
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uptake dynamics of graphene quantum dots into primary human blood cells following in vitro exposure†
Stefan Fasbender a,
Sonja Allania,
Christian Wimmenauera,
Ron-Patrick Cadeddub,
Katharina Rabac,
Johannes C. Fischerc,
Bekir Bulatd,
Martina Luysberge,
Claus A. M. Seideld,
Thomas Heinzel*a and
Rainer Haas*b
a,
Sonja Allania,
Christian Wimmenauera,
Ron-Patrick Cadeddub,
Katharina Rabac,
Johannes C. Fischerc,
Bekir Bulatd,
Martina Luysberge,
Claus A. M. Seideld,
Thomas Heinzel*a and
Rainer Haas*b
aCondensed Matter Physics Laboratory, Heinrich-Heine-University, 40204 Düsseldorf, Germany. E-mail: Thomas.Heinzel@hhu.de
bDepartment of Haematology, Oncology and Clinical Immunology, Heinrich-Heine-University, 40204 Düsseldorf, Germany. E-mail: Haas.med@uni-duesseldorf.de
cInstitute for Transplantation Diagnostics and Cell Therapeutics, Heinrich-Heine-University, 40204 Düsseldorf, Germany
dInstitute of Molecular Physical Chemistry, Heinrich-Heine-University, 40204 Düsseldorf, Germany
eErnst Ruska-Centre, Jülich Research Centre, 52425 Jülich, Germany
First published on 21st February 2017
Abstract
Human leukocytes obtained from samples of leukapheresis products of three healthy donors stimulated by granulocyte colony stimulating factor (G-CSF) were exposed to graphene quantum dots. A time- and concentration dependent uptake was observed with a significantly greater uptake into monocytes and granulocytes in comparison to lymphocytes, suggesting a better incorporation ability of cells with phagocytotic properties. The uptake rates also correlate with the cell membrane area. Looking at the different lymphoid subsets a greater uptake was found into CD19+ B-, CD56+ natural killer cells and CD34+ hematopoietic stem cells (HSC) in comparison to CD4+ T- and CD8+ T cells. Independent of the cell type studied, the observed uptake dynamics is consistent with a diffusion-driven process, which allows the determination of cell-specific membrane permeabilities for the graphene quantum dots. The toxicity of the quantum dots is relatively low resulting in a 90% viability of the entire leukocyte population after 36 hours of exposure to GQDs at a concentration of 500 μg ml−1.
1 Introduction
Semiconductor quantum dots are used in many fields of biology and medicine, like long term imaging of various normal and malignant cells in vivo and in vitro,1,2 cancer diagnostics3,4 and therapeutic tumor cell targeting.5,6 Their application is advantageous because of their long-term photostability, tunable color and high photoluminescence quantum yield. Still, potential toxic side effects are an important issue prompting the search for suitable alternatives which also necessitate comprehensive studies as far as biocompatibility, toxicity and applicability are concerned. Graphene quantum dots (GQDs) are promising candidates sharing the advantages of semiconductor quantum dots without their intrinsic toxicity.7 In addition, their production is relatively easy, reproducible and inexpensive. The spectrum of applications includes photocatalysts, ion detectors, solar cells with improved light-to-energy conversion, drug delivery and biomarkers.8 Due to their solubility in water, additional coatings are not necessary for studies in bio-environments. GQDs enter into the cytoplasm of different human cell lines as well as in human neural stem cells most likely via endocytosis without obvious negative effects on cell proliferation.9–12 Their fluorescence intensity depends on the environmental pH value,13 a relevant parameter related to membrane permeability, cell–cell coupling, metabolism and fertilization.14 In vivo studies have shown that GQDs accumulate in the kidney with a time constant of approximately 3 hours after subcutaneous and intravenous injection in mice, before they are excreted via the urine.15 As far as cancer therapy is concerned, it was interesting to observe that GQDs are able to pass the blood–brain barrier with enhanced uptake in glioma tissue in comparison to normal brain cells.16 GQDs are also believed to enhance the potency and selectivity of cancer drugs like cisplatin and doxorubicin.17,18 While larger sheets of graphene oxide show significant toxicity,19 GQDs are often considered to be less toxic with a dependency on particle size, charge and impurities.20 We are interested in the interaction of GQDs with blood cells and its stem and progenitor cells, because the later ones have a great potential of differentiation and are particularly sensitive to all kind of genotoxic effects. For instance, in the treatment of patients with cytotoxic chemotherapy, the hematopoietic system represents most often the dose-limiting organ. For our in vitro studies, we used the preparation process of Wu et al. due to its excellent reproducibility of the quantum yield and the fluorescence spectra in our laboratory. The obtained GQDs with a diameter of approximately 2 nm are not only excellent fluorescence markers, but are also suitable for drug delivery as well as for functional studies inside cells.13 We also view GQDs as model particles for other kinds of graphitic particles, which are abundantly present in the environment due to e.g. combustion.2 Results and discussion
2.1 Preparation and characterization of the graphene quantum dots
