 1371, 1260, 1136, 1012, 799 cm−1. ESI-HRMS: m/z calcd for C21H28NaO4Si 395.1649. Found: 395.1647 [M + Na]+.Acknowledgements
S. K. G. sincerely thanks NIPER, S. A. S. Nagar and the Ministry of Chemicals and Fertilizers, Govt. of India for permission to pursue PhD under its staff development scheme and R. G. sincerely thanks NIPER, S. A. S. Nagar for the Senior Research Fellowship.Notes and references
- S. K. Giri, N. Yadav and K. P. R. Kartha, Trends Carbohydr. Res., 2015, 7, 6–27 CAS.
- (a) E. Vedejs and S. T. Diver, J. Am. Chem. Soc., 1993, 115, 3358–3359 CrossRef CAS; (b) E. Vedejs, N. S. Bennett, L. M. Conn, S. T. Diver, M. Gingras, S. Lin, P. A. Oliver and M. J. Peterson, J. Org. Chem., 1993, 58, 7286–7288 CrossRef CAS.
- J. Iqbal and R. R. Srivastava, J. Org. Chem., 1992, 57, 2001–2007 CrossRef CAS.
- S. Chandrasekhar, T. Ramachander and M. Takhi, Tetrahedron Lett., 1998, 39, 3263–3266 CrossRef CAS.
- P. A. Procopiou, S. P. D. Baugh, S. S. Flack and G. G. A. Inglis, J. Org. Chem., 1998, 63, 2342–2347 CrossRef CAS.
- P. Ilankumaran and J. G. Verkade, J. Org. Chem., 1999, 64, 9063–9066 CrossRef CAS.
- A. Orita, K. Sakamoto, Y. Hamada, A. Mitsutome and J. Otera, Tetrahedron, 1999, 55, 2899–2910 CrossRef CAS.
- D. Lloyd, R. H. McDougall and D. R. Marshall, J. Chem. Soc., 1965, 3785–3792 RSC.
- (a) D. Lloyd and D. R. Marshall, J. Chem. Soc., 1956, 2597–2600 RSC; (b) D. Lloyd and D. R. Marshall, J. Chem. Soc., 1958, 118–120 RSC; (c) A. M. Gorringe, D. Lloyd and D. R. Marshall, J. Chem. Soc., 1969, 1081–1084 CAS.
- Compound 2 is a colourless crystalline solid with neutral pH and can be easily prepared in the laboratory on multi-gram quantities and for which the starting materials required are cheap and commercially available. Moreover, no major toxicities have been reported for 2 in the literature in spite of the fact that many structurally diverse diazepines have been known to be variously pharmacologically active.
- K. P. R. Kartha and R. A. Field, Tetrahedron, 1997, 53, 11753–11766 CrossRef CAS.
- S. K. Giri, M. Verma and K. P. R. Kartha, J. Carbohydr. Chem., 2008, 27, 464–478 CrossRef CAS.
- S. K. Giri and K. P. R. Kartha, Synth. Commun., 2010, 40, 3378–3383 CrossRef CAS.
- Other metal perchlorates investigated for acetylation of alkyl/aryl alcohols of various descriptions are: (a) [Mg(ClO4)·2.6H2O]: G. Bartoli, M. Bosco, R. Dalpozzo, E. Marcantoni, M. Massaccesi, S. Rinaldi and L. Sambri, Synlett, 2003, 39–42 CAS; (b) [Zn(ClO4)·2.6H2O]: G. Bartoli, M. Bosco, R. Dalpozzo, E. Marcantoni, M. Massaccesi and L. Sambri, Eur. J. Org. Chem., 2003, 4611–4617 CrossRef CASShivani, R. Gulhane and A. K. Chakraborti, J. Mol. Catal. A: Chem., 2007, 264, 208–213 CrossRef CAS.
- (a) Y. Nakae, I. Kusaki and T. Sato, Synlett, 2001, 1584–1586 CrossRef CAS; (b) K.-C. Lu, S.-Y. Hsieh, L. N. Patkar, C.-T. Chen and C.-C. Lin, Tetrahedron, 2004, 60, 8967–8973 CrossRef CAS; (c) L.-C. Huang, P.-H. Liang, C.-Y. Liu and C.-C. Lin, J. Carbohydr. Chem., 2006, 25, 303–313 CrossRef.
- T. Steiner, Crystallogr. Rev., 2003, 9, 177–228 CrossRef CAS.
- T. Steiner and G. R. Desiraju, Chem. Commun., 1998, 891–892 RSC.
- S. K. Giri and K. P. R. Kartha, RSC Adv., 2015, 5, 11687–11696 RSC.
- R. Dumeunier and I. E. Marko, Tetrahedron Lett., 2004, 45, 825–829 CrossRef CAS.
- G.-T. Ong, K.-Y. Chang, S.-H. Wu and K.-T. Wang, Carbohydr. Res., 1994, 265, 311–318 CrossRef CAS PubMed.
- F. Brisse, R. H. Marchessault, S. Perez and P. Zugenmaier, J. Am. Chem. Soc., 1982, 104, 7470–7476 CrossRef CAS.
- P. Tiwari and A. K. Misra, Carbohydr. Res., 2006, 341, 339–350 CrossRef CAS PubMed.
- B. K. Gadakh, P. R. Patil, S. Malik and K. P. R. Kartha, Synth. Commun., 2009, 39, 2430–2438 CrossRef CAS.
