
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the catalytic activity of Lewis-acidic uranyl complexes in the nucleophilic acyl substitution of acid anhydrides†
Koichiro Takao * and
Shin Akashi
* and
Shin Akashi
Laboratory for Advanced Nuclear Energy, Institute of Innovative Research, Tokyo Institute of Technology, 2-12-1 N1-32, O-okayama, Meguro-ku, 152-8550 Tokyo, Japan. E-mail: ktakao@lane.iir.titech.ac.jp
First published on 21st February 2017
Abstract
The catalytic activities of several uranyl complexes, such as N,N′-disalicylidene-o-phenelyenediaminato(ethanol)dioxouranium(VI) (UO2(salophen)EtOH), bis(dibenzoylmethanato)(ethanol)dioxouranium(VI) (UO2(dbm)2EtOH), pentakis(N,N-dimethylformamide)dioxouranium(VI) ([UO2(DMF)5]2+), and tetrakis(triphenylphosphine oxide)dioxouranium(VI) ([UO2(OPPh3)4]2+), were examined in the nucleophilic acyl substitution of acid anhydrides. Among them, [UO2(OPPh3)4]2+ was the most efficient to give ethyl acetate and acetic acid from acetic anhydride (Ac2O) and ethanol, and was resistant towards decomposition during the catalytic reaction. Several nucleophiles were also subjected to the catalytic acylation reaction using acetic and pivalic anhydride. Kinetic and spectroscopic studies suggested that [UO2(OPPh3)4]2+ interacts with Ac2O to form [UO2(Ac2O)(OPPh3)3]2+. Interaction of this actual catalyst with additional Ac2O determines the rate of the overall nucleophilic acyl substitution reaction.
Introduction
Although uranium is undoubtedly the most important element in nuclear engineering, the less abundant 235U isotope is primarily employed in the practical utilization of atomic power. Accordingly, this fissile isotope has to be enriched in the nuclear fuel process, particularly in light water reactors. As a result, a huge amount of depleted uranium is generated and stored.1Recently, there have been numerous studies towards the use of uranium compounds as catalysts in organic syntheses to develop a sophisticated use for depleted uranium that has already been refined.2 In the former reports, three main methods appear to be present for organic syntheses: (i) catalysis using organouranium complexes,3–8 (ii) the activation of small molecules by low valent uranium complexes,8 and (iii) the activation of axial (namely, “-yl”) oxygen atoms in the [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO]n+ moiety (n = 1, 2);9–14 thus far, the first research direction has succeeded in the coupling reactions of alkynes and the polymerization of lactones, but the latter two do not attain actual catalytic systems and remain as stoichiometric reactions. Furthermore, these reaction systems always require dry anaerobic conditions because of the low stability of these (pre)catalysts towards oxygen and moisture.
UO]n+ moiety (n = 1, 2);9–14 thus far, the first research direction has succeeded in the coupling reactions of alkynes and the polymerization of lactones, but the latter two do not attain actual catalytic systems and remain as stoichiometric reactions. Furthermore, these reaction systems always require dry anaerobic conditions because of the low stability of these (pre)catalysts towards oxygen and moisture.
Using the Lewis acidity of uranium should be another alternative for exploring its catalytic activity. Particularly, the uranyl ion ([OUO]2+, UO22+) is highly Lewis acidic and exhibits strong hardness according to Pearson's HSAB principle.15 Therefore, any undesired side reactions arising from the organometallic behaviour of the uranium centre can be avoided, although some organouranyl compounds have been known to be formed in exceptional circumstances.16–19 As a matter of fact, several examples of uranyl-catalysed reactions including the alcoholysis of esters,20 Michael additions,21–26 Diels–Alder reactions27 and polymerizations28–30 have been reported to date. Furthermore, its robust axial structure only allows a planar ligand arrangement in the equatorial coordination plane. Such a ligand set may provide some regio- or chemoselectivity to the reaction of interest.
Although the Lewis basicity of “-yl” oxygen atoms is not very high, they can interact with a strong Lewis acid such as B(C6F5)3.31,32 Previously, Chen and co-workers studied the catalytic activity of the oxo ions of V(IV), Mo(VI), W(VI) and Cr(VI) in the nucleophilic acyl substitution of acid anhydrides.33,34 Among them, a high efficiency was recorded when using VO(OTf)2 and MoO2Cl2. In their catalytic systems, these oxo ions are believed to be amphoteric and can efficiently activate acid anhydrides. This information strongly motivated us to examine the catalytic activity of the uranyl ion, although the Lewis basicity of its “-yl” oxygen atoms is rather weak.
In this study, several uranyl complexes were tested as Lewis acid catalysts in the nucleophilic acyl substitution to clarify their requirements as a catalyst. Several nucleophiles were also subjected to the catalytic acylation reaction using acetic and pivalic anhydride. Furthermore, kinetic and spectroscopic studies were also performed to understand the catalytic mechanism in detail.