The GQDs were prepared by pyrolysis of citric acid21 under high pressure and in the presence of dicyandiamide to ensure nitrogen adsorbates at their edges.13 The dots were purified by dialysis yielding particles with a minimum mass of 500 Da and a maximum mass of 20 kDa. Some of these GQDs were placed on a silicon dioxide substrate for characterization by scanning force microscopy, showing a height of 0.7 nm to 1.3 nm (see Fig. S1†) which is consistent with one to three layers of graphene.22,23 TEM images of the GQDs (see Fig. S2†) reveal almost circular particles with a diameter around 2 nm (ranging from 1–3 nm). We used X-ray photoelectron spectroscopy (XPS) to determine the chemical composition. The relative strength of the C 1s, N 1s and O 1s peaks indicate that the GQDs are composed of 50% carbon, 30% nitrogen and 20% oxygen atoms (see Fig. S3†). The carbon 1s XPS spectrum is the most relevant (see Fig. 1A). According to a best fit analysis, the C 1s signal around an energy of 287 eV is composed of five significant terms, plus three terms of low weight which will not be discussed here. Approximately 36% of the total signal strength originates from a peak centered at 285.0 eV, corresponding to C–C bonds of sp2 hybridized carbon as it is characteristic for carbon atoms in graphene.24 The peak at 288.2 eV, contributing a weight of 19.4%, stems from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond, which is interpreted as oxidized bulk graphene that generates a local sp3-hybridization. The CO double bond also appears in combination with nitrogen with a similar weight of 16.5% at an energy of 289.0 eV. We attribute this arrangement to C atoms at the GQD edge which have only a single bond to one neighboring C atom of the graphene layer. Furthermore, another 11.0% is contributed by the O–CO group, which is also a plausible conformation at the GQD edge. Finally, 8% of the total signal is due to the C–O single bond. This analysis suggests that about one third of the carbon atoms in the bulk of the GQDs is oxidized, and oxygen as well as nitrogen are engaging the loose bonds of the carbon atoms at the edge. Our results differ somewhat from those obtained by Wu et al.13 for GQDs prepared by a very similar method. They observed a larger fraction of nitrogen bonds, which may be explained by the different filter parameters they used. Absorption- and fluorescence spectroscopy was carried out for optical characterization (see Fig. 1B and C). A prominent absorbance peak is observed at a wavelength of 340 nm, which can be attributed to the n–π* transition the CO site.25 Along with a reduction of the wavelength below 280 nm, a strong increase in absorption is observed, with a superimposed peak at ≈235 nm, which is usually attributed to the π–π* – transition of the GQD.24 Furthermore, a small absorption peak of unknown origin is seen at 420 nm. The absorption shows a long tail towards larger wavelengths as reported earlier.13 This may be due to other graphitic particles but is of minor relevance here. The absorbance is a linear function of the GQD concentration, which is characteristic for a homogeneous solution of GQDs without absorption by the solvent itself (see Fig. S4†). The fluorescence spectrum depends, in qualitative agreement with Wu et al., on the excitation wavelength. Absorption around the peak at 340 nm results in strong fluorescence at approximately 450 nm, while a smaller excitation wavelength generates a second, weaker fluorescence resonance at 380 nm. Absorption around 420 nm induces a fluorescence peak close to 530 nm. The strongest fluorescence around 450 nm results from the n–π* – transition, while the fluorescence at larger wavelengths may be due to groups at the GQD edge containing nitrogen.26 The quantum yield was calculated to be 17% using quinine sulfate as reference (see inset in Fig. 1B). We note that this is an estimate of the lower bound since the long wavelength tail of the absorbance has been attributed to the GQDs. In general, the relation between the optical spectra and electronic states in GQDs is not yet well understood.8
O double bond, which is interpreted as oxidized bulk graphene that generates a local sp3-hybridization. The CO double bond also appears in combination with nitrogen with a similar weight of 16.5% at an energy of 289.0 eV. We attribute this arrangement to C atoms at the GQD edge which have only a single bond to one neighboring C atom of the graphene layer. Furthermore, another 11.0% is contributed by the O–CO group, which is also a plausible conformation at the GQD edge. Finally, 8% of the total signal is due to the C–O single bond. This analysis suggests that about one third of the carbon atoms in the bulk of the GQDs is oxidized, and oxygen as well as nitrogen are engaging the loose bonds of the carbon atoms at the edge. Our results differ somewhat from those obtained by Wu et al.13 for GQDs prepared by a very similar method. They observed a larger fraction of nitrogen bonds, which may be explained by the different filter parameters they used. Absorption- and fluorescence spectroscopy was carried out for optical characterization (see Fig. 1B and C). A prominent absorbance peak is observed at a wavelength of 340 nm, which can be attributed to the n–π* transition the CO site.25 Along with a reduction of the wavelength below 280 nm, a strong increase in absorption is observed, with a superimposed peak at ≈235 nm, which is usually attributed to the π–π* – transition of the GQD.24 Furthermore, a small absorption peak of unknown origin is seen at 420 nm. The absorption shows a long tail towards larger wavelengths as reported earlier.13 This may be due to other graphitic particles but is of minor relevance here. The absorbance is a linear function of the GQD concentration, which is characteristic for a homogeneous solution of GQDs without absorption by the solvent itself (see Fig. S4†). The fluorescence spectrum depends, in qualitative agreement with Wu et al., on the excitation wavelength. Absorption around the peak at 340 nm results in strong fluorescence at approximately 450 nm, while a smaller excitation wavelength generates a second, weaker fluorescence resonance at 380 nm. Absorption around 420 nm induces a fluorescence peak close to 530 nm. The strongest fluorescence around 450 nm results from the n–π* – transition, while the fluorescence at larger wavelengths may be due to groups at the GQD edge containing nitrogen.26 The quantum yield was calculated to be 17% using quinine sulfate as reference (see inset in Fig. 1B). We note that this is an estimate of the lower bound since the long wavelength tail of the absorbance has been attributed to the GQDs. In general, the relation between the optical spectra and electronic states in GQDs is not yet well understood.8