- L. Bomaghi, G. Dekany, N. B. Drinnan, J. Papageorgiou and M. L. West, PCT Int. Appl. WO 2001051499 A1, 2001.
- B. Mukhopadhyay, D. A. Russell and R. A. Field, Carbohydr. Res., 2005, 340, 1075–1080 CrossRef CAS PubMed.
- A. Orita, C. Tanahashi, A. Kakuda and J. Otera, J. Org. Chem., 2001, 66, 8926–8934 CrossRef CAS PubMed.
- C. D. Hurd and R. W. Liggett, J. Am. Chem. Soc., 1941, 63, 2659–2662 CrossRef CAS.
- B. Kashyap and P. Phukan, RSC Adv., 2013, 3, 15327–15336 RSC.
- (a) A. K. Chakraborti, L. Sharma, R. Gulhane and Shivani, Tetrahedron, 2003, 59, 7661–7668 CrossRef CAS; (b) A. A. Ibrahim and E. A. Abdalrazaq, Am. J. Appl. Sci., 2009, 6, 1385–1389 CrossRef CAS.
- U. Mandi, A. S. Roy, B. Banerjee and S. M. Islam, RSC Adv., 2014, 4, 42670–42681 RSC.
- S. Mark, E. Christiaan, H. Ahmed, D. Wim, S. Celest and P. Andre, Proceedings of SPIE-The International Society for Optical Engineering, 2003, vol. 5216, pp.71–82 Search PubMed.
- S. Yaragorla, G. Singh, P. L. Saini and M. K. Reddy, Tetrahedron Lett., 2014, 55, 4657–4660 CrossRef CAS.
- M. Adinolfi, G. Barone, A. Iadonisi and M. Schiattarella, Tetrahedron Lett., 2003, 44, 4661–4663 CrossRef CAS.
- P.-H. Liang, Y.-J. Lu and T.-H. Tang, Tetrahedron Lett., 2010, 51, 6928–6931 CrossRef CAS.
- R. Dalpozzo, A. D. Nino, L. Maiuolo, A. Procopio, M. Nardi, G. Bartoli and R. Romeo, Tetrahedron Lett., 2003, 44, 5621–5624 CrossRef CAS.
- J.-C. Lee, C.-A. Tai and S.-C. Hung, Tetrahedron Lett., 2002, 43, 851–855 CrossRef CAS.
- S. K. Das, K. A. Reddy, V. L. N. R. Krovvidi and K. Mukkanti, Carbohydr. Res., 2005, 340, 1387–1392 CrossRef CAS PubMed.
- I. Lopez, J. L. Bravo, M. Caraballo, J. L. Barneto and G. Silvero, Tetrahedron Lett., 2011, 52, 3339–3341 CrossRef CAS.
- D. Chatterjee, A. Paul, Rajkamal and S. Yadav, RSC Adv., 2015, 5, 29669–29674 Search PubMed.
- P. M. Bhaskar and D. Loganathan, Tetrahedron Lett., 1998, 39, 2215–2218 CrossRef CAS.
- P. Tiwari, R. Kumar, P. R. Maulik and A. K. Misra, Eur. J. Org. Chem., 2005, 4265–4270 CrossRef CAS.
- (a) R. Behrend and P. Roth, Justus Liebigs Ann. Chem., 1904, 231, 369 Search PubMed; (b) C. S. Hudson and J. K. Dale, J. Am. Chem. Soc., 1915, 37, 1264–1270 CrossRef CAS.
- P. Tiwari and A. K. Misra, Carbohydr. Res., 2006, 341, 339–350 CrossRef CAS PubMed; V. K. Yadav and K. G. Babu, J. Org. Chem., 2004, 69, 577–580 CrossRef PubMed.
- R. Ch, M. Tyagi, P. R. Patil and K. P. R. Kartha, Tetrahedron Lett., 2011, 52, 5841–5846 CrossRef CAS.
- C. L. Hillemann, US Pat., 4 666 506, 1987.
- (a) A. Kamal, M. N. A. Khan, K. S. Reddy, Y. V. V. Srikanth and T. Krishnaji, Tetrahedron Lett., 2007, 48, 3813–3818 CrossRef CAS; (b) M. Yadegari, M. Moghadam, S. Tangestaninejad, V. Mirkhani and I. Mohammadpoor-Baltork, Polyhedron, 2011, 30, 2237–2243 CrossRef CAS.
- Q.-H. Fan, S. Striegler, R. G. Langston and J. D. Barnett, Org. Biomol. Chem., 2014, 12, 2792–2800 CAS.
- K. V. Rao, P. R. Patil, S. Atmakuri and K. P. R. Kartha, Carbohydr. Res., 2010, 345, 2709–2713 CrossRef CAS PubMed.
- D. Delay and F. Delmotte, Carbohydr. Res., 1990, 198, 223–224 CrossRef CAS.
- M. Adinolfi, G. Barone, A. Iadonisi and M. Schiattarella, Tetrahedron Lett., 2003, 44, 3733–3735 CrossRef CAS; A. S. M. Sofian and C. K. Lee, J. Carbohydr. Chem., 2001, 20, 431–440 CrossRef.
- H.-X. Liu, Q.-P. Wu, Y.-N. Shu, X. Chen, X.-D. Xi, T.-J. Du and Q.-S. Zhang, Carbohydr. Res., 2009, 344, 2342–2348 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H/13C NMR spectra of crude products. See DOI: 10.1039/c6ra28882k |
| This journal is © The Royal Society of Chemistry 2017 |