Results and discussion
Catalyst screening
At least one of the equatorial coordination sites of a uranyl complex must be offered for a substrate to be activated. Therefore, one or more leaving ligand(s) should be present in the uranyl catalyst. In this study, N,N′-disalicylidene-o-phenelyenediaminato(ethanol)dioxouranium(VI) (UO2(salophen)EtOH, 1), bis(dibenzoylmethanato)(ethanol)dioxouranium(VI) (UO2(dbm)2EtOH, 2), pentakis(N,N-dimethyl-formamide)dioxouranium(VI) ([UO2(DMF)5]2+, 3), and tetrakis(triphenylphosphine oxide)dioxouranium(VI) ([UO2(OPPh3)4]2+, 4) were employed as the catalyst candidates. Fig. 1 shows the schematic structures of these uranyl complexes. Acetic anhydride (Ac2O) and ethanol (EtOH) were selected as substrates to test these complexes in the acyl substitution reaction (eqn (1)).‡ The results obtained from the catalyst screening are summarized in Table 1.
 | (1) |
 | ||
| Fig. 1 A schematic structure of the uranyl(VI) complexes tested in this study (EtOH = ethanol). | ||
| Catalyst (mol%) | Solvent | T/°C | Time/h | Yield/% |
|---|---|---|---|---|
| a Reaction conditions: 0.50 M Ac2O, 0.50 M EtOH and the uranyl complex were loaded in CD2Cl2. | ||||
| 1 (0.5) | CDCl3 | 50 | 7 | 90 |
| 2 (0.5) | CDCl3 | 50 | 4 | 90 |
| 3 (2.3) | CD3CN | 22 | 6 | 87 |
| 4 (1.3) | CD2Cl2 | 22 | 2 | 97 |
First, complexes 1 and 2 were examined in CDCl3. Previously, our group reported that 1 and its related complexes tend to release a monodentate ligand, such as EtOH, to form a dimeric complex, [UO2(salophen)]2, in chlorinated, non-coordinating solvents such as CHCl3 and CH2Cl2.35 Therefore, we expected that a substrate will be efficiently activated through this substitution reaction with the initially coordinated EtOH molecule. Nevertheless, no progress of eqn (1) was observed at 22 °C in both systems. When the sample solutions were warmed to 50 °C, the desired reactions started, as shown in Fig. S1 (ESI†). To reach an yield >90%, it took 7 h and 4 h at 50 °C when using 1 and 2, respectively. The obtained products were EtOAc and AcOH only, demonstrating the high selectivity of these systems. On the other hand, the 1H NMR spectra revealed that both complexes underwent partial or full decomposition. This would be due to AcOH that was generated, as shown in eqn (1). Complex 1 appeared to be more resistant towards the decomposition with acetic acid because of its more stable tetradentate chelate, although new signals arising from several salophen2− species including a free N,N′-disalicylidene-o-phenylenediamine (H2salophen) were also detected, as seen in Fig. S2 (ESI†). In contrast, 2 was completely decomposed within 2 h as free dibenzoylmethane (Hdbm) was observed, as seen in Fig. S3 (ESI†). Through these experiments, it has been evidenced that the Lewis acidity of these uranyl complexes actually catalyses the acyl substitution reaction of Ac2O. However, it is better to shift to other uranyl complexes because the mechanism and kinetics cannot be discussed in detail.
As another alternative for the catalyst in the acyl substitution reaction, complex 3 was investigated. Although the equatorial plane of the uranyl ion of this complex is fully occupied with 5-fold coordination offered by the DMF molecules, the exchange or substitution reaction of these monodentate ligands is expected to occur in high frequency because of the labile character of the uranyl ion.35–37 Therefore, one or both of the substrates in eqn (1) may enter the uranyl coordination sphere. The reaction progress at 22 °C in the presence of 3 (2.3 mol%) in CD3CN is displayed in Fig. S4 (ESI†). On one hand, the substrates shown in eqn (1) are actually converted to the desired products in 87% yield after 6 h. On the other hand, the 1H NMR spectrum in Fig. S5 (ESI†) indicates the occurrence of unknown products at 8.27 ppm (cf. CHO– of DMF at 8.60 ppm). These substances cannot be a result of the reaction between Ac2O and DMF. The peak integrals indicate that the concentrations of these undesired products are rather small when compared with that of the desired ester, whereas the uranyl complex (2.3 mol% in total) suffered from this undesired reaction. This means that several mixed ligand complexes other than 3 were also present in the reaction mixture. These in situ generated uranyl complexes may also exhibit some catalytic activity in the reaction shown in eqn (1). The occurrence of unknown catalysts makes further discussion on the kinetics and mechanism complicated. Therefore, it is necessary to find a monodentate ligand with high stability towards acid anhydrides. Nevertheless, it should be emphasized that the higher catalytic activity of a uranyl complex was successfully attained in use of the solvate complex 3 as demonstrated by the lower reaction temperature.