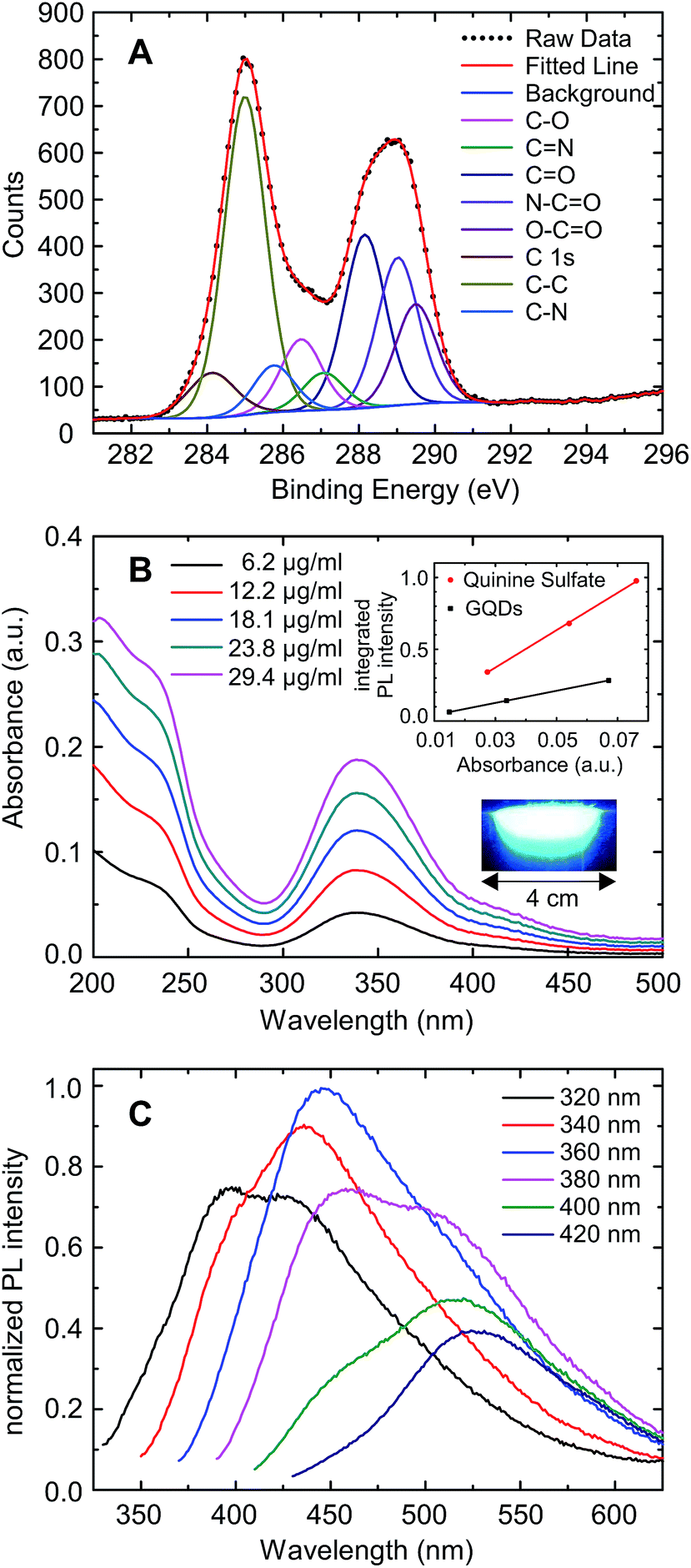 | ||
| Fig. 1 (A) XPS spectrum of the GQDs at the carbon 1s state (dots) and its composition, based on a best fit analysis. (B) Absorption spectra of the GQDs in aqueous solution. The upper inset shows the integrated PL intensity vs. absorbance of quinine sulfate and GQDs at concentrations of 3 μg ml−1, 6 μg ml−1 and 12 μg ml−1 and the lower inset shows an image of the GQD solution excited with UV light. (C) Fluorescence spectra of the quantum dots as a function of the excitation wavelength. | ||
2.2 Uptake studies in blood cells from leukapheresis products of normal donors
In a first step we examined the uptake of GQDs into primary human cells obtained from the leukapheresis product (LP) of 3 normal donors who had received granulocyte-colony stimulating factor (G-CSF) to mobilize CD34+ human progenitor and stem cells (HSCs) into the peripheral blood for allogeneic HSC transplantation. Samples of this kind of LPs are enriched for mononuclear blood cells (MNC) including T and B cells, natural killer cells monocytes and CD34+ HSCs.27–29 Still, they also contain a substantial number of granulocytes which are functionally activated as a result of the exposure to G-CSF for at least 4 days.30 To look for a potentially different uptake of the various white blood cell subpopulations a scatter plot was created based on the expression of the pan leukocyte antigen CD45 and the side scatter properties of the cells defining three gates: (1) lymphoid, (2) monocyte, and (3) granulocyte. Monoclonal antibodies directed against lineage specific antigens were used to differentiate between the lymphoid subpopulations. The gating procedure is shown in Fig. 2A–D. Looking at the samples of the donors we found the usual variation among individuals as far as the composition of the cell types is concerned. Reflecting the mode of collection the major population consists of lymphoid cells with a proportion between 80% and 89%, while the proportion of monocytes is relatively small ranging between 1% and 3%. The number and proportion contained within the samples may also vary depending on the total number of leukocytes induced following the stimulation by G-CSF, with a fraction of granulocytes between 7% and 18% in our series. The median values of leukapheresis products collected for allogeneic HSC transplantation are 60% leukocytes, 28% monocytes and 12% granulocytes. The deviations in the samples under study reflect the typical changes observed after keeping them in cell culture for 36 hours. The majority of lymphoid cells belongs to the CD3+ T cell fraction with a variable proportion of either CD4+ T helper or CD8+ cytotoxic/suppressor T cells. There is also a considerable number of CD19+ B cells – varying between 9% and 24% – while the proportion of CD56+ natural killer cells is relatively small. The composition of the three different blood samples is summarized in Fig. 2E. It should be noted that in comparison to “steady-state hematopoiesis” the proportion of CD34+ cells representing HSCs is greatly enhanced as a consequence of a successful G-CSF induced in vivo mobilization.27 Different from blood samples obtained from normal volunteers during steady-state hematopoiesis, this kind of sample from cytokine-stimulated blood allows us to perform uptake studies in an otherwise extremely rare cell population which is usually residing in the bone marrow requiring a biopsy for sufficient yield.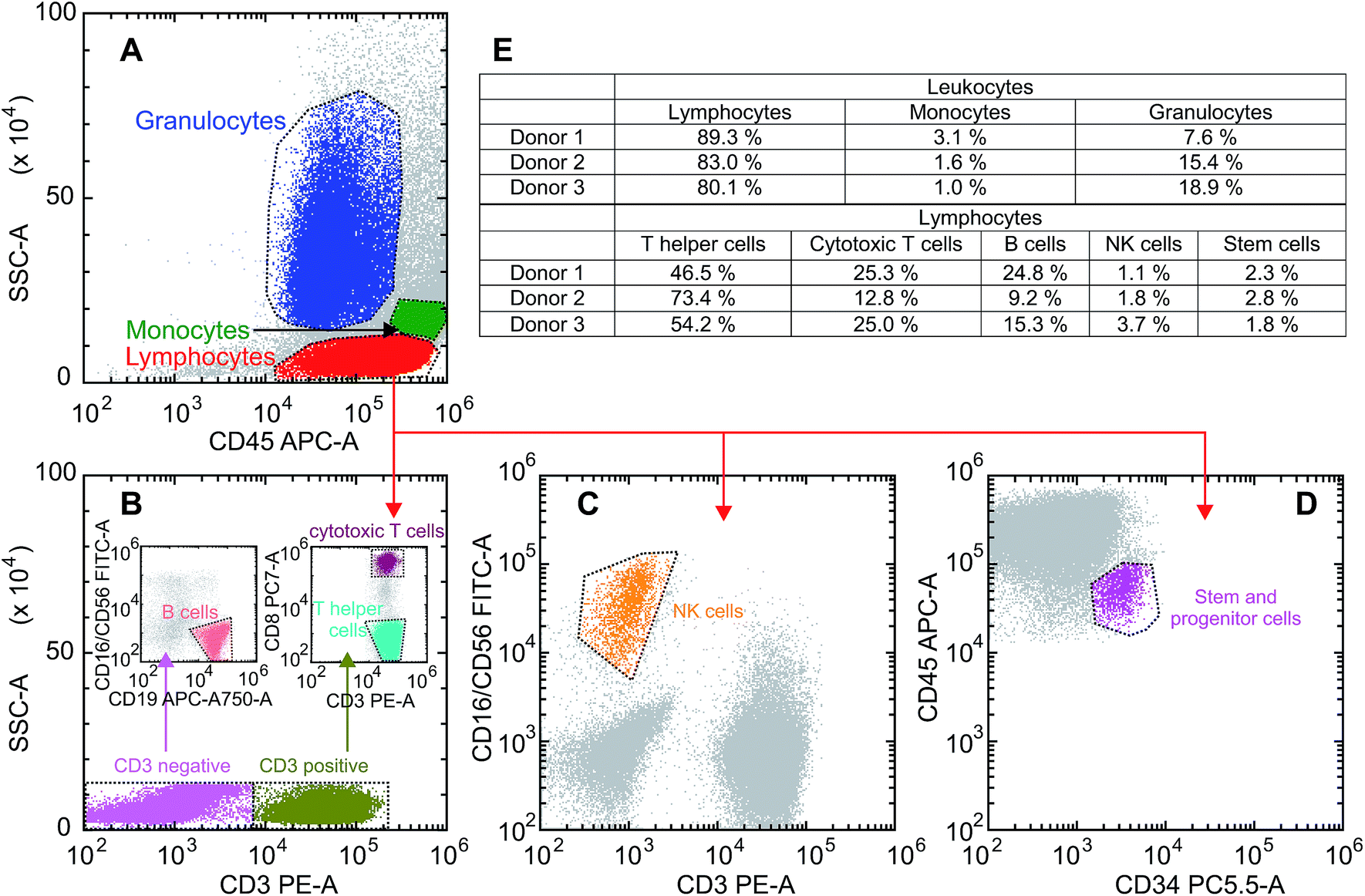 | ||
| Fig. 2 Gating strategy and composition of the three blood samples: (A) scatter plot of the leukocyte population of donor 2 to differentiate between lymphocytes, monocytes and granulocytes. (B) Discrimination between CD3+ and CD3neg lymphocytes to assess CD3neg/CD19+ B cells (left inset) and CD3+/CD8+ cytotoxic T cells as well as CD3+/CD8neg T helper cells (right inset). (C) Differentiation of CD3neg/CD16+/CD56+ NK cells out of the lymphocyte population. (D) Gating of the CD34+ stem and progenitor cells. (E) Composition of the three different blood samples. | ||
The donor samples were exposed to GQD solutions with concentrations of 200 μg ml−1 as well as 500 μg ml−1, and the uptake was studied as a function of time via the fluorescence intensity emitted by the cells. Typical microscopic images of the fluorescent cells are shown in the insets in Fig. 3A–C following an exposure time of 36 hours. As seen previously for adherent cells,9–12 the GQDs accumulate in the cells without entering the nuclei. Following the uptake microscopically, we noted an increase of the fluorescence intensity over time (see Fig. S5†). For a quantitative analysis, the fluorescence intensity distribution was measured over a period of 36 h for all donor samples in intervals of 2 hours. A representative example for that kind of measurement is given separately for lymphocytes, granulocytes and monocytes in Fig. 3A–C. Starting the incubation at time t = 0 h, the peaks represent the distribution of the autofluorescence. With time elapsing, the mean value of the intensity increases along with a broadening of the distributions, as illustrated here for an incubation time of 36 h. It is also noted that the time dependence of the intensity distribution is cell-type specific.
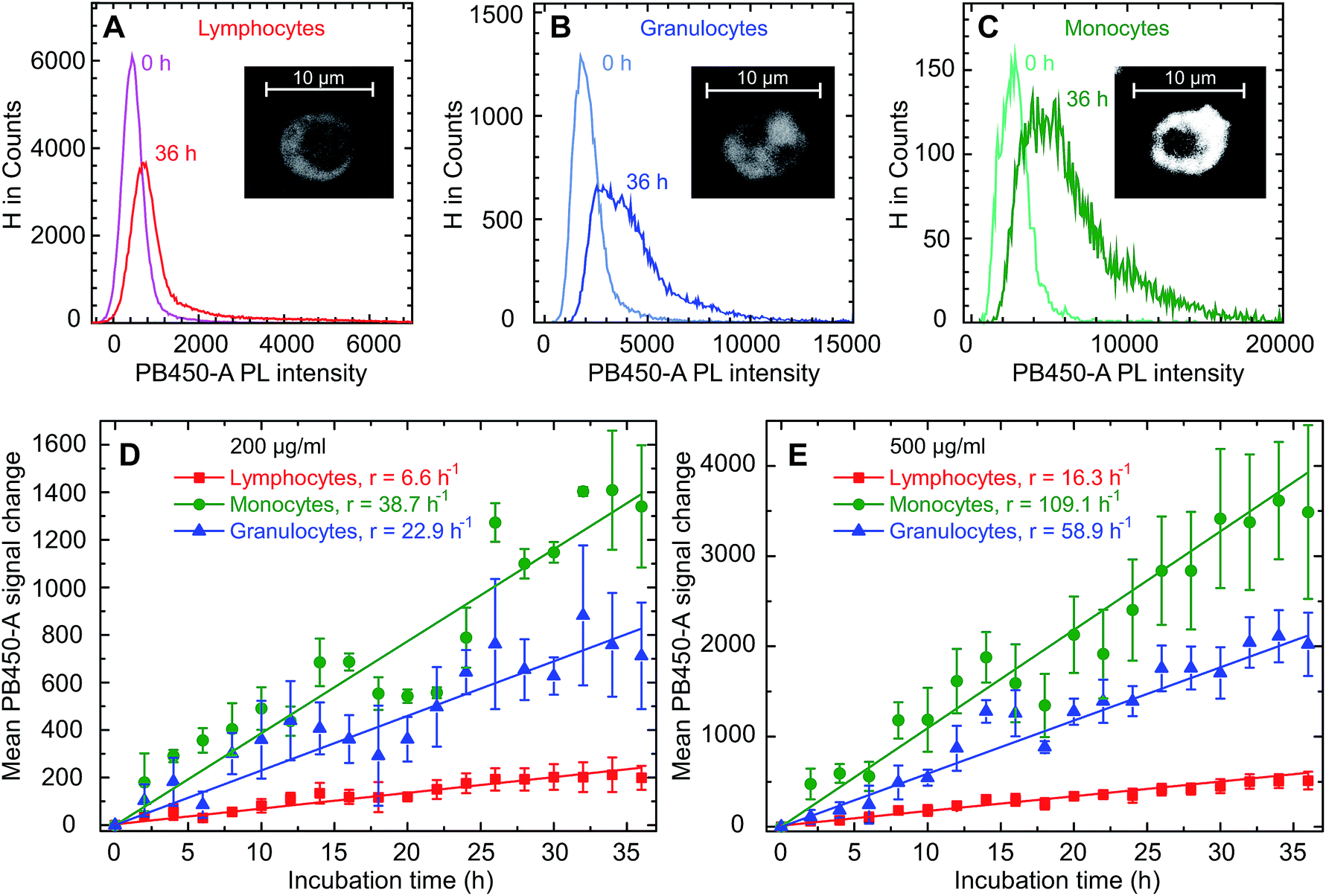 | ||
| Fig. 3 Changes of the fluorescence intensity histogram in lymphocytes (A), monocytes (B) and granulocytes (C) for donor 2, where the number counts of cells is plotted as a function of the intensity measured in the Cytoflex PB450-A channel following the exposure to GQDs at a concentration of 500 μg ml−1. The insets show typical confocal microscope pictures of the various cell types after 36 h exposure to GQDs. The change of intensity mean values in these three cell types for a GQD concentration of 200 μg ml−1 and 500 μg ml−1 is shown in (D) and (E). Linear fits were used to determine the uptake rates r as given in the legend. The error bars represent the standard errors obtained for the three samples. | ||
These changes of the immunofluorescence distributions as a function of time were recorded for the three donor samples using a GQD concentration of 200 μg ml−1 and 500 μg ml−1 (see Fig. 3D and E). The mean values of the intensity – with the error bars representing the corresponding standard errors obtained for the three samples – show an approximately linear increase for all three cell types. For all cell types the mean GQD uptake was approximately 2.5-fold greater when the cells were exposed to 500 μg ml−1 instead of 200 μg ml−1, while the mean uptake by granulocytes and monocytes is 3.6 and 6.7 times greater compared to lymphocytes, respectively. This implies a cell-specific GQD uptake with a time independent rate r (given in the figure) which depends linearly on the GQD concentration in solution. Assuming that an incubation period of 36 h – particularly as far as the viability of cells in culture is concerned – at a concentration of 500 μg ml−1 might be an optimal condition for the signal strength obtainable from the entire population of leukocytes, a more detailed uptake analysis was performed based on lineage-specific antigens such as CD34 (HSC), CD3 (pan T cell), CD19 (pan B cell), CD56 (natural killer cell). As shown in Fig. 4, similar to the findings made with the leukocytes an uptake of GQDs into the various lymphoid subpopulations was observed which increases linearly with incubation time. Representative examples of the evolution of the intensity distribution as observed for the sample of donor 2 are given for the different subsets. In general, a shift of the mean immunofluorescence intensity distribution towards larger mean values is noted. A greater enhancement was found within CD34+ cells, NK- and B cells in comparison to the cytotoxic and helper T cells. The combined results observed in our 3 normal donors following the various incubation periods are shown in Fig. 4F. Again as for the leukocytes in total, the mean value of the intensity is specific for each lymphoid subset and increases linearly with time, with rates rTh = 8.3 h−1 for T helper cells, rcT = 9.4 h−1 for cytotoxic T cells, rNK = 17.7 h−1 for NK cells, rB = 20.1 h−1 for B cells and rstem = 22.2 h−1 for stem and progenitor cells.
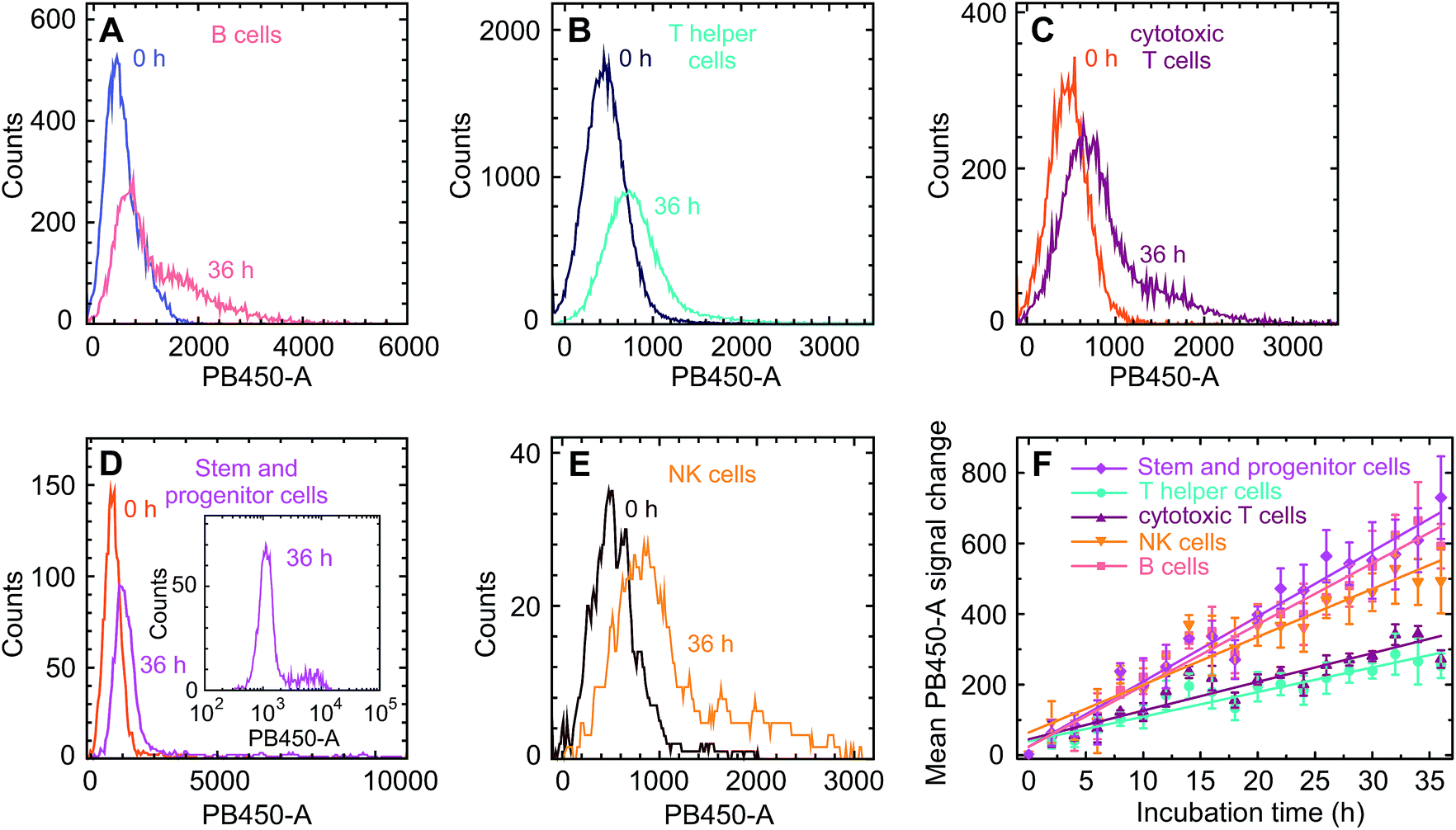 | ||
| Fig. 4 (A–E) Fluorescence intensity distributions and their changes with incubation time for the five lymphoid subsets under study as seen in the PB 450-A channel, exemplified at samples from donor 2. We note the particularly pronounced tail towards large GQD-induced fluorescence intensities observed in stem and progenitor cells (inset in (D)). (F) Temporal evolution of the ensemble – averaged intensity mean values. The GQD concentration was 500 μg ml−1 in all experiments. | ||
We proceed with an analysis of the uptake dynamics. Due to the approximately linear dependence of the uptake as a function of both time and GQD concentration in solution csol, its dynamics is qualitatively consistent with a diffusion process, i.e. a process driven by a concentration gradient across the cell membrane, which can be described by the diffusion equation j = PΔc, where j denotes the particle current density into the cell, P the permeability of the cell membrane for the GQDs and Δc ≡ csol − ccell the difference of the GQD particle concentration between the solution and the intracellular space. The diffusion model allows an order-of-magnitude estimate of P. For the GQD mass concentration in solution of 500 μg ml−1, csol can be estimated using the GQD size of approximately 2 nm,13 an average of two graphene layers per quantum dot and their composition as given on