Triphenylphosphine oxide (OPPh3) is one of strong monodentate ligands for uranyl and could be durable enough to remain unchanged under the reaction conditions shown in eqn (1) because of its small potential to react with acid anhydrides and carboxylic acid. An isolable form of the uranyl–OPPh3 complex is [UO2(OPPh3)4]2+ (4), which is usually obtained as a salt of a poorly coordinating anion such as ClO4− and TfO−.38–40 The catalytic activity of 4 was examined in a similar manner to the abovementioned test systems. Fig. 2 shows the reaction progress in the presence of 4 (1.3 mol%) in CD2Cl2 at 22 °C. As a consequence, the products in eqn (1) were formed in 97% yield within 2 h. It is noteworthy that OPPh3 remains unchanged in this system. In summary, the catalyst screening in Table 1 concluded that 4 is, in hand, the best uranyl complex to catalyse the reaction shown in eqn (1) from the viewpoints of activity and durability. Usually, the most popular coordination number of uranyl complexes in its equatorial plane is 5, as revealed by 1, 2, and 3 (Fig. 1). Even in the complexation of uranyl with OPPh3, the pentacoordinate complex, [UO2(OPPh3)5]2+ (5), tends to be formed below −40 °C (vide infra), where the thermal vibrations of the bulky OPPh3 ligands decrease. When the solution is warmed, complex 5 tends to release OPPh3 to give the 4-fold complex 4 because of the steric hindrance between the neighboring OPPh3 molecules in the coordination sphere. As a result, some Lewis-acidic vacancies would be still left in the uranyl equatorial plane of 4, which can be offered to the activation of a substrate in the current catalytic system.
 | ||
| Fig. 2 The progress and efficiency of the acetylation of ethanol catalysed by [UO2(OPPh3)4]2+ (4, 6.5 mM, 1.3 mol%) in CD2Cl2 at 22 °C. Initial conditions: [Ac2O] = 0.50 M and [EtOH] = 0.50 M. | ||
Acyl substitution reactions catalyzed by complex 4
With the optimal catalyst 4, the catalytic acetylation and pivalation of several nucleophiles, including alcohols and thiophenol, were examined. The results are summarized in Table 2. In most cases except for ternary alcohols, the desired acylation products were obtained in good yield together with the carboxylic acids corresponding to the anhydride used. As a general trend, the acetylation was much faster than the corresponding pivalation, implying that the steric hindrance of an anhydride affects the reaction rate. When compared to entry 1, a much shorter reaction time was required for both the acetylation and pivalation of 2-phenylethyl alcohol (Ph(CH2)2OH, entry 2). This suggests that coordination of less sterically-hindered nucleophiles, such as EtOH, inhibits the catalytic activity of the uranyl ion. As a matter of fact, the reaction rate decreased, when extra EtOH was added to the reaction (Fig. S6, ESI†). Such inhibition could also be the case in entry 3, where the carbonyl and hydroxyl groups of hydroxyacetone may interact with or chelate to the uranyl ion to prevent the desired acylation reaction. Nevertheless, this nucleophile is still amenable to acylation in quantitative yield, although a longer reaction time than Ph(CH2)2OH is required.
|
|||
|---|---|---|---|
| Entry | Nucleophile | Time/h | Yieldb/% |
| a Reaction conditions: 0.50 M anhydride, 0.50 M nucleophile and 6.5 mM (1.3 mol%) complex 4 were loaded in CH2Cl2 or CD2Cl2 at 22 °C, unless stated otherwise. Data in the parentheses correspond to the pivalation reaction.b Determined by 1H NMR peak integrals.c Catalyst: 2.0 mol%. | |||
| 1 | C2H5OH | 2 (24) | 97 (90) |
| 2 | Ph(CH2)2OH | 0.75 (3) | 95 (96) |
| 3c | H3CC(O)CH2OH |
2.5 | >99 |
| 4 | tert-BuOH | 4 | 68 |
| 5 | Ph3COH | 72 | 63 |
| 6 | PhOH | 2 | 95 |
| 7 | PhSH | 6 | 85 |

Entry 4 shows that tert-butyl alcohol is actually reactive in the current system even without any additional treatments such as raising the temperature and the addition of ternary amines. This point is distinguishable from the acylations catalyzed by vanadyl and molybdenyl ions, where this tertiary alcohol was completely inert towards the acylation reaction.33,34 The yield of tert-butyl acetate in entry 4 was 68% after 4 h, while Ac2O already disappeared in the sample solution. This result was reproducible. Except for the expected products and leftover tert-butyl alcohol, the 1H NMR spectrum of this sample (Fig. S7, ESI†) revealed the presence of isobutene [δ/ppm: 1.73 (t, JHH = 1.2 Hz, 6H, H2CC(CH3)2), 4.66 (septet, JHH = 1.2 Hz, 2H, H2CC(CH3)2)], which is the product resulting from the E1 reaction of tert-butyl alcohol. Another product in this side reaction was water, which also consumes the anhydride to give the corresponding carboxylic acid. Trityl alcohol was also tested as another sterically hindered nucleophile (entry 5). As a result, the progress of the acetylation of this bulky nucleophile was actually observed, although the reaction was so sluggish that it took 3 days to reach 63% yield. This is also much different from the inertness of vanadyl and molybdenyl catalysts in the acylation of trityl alcohol.33,34 Such a difference may be related to the size of the metal center because the ionic radius of U6+ (0.73 Å at 6-coordinated) is much greater than those of V4+ (0.53 Å at 5-coordinated) and Mo6+ (0.59 Å at 6-coordinated).41
Phenol and thiophenol (entries 6 and 7) provide a good contrast of the difference in nucleophilic moieties. The nucleophilicity of PhS− is widely known to be much greater than that of PhO−. Nevertheless, phenol as a nucleophile exhibited a higher efficiency than thiophenol in terms of the reaction rate and yield. This difference appears to be related to the hardness of the nucleophilic atoms, although the details are still unclear.