the basis of the XPS analysis to 4 × 1019 cm−3. By comparing the fluorescence light intensity of the GQD solution and the cells prior to and after the uptake experiments (see Fig. S6†), we find that the 106 cells per sample take up approximately 0.7% of the GQDs in the solution. This leads to an estimation of the extent of the uptake. After 36 h incubation time, the number of GQDs per cell amounts to 1.9 × 108, 6.9 × 108 and 1.3 × 109 for lymphocytes, granulocytes and monocytes, respectively. As can be seen from the fluorescence images in Fig. 3A–C, the particles distribute homogeneously in the cytoplasm but do not enter the nucleus. For the cytoplasm volumes of Vcp(L) = 62 μm3, Vcp(G) = 120 μm3, Vcp(M) = 150 μm3,31 we find an average distance between the GQDs in the cytoplasm between 5 nm for monocytes and 7 nm for lymphocytes, under the assumption that the cells do not swell due to the uptake. These values illustrate the leukocyte's large capability for GQD incorporation.
Since the fraction of the GQDs entering the cells is quite small, we assume Δc as time-independent, which is in accordance with the fact that no saturation of the uptake is observed. With the presumption that the GQD uptake is proportional to the GQD-induced fluorescence intensity, we obtain particle uptake rates per cell of 890 s−1 for lymphocytes, 3190 s−1 for granulocytes and 5950 s−1 for monocytes. With the reported average cell membrane areas of 270 μm2 for lymphocytes, 300 μm2 for granulocytes and 430 μm2 for monocytes,31 the GQD influx densities j are ≈3.3 μm−2 s−1, ≈10.6 μm−2 s−1, and ≈13.8 μm−2 s−1, corresponding to membrane permeabilities for the GQDs of P ≈ 8.3 × 10−14 ms−1, ≈2.7 × 10−13 ms−1 and ≈3.5 × 10−13 ms−1 for lymphocytes, granulocytes and monocytes, respectively. These permeabilities are plausible values considering that they are significantly smaller than those for large molecules, like P(erythritol) ≈ 6.7 × 10−11 ms−1 (ref. 32) or P(glycerol) ≈ 1.6 × 10−9 ms−1,33 since it is well-known that P decays approximately exponentially as the size of the particle increases.34 While the permeabilities of the granulocyte and monocyte membranes are comparable, that one of the lymphocytes is a factor of about four smaller. Our results therefore imply that the differences in the uptake rates between lymphocytes and other leukocytes cannot be solely explained by the cell size, even if the relatively large size variation of the monocytes35 is taken into account. The measurements rather suggest that the uptake mechanisms differ. Electrostatic effects may be considered as an explanation, since it is well-known that the membrane potential influences the uptake of charged particles. While typical zeta potentials of GQDs in aqueous solution of −10 mV to −48 mV have been reported in literature,13,23 we are not aware of significant differences among the membrane potentials of the various leukocyte subtypes of approximately −60 mV,36 and such differences are also not to be expected considering that the cells are not excitable. We therefore discard electrostatic effects as possible explanation. It is, however, notable that the preferential uptake is observed for cell types with a preponderance for phagocytosis, a correlation which may be worth further studies.
As can be seen in Fig. 4, the uptake rates of the lymphocyte subspecies fall into two categories. A faster uptake is observed for stem and progenitor, NK and B cells, while that one of T and T helper cells is relatively slow. To the best of our knowledge, corresponding differences in the cell sizes are not reported. Furthermore, this bimodal behavior does not correlate with the cell's phagocytotic activity, even though B cells are capable of absorbing objects. The distribution of the uptake for the stem and progenitor cells is particularly broad and shows a pronounced, long tail towards large values, see the inset of Fig. 4D. Their relatively large uptake rate may therefore indicate a differentiated hierarchical heterogeneity in relation to the cell evolution. In this context, a recent article of D'Aveni et al. is of interest, who describe a particular subset of monocytic CD34+ cells which are only seen in individuals who had received G-CSF like our normal donors.29 It is tempting to speculate that the subset of bright CD34+ cells in our samples could represent that type of monocytic HSC with a more avid uptake of GQDs.
Some further insight regarding the uptake dynamics can be gained by a separate study of the GQD-induced fluorescence intensity distribution Q(I). The measured intensity distribution H(I) is a convolution of the autofluorescence A(I) and of Q(I), expressed by
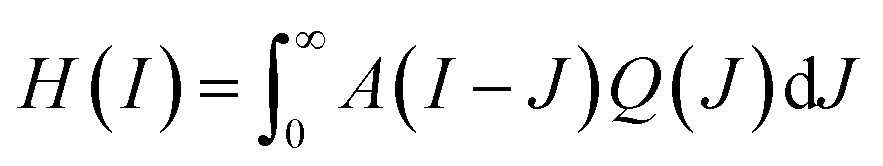
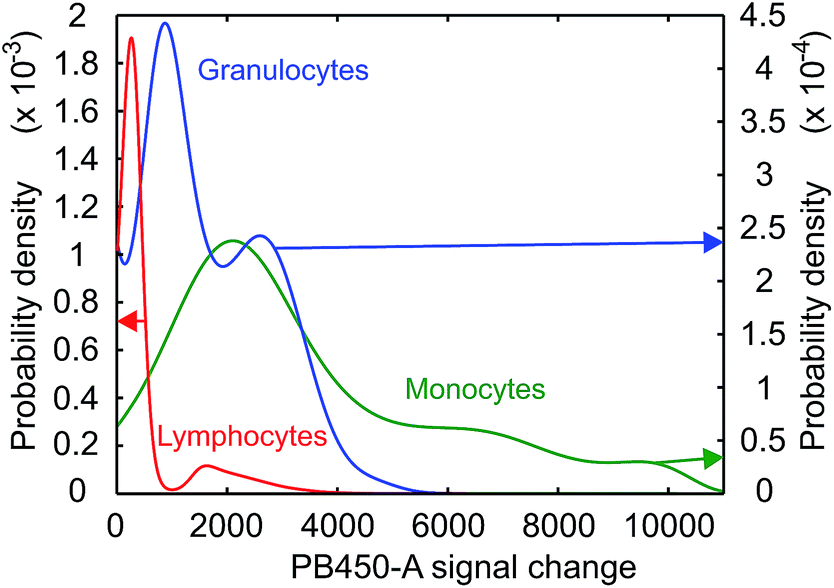 | ||
| Fig. 5 Probability densities of the GQD – induced fluorescence in lymphocytes, granulocytes and monocytes after 36 h of exposure to a GQD concentration of 500 μg ml−1. | ||
Even taking into account a potential decrease of the permeability among the various leukocyte populations during the incubation period this would rather be viewed as a continuous process and therefore does not explain distinct peaks in the uptake rates. As a second possibility, different charge states of the GQDs in solution may be considered, which would result in charge-specific uptake rates. However, since all cells are exposed to the same GQD ensemble, this should lead to a qualitatively identical peak distribution for all three cell types, which is not observed. The reasons for this behavior may be related to different functional states of unidentified character and require further studies. It should be noted that this structure is visible at all exposure times.
2.3 Toxicity studies
The question of toxicity was addressed using the XTT viability assay concentrating on the total population of mononuclear cells without particular subset analysis (see Fig. 6). Over the entire period of 36 h covered by 2 h incubation intervals we observed a time- and concentration dependent approximately linear decrease with regard to the proportion of viable cells. The strongest effect was observed during the last 12 h of incubation at a concentration of 500 μg ml−1 resulting in a proportion of 90% viable cells in comparison to the untreated controls (see Fig. S7† for the control measurement of all intervals).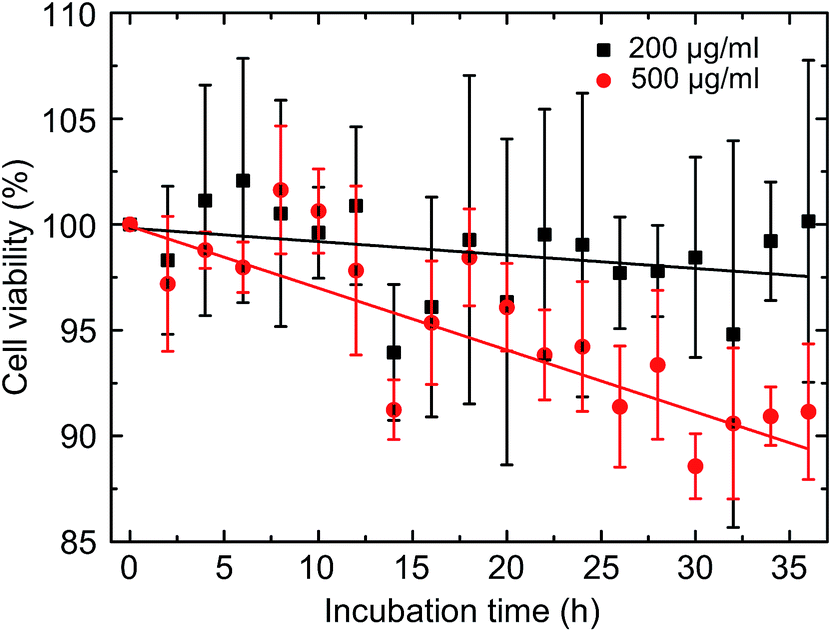 | ||
| Fig. 6 Viability of the cells as a function of time for the two concentrations. | ||
Putting our findings in the context of the current literature, there are only few studies on the effects of GQDs on blood cells. There is one report showing that large graphitic flakes with diameters in the range of 300 nm to 1 μm cause apoptosis in red blood cells and in macrophages, most likely through generation of intracellular reactive oxygen species (ROS),37 which is in line with earlier studies on skin fibroblasts.38 In this study, a GQD-triggered increase of in vivo haemolysis has been observed as well. GQDs also induced apoptosis and inflammatory reactions in macrophages following exposure to GQDs. This was accompanied by expression of characteristic response factors such as interleukin-8 or tumor necrosis factor.39 Interestingly, in an animal model using rats no significant toxicities were observed at different doses up to 10 mg g−1.40 The general cytotoxicity is apparently not influenced by functional groups attached to GQDs.41 As far as the aspect of toxicity is concerned within our experimental setting, the CD34+ cells would be of particular interest for further studies, as they represent the stem cell population responsible for the lifelong self-renewal of the hematopoietic system, whereas granulocytes and monocytes have a half live of only a few hours, thus representing target cells of immediate and not long time toxicity.
3 Conclusions
In our studies we assessed the new generation of quantum dots based on graphene with regard to their uptake properties into primary human blood cells consisting of a broad spectrum of leukocyte subsets which include the rare population of CD34+ hematopoietic stem and progenitor cells. The in vitro exposure resulted in a time- and concentration dependent cellular uptake leading to significant intracellular concentrations of quantum dots without undue toxicity. We found that the uptake rate in granulocytes and monocytes is significantly greater as compared to lymphocytes, a difference that cannot solely be explained by the variations in cell size alone but rather implies an intrinsic difference of the effective cell membrane permeabilities. As the leukocytes of the same species show multi-peaked uptake rates we suggest that some functional diversity among the subsets exists which requires further assessment. The open questions in mind we consider these readily available GQDs as promising compounds for in vivo an in vitro cell tracking studies as well as low weight carriers for different molecules such as mRNA for in vitro vaccination CD34+ cell derived dendritic cells.4 Experimental section
4.1 Materials
Citric acid (ACS reagent, ≥99.5%), dicyandiamide (99%), Fetal Bovine Serum (FBS), Iscove's Modified Dulbecco's Medium (IMDM), L-glutamine–penicillin–streptomycin solution, Dulbecco's Phosphate Buffered Saline (PBS) and the Cell Proliferation Kit II (XTT) were purchased from Sigma Aldrich. Float-A-Lyzer dialysis devices (100–500 Da and 20 kDa) and sterile filters (200 nm) were obtained from VWR and antibodies against CD45-APC, CD16/CD56-FITC, CD3-PE, CD34-PerCP-Cy5.5, CD8-PE-Cy7 and CD19-APC-H7 were purchased from BD biosciences.4.2 Synthesis of graphene quantums dots (GQDs)
Fluorescent GQDs were synthesized according to the method of Wu et al.13 with slight modifications. 70 mg citric acid and 250 mg dicyandiamide were dissolved in 4 ml DI water and heated to 180 °C on a hot plate for 3 h under continuous stirring in a 10 ml stainless steel autoclave. The obtained aqueous solution was centrifuged with an Eppendorf MiniSpin® at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 rpm for 15 minutes to remove insoluble residual. Citric acid and dicyandiamide waste were removed by dialyzing 10 ml of GQD solution against 2 l of DI water for 48 h with one water exchange after 24 h using a 100–500 Da dialysis membrane. Afterwards larger particles were filtered out with a 20 kDa membrane. Finally, the GQDs were dried and weighed using a Sartorius A 200S electronic analytical balance.
400 rpm for 15 minutes to remove insoluble residual. Citric acid and dicyandiamide waste were removed by dialyzing 10 ml of GQD solution against 2 l of DI water for 48 h with one water exchange after 24 h using a 100–500 Da dialysis membrane. Afterwards larger particles were filtered out with a 20 kDa membrane. Finally, the GQDs were dried and weighed using a Sartorius A 200S electronic analytical balance.