Reaction kinetics and catalysis mechanism
Mechanistic insights into the nucleophilic acyl substitution reaction catalyzed by 4 were obtained by investigating how the concentrations of the substrates affected the reaction rate. Herein, the acetylation of Ph(CH2)2OH (entry 2, Table 2) was employed as a model reaction. Fig. 3a shows the progress of this reaction in CD2Cl2 at 22 °C under different initial concentrations of Ac2O ([Ac2O]ini). Upon increasing [Ac2O]ini, the reaction rate clearly increased. The initial rate (vini) estimated from Fig. 3a was plotted against [Ac2O]ini as shown in Fig. S8 (ESI†). As a result, vini versus [Ac2O]ini2 shows a linear relationship with a slope equal to 1.04 × 10−3 M−1 s−1. We also examined the dependency of vini on the initial concentration of the nucleophile ([Ph(CH2)2OH]ini). However, the progress of the reaction was almost the same regardless of the different concentrations of [Ph(CH2)2OH]ini, as shown in Fig. S9 (ESI†) at least under the tested conditions ([Ph(CH2)2OH]ini = 0.25–1.00 M). This means that Ph(CH2)2OH does not participate in the rate-determining step of this catalytic acyl substitution reaction.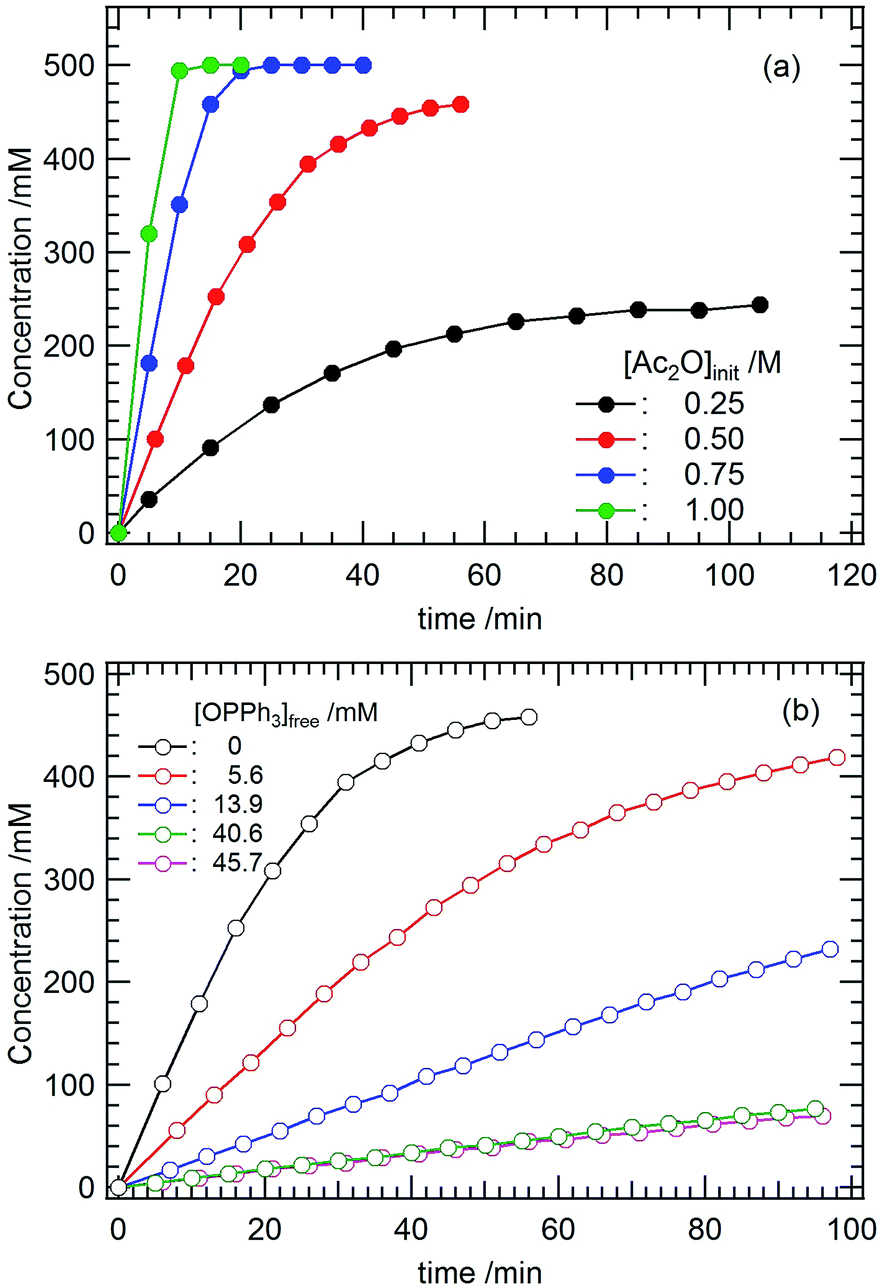 | ||
| Fig. 3 The progress of the acetylation of Ph(CH2)2OH catalysed by [UO2(OPPh3)4]2+ (4) in CD2Cl2 at 22 °C. (a) [Ac2O] dependency; initial condition: 0.50 M Ph(CH2)2OH, 0.25–1.00 M Ac2O and 6.5 mM 4, and (b) [OPPh3] dependency; initial condition: 0.50 M Ph(CH2)2OH, 0.50 M Ac2O, 6.5 mM 4 and 0–45.7 mM OPPh3. | ||
When Ac2O interacts with 4, its OPPh3 ligand(s) may dissociate from UO22+. Therefore, we also studied the dependency of vini on the concentration of free OPPh3 ([OPPh3]free). The results are displayed in Fig. 3b. With increasing [OPPh3]free, the reaction rate decreases. In Fig. S10 (ESI†), vini is proportional to [OPPh3]free−1 with a slope equal to 6.54 × 10−7 M2 s−1.
In conclusion, the following is the rate equation for the acetylation of Ph(CH2)2OH catalyzed by 4.
| vini = k[Ac2O]2[OPPh3]−1[4] | (2) |
The rate constant (k) of eqn (2) was estimated to be 4.02 × 10−4 M−1 s−1 at 22 °C.
Further mechanistic information should be included in eqn (2) because multi-body collision is unlikely to occur. Thus, there should be an equilibrium preceding the rate-determining step. When Ac2O was added to the CD2Cl2 solution containing 4, the yellow color arising from the uranyl compound immediately diminished, implying ligand substitution between the coordinated OPPh3 and Ac2O. In contrast, any interaction of Ph(CH2)2OH with 4 was unlikely to occur despite its nucleophilicity because vini is independent of [Ph(CH2)2OH]ini, as shown in Fig. S9 (ESI†). This could be ascribed to steric hindrance arising from the phenyl groups. The complexation reaction between 4 and Ac2O was studied in detail using a UV-vis absorption spectrophotometric titration study. The obtained spectral series recorded at different [Ac2O] is shown in Fig. 4. Upon increasing [Ac2O], a decrease in the intensity of the finely structured absorption bands arising from ligand-to-metal charge transfer (LMCT) in the UO22+ moiety was observed together with a slight red shift in the peak maxima. This observation indicates that OPPh3 in 4 was substituted by Ac2O as shown in eqn (3).
| [UO2(OPPh3)4]2+ (4) + Ac2O = [UO2(Ac2O)(OPPh3)4−n]2+ + nOPPh3 | (3) |
 | ||
| Fig. 4 UV-vis absorption spectra of a CH2Cl2 solution of 4(ClO4)2 (10 mM) at 22 °C. [Ac2O] varied from 0 to 0.508 M. | ||
Although no isosbestic points were observed in Fig. 4, principal component analysis (PCA)42,43 has suggested that a total of 2 components are involved in this spectral series. This result corroborates the occurrence of eqn (3) in the current system. In addition, the Job plot shown in Fig. S11 (ESI†) also suggests a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 interaction between 4 and Ac2O in eqn (3).
:1 interaction between 4 and Ac2O in eqn (3).
Although additional coordination to the fifth equatorial site of 4 is possible in a cooled solution (below −40 °C) as we have demonstrated the occurrence of [UO2(OPPh3)5]2+ (5) in the former,40 there is not enough room for another ligand in the equatorial plane of 4 at 22 °C because of the thermal motion of the coordinated OPPh3 molecules.
| [UO2(OPPh3)4]2+ (4) + OPPh3 = [UO2(OPPh3)5]2+ (5) | (4) |
Therefore, the exclusion of OPPh3 from the first coordination sphere of UO22+ should be convincing. For the equilibrium analysis of the data in Fig. 4, it is necessary to know the number of released OPPh3, n, in eqn (3). Fig. 5 displays the 31P{1H} NMR spectrum of the CD2Cl2 solution containing 4 (14.7 mM) and Ac2O (0.40 M) at −70 °C. The sample had to be cooled to suppress the frequency of the chemical exchange of the ligands bound to UO22+. In accordance with our former findings,40 the 31P signals at 53.1 ppm and 41.4 ppm were attributed to 4 and 5, respectively. The new signals at 47.6 and 46.6 ppm appear upon the addition of Ac2O. Upon increasing [Ac2O], the peak integrals of these signals increase, while the ratio between them remains constant at 2:1. Therefore, these signals should arise from the OPPh3 ligands in the different environments of a single uranyl complex other than 4 and 5. The most plausible species occurring in this test solution is [UO2(Ac2O)(OPPh3)3]2+ (6) as shown in Scheme 1, where two of the OPPh3 are chemically equivalent (red) and there is also another OPPh3 in a different environment (blue). The coordination mode of Ac2O may be bidentate in a similar manner to a β-diketonato ligand.