4.3 Characterization of GQDs
Photoluminescence properties were measured using a Horiba FluoroMax®-4 spectrofluorometer and absorbance spectra were taken with an Agilent Cary 4000 spectrophotometer. The quantum yield (QY) was obtained using quinine sulfate (QS) dissolved in 0.5 M H2SO4 as reference (QYQS = 54%). The concentrations 3 μg ml−1, 6 μg ml−1 and 12 μg ml−1 of OS and of the GQDs were excited at a wavelength of 346 nm and 360 nm, respectively. The integrated fluorescence intensities were plotted vs. the corresponding absorbance values from which the slopes were determined. The quantum yield was calculated using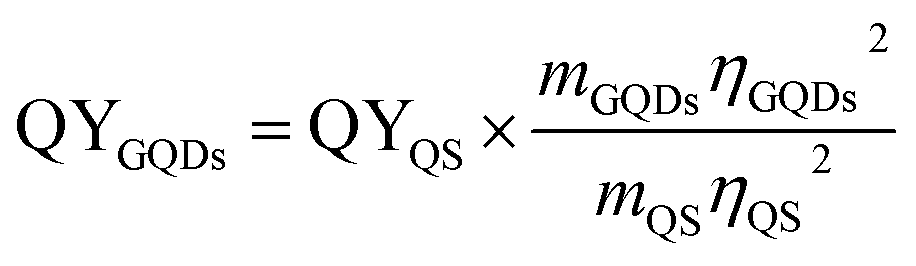 where m is the slope and η the refractive index of the solvent. Confocal fluorescence microscopy was performed on a Zeiss LSM710 laser scanning system and AFM measurements were executed using the tapping mode of a Veeco 3100 with SiO2 as substrate. XPS data were collected on a PHI 5000 Versaprobe II XPS microprobe instrument using Au as substrate and TEM images were taken with the Titan G3 50-300 PICO42 on amorphous carbon TEM grids.
where m is the slope and η the refractive index of the solvent. Confocal fluorescence microscopy was performed on a Zeiss LSM710 laser scanning system and AFM measurements were executed using the tapping mode of a Veeco 3100 with SiO2 as substrate. XPS data were collected on a PHI 5000 Versaprobe II XPS microprobe instrument using Au as substrate and TEM images were taken with the Titan G3 50-300 PICO42 on amorphous carbon TEM grids.
4.4 Collection of leukapheresis derived blood samples from normal donors
The primary human cells for our study were obtained from leukapheresis products (LP) of three normal individuals who served as HLA-identical donors for an allogeneic blood stem cell transplantation (SCT) in patients with hematological malignancies. For the purpose of blood stem cell collection, the normal donors had received granulocyte colony stimulating factor (G-CSF) at the usual dose of 480 μg per day over a time period of 4 to 5 days in order to increase the number of circulating human progenitor and stem cells (HSC) – as defined by the expression of CD34 on the cell surface – in the peripheral blood for allogeneic HSC transplantation. On average, the treatment with G-CSF in normal individuals leads to a 100-fold increase in the concentration of CD34+ blood stem cells in comparison to steady-state hematopoiesis. Samples of this kind of LPs are enriched for mononuclear blood cells (MNC) including T and B cells, natural killer cells monocytes and CD34+ HSC. In that respect, the samples that we used for our experiments are unique with regard to their content of early hematopoietic stem and progenitor cells. Still, they also contain a substantial number of granulocytes which are functionally activated as they had been exposed to G-CSF for at least 4 days.4.5 Cell preparation
Blood samples from leukapheresis products of three healthy donors were used for the in vitro studies. From each of them a sample of 1 ml was obtained and lysed two times using 50 ml ammonium chloride to remove erythrocytes. The remaining leukocytes were resuspended in Iscove's Minimal Dulbeco Medium (IMDM) containing 20% Fetal Bovine Serum (FBS) and 1% L-glutamine–penicillin–streptomycin solution and cultivated in a Heracell TM 150i incubator in a humidified atmosphere at 5% CO2 and 37 °C. 500 μl per well of cell suspension were dispensed in 24 well plates at a final concentration of 2 × 106 cells per ml.4.6 Cell cultivation for GQD uptake studies
GQDs were dissolved in cell culture medium at concentrations of 1.2 mg ml−1 and 3.0 mg ml−1 and the obtained solutions were sterile filtered. From the beginning of the cultivation period – termed as time point 0–100 μl of the GQD solutions were added in 2 h intervals into the wells to achieve GQD concentrations of 200 μg ml−1 and 500 μg ml−1. The cultivation was stopped after 36 hours. As a result, uptake measurements for a total of 18 different incubation periods are available. In parallel, the same amount of sterile filtered cell culture medium without containing GQDs was added to the wells serving as control. After 36 hours all samples were washed two times with PBS (centrifugation for 5 minutes at 470g) and stained with antibodies as detailed below.4.7 Immunocytological characterization of blood-derived mononuclear cells
To look for a potentially different uptake of the various MNC subpopulation monoclonal antibodies directed against lineage- and differentiation specific antigens, i.e. CD45-APC, CD16/CD56-FITC, CD3-PE, CD34-PerCP-Cy5.5, CD8-PE-Cy7 and CD19-APC-H7 were used. The cells were incubated for 15 minutes with 5 μl of each antibody per sample and fixed with 150 μl 4% formaldehyde solution, before the cells were transferred to a 96 well plate for flow cytometry analysis. Debris was removed by gating the living cells in a forward vs. sideward scatter plot (FSC vs. SSC). Out of all living cells, gates were set individually for each sample, in a CD45 vs. SSC plot to assess lymphocytes, monocytes and granulocytes as established in immune-phenotyping of leukemia43 and the analysis of human bone marrow specimens.44 To distinguish between lymphocyte subpopulations, CD3+ and CD3neg cells were gated in a SSC vs. CD3 plot. NK cells (CD3neg/CD16+/CD56+) were gated out of the lymphocyte population in a CD3 vs. CD16/CD56 plot and B-cells (CD3neg/CD16neg/CD56neg/CD19+) were gated out of the CD3 negative cells in a CD16/CD56 vs. CD19 plot. Cytotoxic T cells (CD3+/CD8+) and T helper cells (CD3+/CD8neg) were differentiated out of the CD3+ population in a CD3 vs. CD8 plot. Finally, stem and progenitor cells (CD45+/CD34+) were assessed with a CD34 vs. CD45 plot. The gating strategy is shown in Fig. 2. Information regarding the occurrence of NK cells, cytotoxic T cells, T helper cells, B-cells and stem and progenitor cells is given in relation to the lymphocyte population out of the CD45/SSC plot, whereas the occurrence of lymphocytes, monocytes and granulocytes is given with respect to the whole leukocyte population, i.e., the sum of lymphocytes, monocytes and granulocytes out of the CD45/SSC plot. FACS analysis was performed using a Beckmann Coulter CytoFLEX flow cytometer with an automatic 96-well-plate loader. The FACS is equipped with a 488 nm and a 638 nm laser beam, measuring the antibody-coupled fluorescence dyes in the APC, PE, PC5.5, APC-A750, PC7 and FITC channels and a laser beam with an excitation wavelength of 405 nm, measuring the GQD induced fluorescence in the PB450 channel. For each sample at least 100000 events were recorded permitting a statistically valid evaluation with a minimum of at least 1000 cellular events per sample and cell type. The mean fluorescence intensity recorded for a particularly labeled blood cell was subtracted by the respective autofluorescence observed in the control and the resulting “true fluorescence activity” taken as the parameter reflecting the cellular uptake of GQDs. The analysis was carried out using the Beckmann Coulter CytExpert software.