 | ||
| Fig. 5 31P{1H} NMR spectrum of CD2Cl2 solution dissolving 4 (14.7 mM) and Ac2O (0.40 M) at −70 °C. | ||
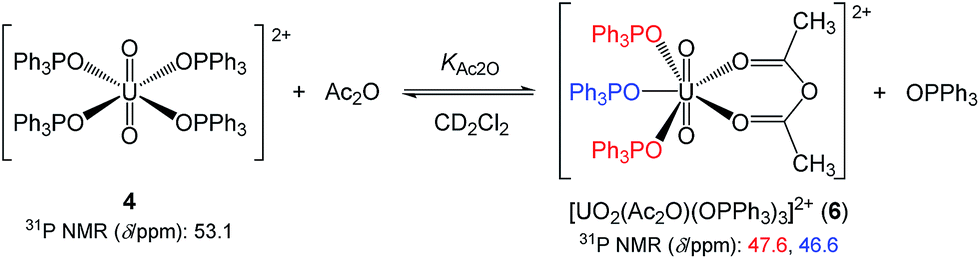 | ||
| Scheme 1 The ligand substitution equilibrium between 4 and [UO2(Ac2O)(OPPh3)3]2+ (6) in CD2Cl2 at −70 °C. | ||
Note that no signals were observed at 29 ppm, where the 31P signal of free OPPh3 appeared. This implies that the concentration of free OPPh3 was negligible. It is necessary to consider that OPPh3 in the product part of Scheme 1 is also involved in eqn (4). In accordance with our previous report,40 the logarithmic equilibrium constant of eqn (4) (logK45) at −70 °C is 3.33, which reveals that [OPPh3]free is as small as 7 × 10−6 M under the current conditions. This is the reason why the free OPPh3 was undetectable in Fig. 5.
The reaction in Scheme 1 was taken into account to analyze the UV-vis absorption spectral series shown in Fig. 4. Note that this treatment assumes that the equilibrium between 4 and 6 is also present in the current system at 22 °C. It was also necessary to consider the following complexation to form 4.
| UO22+ + 4OPPh3 = [UO2(OPPh3)4]2+ (4) | (5) |
A sufficiently large gross stability constant of 4 (β4 = [4]/[UO22+][OPPh3]free4, e.g., logβ4 = 20) was assumed to simulate the predominant formation of 4 and practically negligible concentrations of free UO22+. Formation of 5 can be ruled out at 22 °C because logK45 is as low as −1.31. Analysis using HypSpec software44 resulted in the logarithmic equilibrium constant of Scheme 1 (logKAc2O) equal to −0.513 ± 0.013 at 22 °C. The characteristic LMCT bands arising from the UO22+ moiety were assigned to both 4 and 6 as shown in Fig. S12 (ESI†), implying that the equilibrium analysis was successfully completed. It was also confirmed that the value of KAc2O was independent of the assumption of β4 unless the occurrence of free UO22+ was significant in the simulation (where logβ4 < 12).
Assuming that complex 6 is an actual catalyst in the acetylation of Ph(CH2)2OH, eqn (2) can be modified using KAc2O = [6][OPPh3]free/[4][Ac2O] as follows:
| vini = (k/KAc2O)[Ac2O][6] | (6) |
The rate constant k/KAc2O is equal to 1.31 × 10−3 M−1 s−1 at 22 °C. This rate equation indicates that the rate-determining step was the interaction between 6 and Ac2O. Thus, [UO2(Ac2O)2(OPPh3)3]2+ (7) will be formed as an intermediate to activate additional Ac2O, followed by nucleophilic attack of Ph(CH2)2OH to the electrophilic carbon of the carbonyl moiety in the activated Ac2O. Taking into account the following points, (i) the stoichiometry of 7, (ii) the absence of leaving OPPh3 supported by eqn (6) and (iii) the potential bidentate coordination manner of Ac2O, UO22+ has to offer 7 coordination sites in its equatorial plane, being too crowded. Therefore, one or both of the coordinated Ac2O molecules should be monodentate. In the latter case, the coordination number in the equatorial plane is 5, which is most commonly found in uranyl complexes. Although 6-fold uranyl complexes are also often observed, such a structure is allowed only by the chelating ligands bearing narrow bite angles such as CO32− and NO3−.45,46 In summary, the proposed catalytic cycle of the nucleophilic acyl substitution is shown in Scheme 2. This catalysis mechanism is plausible, but still somewhat hypothetical. Our trials to crystallize 6 and uranyl complexes related to intermediate 7 have not been successful to date.
 | ||
| Scheme 2 The proposed cycle for the nucleophilic acyl substitution reaction of Ac2O catalyzed by 4 (R = Ph(CH2)2–). The charges on the uranyl complexes are omitted for clarity. | ||
Lewis-acid catalysis is widely employed in various organic syntheses. A typical example is the series of triflate salts of several d-block metals and trivalent rare earths used in aldol reactions, where the Lewis-acid catalysts are much easier to handle than those traditionally employed, such as AlCl3 and BF3·Et2O, because of their resistance towards hydrolysis.47 The uranyl species also does not show a strong tendency towards hydrolysis and therefore, can be present stably in aqueous or aqueous/organic biphasic reaction systems. Furthermore, the dioxo structure of uranyl may result in the equatorial coordination shown in this study, which is much distinguishable from the known spherical metal ions as water-compatible Lewis-acid catalysts. We intend to expand the coordination chemistry of uranyl to explore its catalytic functions.
Conclusions
In this study, we performed screening of several uranyl complexes as Lewis-acid catalysts used in the nucleophilic acyl substitution reaction. As a result, all the uranyl species tested were found to more or less promote this reaction to afford an ester and carboxylic acid. The most active uranyl complex in hand was [UO2(OPPh3)4]2+ (4), which gives ethyl acetate in 97% yield within 2 h at 22 °C in CD2Cl2. Several nucleophiles were used in the acetylation and pivalation reactions using the corresponding acid anhydrides in the presence of 4. In all the entries reported herein, the progress of these reactions was successfully enhanced by 4. Even when using ternary alcohols such as tert-BuOH and trityl alcohol, 4 still shows its activity, although progress of the reaction in both systems is somewhat slow. According to the kinetic and mechanistic studies, 4 is in an equilibrium with [UO2(Ac2O)(OPPh3)3]2+ (6). Therefore, the former is a pre-catalyst and the latter is an actual catalyst in the current nucleophilic acyl substitution reaction.Experimental
Materials
The uranyl complexes UO2(salophen)EtOH (1),48 UO2(dbm)2EtOH (2),49 [UO2(DMF)5](ClO4)2 (3),50 and [UO2(OPPh3)4](ClO4)2 (4)40 (Fig. 1) were prepared by the methods described elsewhere. All the chemicals used in this study were of reagent grade and used as received.Kinetic and mechanistic studies using NMR spectroscopy
1H and 31P{1H} NMR spectra of the sample solutions were recorded on a JEOL ECX-400 NMR spectrometer (1H: 399.78 MHz, 31P: 161.83 MHz). Tetramethylsilane (TMS) and 85% H3PO4 were employed as reference materials for the 1H and 31P chemical shifts, respectively. A typical procedure used to study the reaction kinetics of the catalytic acyl substitution reaction is described below. The uranyl catalyst was loaded in an NMR sample tube (5 mm O.D.) together with CD2Cl2 and acid anhydride. After optimization of the NMR shim, a nucleophile was injected to the NMR tube. The sample solution was vigorously shaken, followed by starting the kinetic experiment. The 1H NMR spectrum was recorded every 5–30 min. The concentrations of the reactants and products were estimated from the peak integrals of the species occurring in the reaction mixture.UV-vis titration experiments
A CH2Cl2 solution containing complex 4 (10 mM) was prepared. This solution was titrated with neat Ac2O. At each titration step, the UV-vis absorption spectrum of the sample solution was recorded on a Agilent 8453 photodiode array spectrophotometer. The obtained spectral series was analyzed using HypSpec software.44Acknowledgements
We thank Prof. Emeritus Yasuhisa Ikeda and Prof. Emeritus Hirotake Moriyama for their stimulated discussions. This study was partially supported through the Assistant Staffing Program by the Gender Equality Center, Tokyo Institute of Technology.Notes and references
- M. Benedict, T. H. Pigford and H. W. Levi, Nuclear Chemical Engineering, McGraw-Hill, United States, 2nd edn, 1981 Search PubMed.
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341–349 CrossRef CAS PubMed.
- E. Barnea and M. Eisen, Coord. Chem. Rev., 2006, 250, 855–899 CrossRef CAS.
- E. Barnea, T. Andrea, M. Kapon and M. S. Eisen, J. Am. Chem. Soc., 2004, 126, 5066–5067 CrossRef CAS PubMed.
- E. Barnea, T. Andrea, M. Kapon, J. C. Berthet, M. Ephritikhine and M. S. Eisen, J. Am. Chem. Soc., 2004, 126, 10860–10861 CrossRef CAS PubMed.
- E. Barnea, D. Moradove, J.-C. Berthet, M. Ephritikhine and M. S. Eisen, Organometallics, 2006, 25, 320–322 CrossRef CAS.
- W. Ren, N. Zhao, L. Chen and G. Zi, Inorg. Chem. Commun., 2013, 30, 26–28 CrossRef CAS.
- H. S. La Pierre and K. Meyer, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Hoboken, New Jersey, 2014, vol. 58, pp. 303–415 Search PubMed.
- S. Fortier and T. W. Hayton, Coord. Chem. Rev., 2010, 254, 197–214 CrossRef CAS.
- P. L. Arnold, A. F. Pecharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, F. J. White, N. Magnani, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed. Engl., 2011, 50, 887–890 CrossRef CAS PubMed.
- J. B. Love, Chem. Commun., 2009, 3154–3165, 10.1039/b904189c.
- P. L. Arnold, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2004, 43, 8206–8208 CrossRef CAS PubMed.
- P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Nature, 2008, 451, 315–317 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- J. Maynadie, J. C. Berthet, P. Thuery and M. Ephritikhine, Chem. Commun., 2007, 486–488, 10.1039/b617700j.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS PubMed.
- J. C. Tourneux, J. C. Berthet, T. Cantat, P. Thuery, N. Mezailles and M. Ephritikhine, J. Am. Chem. Soc., 2011, 133, 6162–6165 CrossRef CAS PubMed.
- V. van Axel Castelli, R. Cacciapaglia, G. Chiosis, F. C. J. M. van Veggel, L. Mandolini and D. N. Reinhoudt, Inorg. Chim. Acta, 1996, 246, 181–193 CrossRef CAS.
- V. van Axel Castelli, A. D. Cort, L. Mandolini and D. N. Reinhoudt, J. Am. Chem. Soc., 1998, 120, 12688–12689 CrossRef CAS.
- V. van Axel Castelli, F. Bernardi, A. Dalla Cort, L. Mandolini, I. Rossi and L. Schiaffino, J. Org. Chem., 1999, 64, 8122–8126 CrossRef CAS PubMed.
- V. van Axel Castelli, A. Dalla Cort, L. Mandolini, D. N. Reinhoudt and L. Schiaffino, Chem.–Eur. J., 2000, 6, 1193–1198 CAS.
- V. van Axel Castelli, A. Dalla Cort, L. Mandolini, D. N. Reinhoudt and L. Schiaffino, Eur. J. Org. Chem., 2003, 2003, 627–633 CrossRef.
- V. van Axel Castelli, A. Dalla Cort, L. Mandolini, V. Pinto and L. Schiaffino, J. Org. Chem., 2007, 72, 5383–5386 CrossRef CAS PubMed.
- A. Dalla Cort, L. Mandolini and L. Schiaffino, J. Org. Chem., 2008, 73, 9439–9442 CrossRef CAS PubMed.
- A. Dalla Cort, L. Mandolini and L. Schiaffino, Chem. Commun., 2005, 3867–3869, 10.1039/b504713g.
- R. J. Baker and A. Walshe, Chem. Commun., 2012, 48, 985–987 RSC.
- A. Walshe, J. Fang, L. Maron and R. J. Baker, Inorg. Chem., 2013, 52, 9077–9086 CrossRef CAS PubMed.
- J. Fang, A. Walshe, L. Maron and R. J. Baker, Inorg. Chem., 2012, 51, 9132–9140 CrossRef CAS PubMed.
- M. J. Sarsfield and M. Helliwell, J. Am. Chem. Soc., 2004, 126, 1036–1037 CrossRef CAS PubMed.
- T. W. Hayton and G. Wu, Inorg. Chem., 2009, 48, 3065–3072 CrossRef CAS PubMed.
- C.-T. Chen, J.-H. Kuo, C.-H. Li, N. B. Barhate, S.-W. Hon, T.-W. Li, S.-D. Chao, C.-C. Liu, Y.-C. Li, I. H. Chang, J.-S. Lin, C.-J. Liu and Y. C. Chou, Org. Lett., 2001, 3, 3729–3732 CrossRef CAS PubMed.
- C. T. Chen, J. H. Kuo, V. D. Pawar, Y. S. Munot, S. S. Weng, C. H. Ku and C. Y. Liu, J. Org. Chem., 2005, 70, 1188–1197 CrossRef CAS PubMed.
- K. Takao and Y. Ikeda, Inorg. Chem., 2007, 46, 1550–1562 CrossRef CAS PubMed.
- Y. Ikeda, H. Tomiyasu and H. Fukutomi, Inorg. Chem., 1984, 23, 1356–1360 CrossRef CAS.
- Y. Ikeda, H. Tomiyasu and H. Fukutomi, Inorg. Chem., 1984, 23, 3197–3202 CrossRef CAS.
- J. C. Berthet, M. Nierlich and M. Ephritikhine, Angew. Chem., Int. Ed., 2003, 42, 1952–1954 CrossRef CAS PubMed.
- G. H. John, I. May, M. J. Sarsfield, H. M. Steele, D. Collison, M. Helliwell and J. D. McKinney, Dalton Trans., 2004, 734–740, 10.1039/b313045b.
- K. Takao, T. Takahashi and Y. Ikeda, Inorg. Chem., 2009, 48, 1744–1752 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- A. Rossberg, T. Reich and G. Bernhard, Anal. Bioanal. Chem., 2003, 376, 631–638 CrossRef CAS PubMed.
- K. Takao, S. Takao, Y. Ikeda, G. Bernhard and C. Hennig, Dalton Trans., 2013, 42, 13101–13111 RSC.
- A. Sabatini, A. Vacca and P. Gans, Coord. Chem. Rev., 1992, 120, 389–405 CrossRef CAS.
- D. L. Clark, D. E. Hobart and M. P. Neu, Chem. Rev., 1995, 95, 25–48 CrossRef CAS.
- K. Takao, K. Noda, Y. Morita, K. Nishimura and Y. Ikeda, Cryst. Growth Des., 2008, 8, 2364–2376 CAS.
- S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209–217 CrossRef CAS PubMed.
- K. Takao and Y. Ikeda, Inorg. Chem., 2007, 46, 1550–1562 CrossRef CAS PubMed.
- K. Mizuoka and Y. Ikeda, Radiochim. Acta, 2004, 92, 631–635 CrossRef CAS.
- K. Takao, S. Takao, Y. Ikeda, G. Bernhard and C. Hennig, Dalton Trans., 2013, 42, 13101–13111 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Progress and efficiency of acylation using uranyl(VI) catalysts, 1H NMR spectra supporting the decomposition of the catalysts and side reactions, plots of the initial rate under several conditions, and the molar absorption spectra of the uranyl(VI) species. See DOI: 10.1039/c6ra27796a |
| ‡ Control experiments in the absence of a uranyl complex did not result in any significant progress of the acylation reaction (e.g., 0.8% yield after 2 h in a CH2Cl2 solution of 0.5 M Ph(CH2)2OH + 0.5 M Ac2O). |
| This journal is © The Royal Society of Chemistry 2017 |