4.8 Cell viability
Cell viability was determined by the standard XTT assay. Briefly, 100 μl per well of cell suspension with a final concentration of 2 × 106 cells per ml were dispensed in 96 well plates. The cells were treated with 20 μl of sterile filtered GQD containing cell culture medium for each concentration and with sterile filtered pure cell culture medium for the controls. 60 μl XTT labeling solution was added and incubated for 4 h before the absorbance at 490 nm was measured with an ELISA microplate reader.4.9 Ethical statement
All experiments were performed in compliance with the relevant laws and institutional guidelines and have been approved by the ethical committee of the Heinrich Heine University (No. 3240 from August/October 2009). All donors had given their informed consent according to the guidelines of the ethical committee specified above.Acknowledgements
The first author acknowledges financial support of his position by the Düsseldorf School of Oncology (DSO). T. H. acknowledges support by the Betz foundation. Support regarding the XPS analysis by A. Besmehn and H. Hartmann (FZ Jülich), as well as access to the confocal microscope at the Center of Advanced imaging (CAi) of HHU Düsseldorf, is gratefully acknowledged. The authors thank S. Wesselborg for his advice regarding the toxicity studies.References
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Nat. Biotechnol., 2002, 21, 47–51 CrossRef PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS PubMed.
- B. A. Kairdolf, A. M. Smith, T. H. Stokes, M. D. Wang, A. N. Young and S. Nie, Annu. Rev. Anal. Chem., 2013, 6, 143 CrossRef CAS PubMed.
- A. M. Smith, S. Dave, S. Nie, L. True and X. H. Gao, Expert Rev. Mol. Diagn., 2006, 6, 231–244 CrossRef CAS PubMed.
- V. Bagalkot, L. Zhang, E. Levy-Nissenbaum, S. Jon, P. W. Kantoff, R. Langer and O. C. Farokhzad, Nano Lett., 2007, 7, 3065 CrossRef CAS PubMed.
- X. Gao, Y. Cui, R. M. Levenson, L. W. K. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726 CrossRef CAS PubMed.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S. Xie and Y. Sun, J. Am. Chem. Soc., 2007, 129, 11318 CrossRef CAS PubMed.
- C. Liu, P. Zhang, F. Tian, W. Li, F. Li and W. Liu, J. Mater. Chem., 2011, 21, 13163 RSC.
- S. Zhu, J. Zhang, C. Qiao, S. Tang, Y. Li, W. Yuan, B. Li, L. Tian, F. Liu, H. Gao, H. Wei, H. Zhang, H. Sun and B. Yang, Chem. Commun., 2011, 47, 6858 RSC.
- W. Shang, X. Zhang, M. Zhang, Z. Fang, Y. Sun, M. Han and L. Fan, Nanoscale, 2014, 6, 5799 RSC.
- Z. L. Wu, M. X. Gao, T. T. Wang, X. Y. Wan, L. L. Zheng and C. Z. Huang, Nanoscale, 2014, 6, 3868 RSC.
- A. Roos and W. F. Boron, Physiol. Rev., 1981, 61, 296–434 CAS.
- X. Huang, F. Zhang, L. Zhu, K. Y. Choi, N. Guo, J. Guo, K. Tackett, P. Anilkumar, G. Liu, Q. Quan, H. S. Choi, G. Niu, Y.-P. Sun, S. Lee and X. Chen, ACS Nano, 2013, 7, 5684 CrossRef CAS PubMed.
- M. Zheng, S. Ruan, S. Liu, T. Sun, D. Qu, H. Zhao, Z. Xie, H. Gao, X. Jing and Z. Sun, ACS Nano, 2015, 9, 11455 CrossRef CAS PubMed.
- X. Sui, C. Luo, C. Wang, F. Zhang, J. Zhang and S. Guo, Nanomedicine, 2016, 12, 1997 CAS.
- X. Gong, Q. Zhang, Y. Gao, S. Shuang, M. M. F. Choi and C. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 11288 CAS.
- M. Kryuchkova, A. Danilushkina and R. F. Y. Lvov, Environ. Sci.: Nano, 2016, 3, 442–452 RSC.
- X. Guo and N. Mei, J. Food Drug Anal., 2014, 22, 105–115 CrossRef CAS PubMed.
- Y. Dong, J. Shao, C. Chen, H. Li, R. Wang, Y. Chi, X. Lin and G. Chen, Carbon, 2012, 50, 4738 CrossRef CAS.
- D. Graf, F. Molitor, K. Ensslin, C. Stampfer, A. Jungen, C. Hierold and L. Wirtz, Nano Lett., 2007, 7, 238 CrossRef CAS PubMed.
- M. Zhang, L. Bai, W. Shang, W. Xie, H. Ma, Y. Fu, D. Fang, H. Sun, L. Fan, M. Han, C. Liub and S. Yang, J. Mater. Chem., 2012, 22, 7461 RSC.
- F. Liu, M.-H. Jang, H. D. Ha, J.-H. Kim, Y.-H. Cho and T. S. Seo, Adv. Mater., 2013, 25, 3657 CrossRef CAS PubMed.
- G. Eda, Y.-Y. Lin, C. Mattevi, H. Yamaguchi, H.-A. Chen, I. S. Chen, C.-W. Chen and M. Chhowalla, Adv. Mater., 2010, 22, 505 CrossRef CAS PubMed.
- D. Qu, M. Zheng, J. Li, Z. Xie and Z. Sun, Light: Sci. Appl., 2015, 4, e364 CrossRef CAS.
- R. Möhle, M. Pförsich, S. Fruehauf, B. Witt, A. Krämer and R. Haas, Bone Marrow Transplant., 1994, 14, 827 Search PubMed.
- K. Theilgaard-Mönch, K. Raaschou-Jensen, H. Palm, K. Schødt, C. Heilmann, L. Vindeløv, N. Jacobsen and E. Dickmeiss, Bone Marrow Transplant., 2001, 28, 1073 CrossRef PubMed.
- M. D'Aveni, J. Rossignol, T. Coman, S. Sivakumaran, S. Henderson, T. Manzo, P. S. E. Sousa, J. Bruneau, G. Fouquet, F. Zavala, O. Alegria-Prvot, M. Garfa-Traor, F. Suarez, H. Trebeden-Ngre, M. Mohty, C. L. Bennett, R. Chakraverty, O. Hermine and M. T. Rubio, Sci. Transl. Med., 2015, 7, 281ra42 CrossRef PubMed.
- S. Murea, S. Fruehauf, W. J. Zeller and R. Haas, J. Hematother., 1996, 5, 351 CrossRef CAS PubMed.
- G. W. Schmid-Schönbein, Cell Biophys., 1990, 17, 107 CrossRef.
- J. O. Wieth, J. Physiol., 1971, 213, 435 CrossRef CAS.
- A. Carlsen and J. O. Wieth, Acta Physiol. Scand., 1976, 97, 501 CrossRef CAS PubMed.
- W. D. Stein, Transport and Diffusion across Cell Membranes, Academic Press, London, 1985 Search PubMed.
- G. W. Schmid-Schönbein, Y. Y. Shih and S. Chien, Blood, 1980, 56, 866 Search PubMed.
- E. K. Gallin, Physiol. Rev., 1991, 71, 775 CAS.
- G. Qu, X. Wang, Z. Wang, S. Liu and G. Jiang, Nanoscale Res. Lett., 2013, 8, 198 CrossRef PubMed.
- K. H. Liao, Y. S. Lin, C. W. Macosko and C. L. Haynes, ACS Appl. Mater. Interfaces, 2011, 3, 2607 CAS.
- Y. Qin, Z.-W. Zhou, S.-T. Pan, Z.-X. He, X. Zhang, J.-X. Qiu, W. Duan, T. Yang and S.-F. Zhou, Toxicology, 2015, 327, 62 CrossRef CAS PubMed.
- M. Nurunnabi, Z. Khatun, K. M. Huh, S. Y. Park, D. Y. Lee, K. J. Cho and Y. Lee, ACS Nano, 2013, 7, 6858–6867 CrossRef CAS PubMed.
- X. Yuan, Z. Liu, Z. Guo, Y. Ji, M. Jin and X. Wang, Nanoscale Res. Lett., 2014, 9, 108 CrossRef PubMed.
- J. Barthel, L. Houben and K. Tillmann, Journal of large scale research facilities, 2015, 1, 57 Search PubMed.
- F. Lacombe, L. Lacoste, A. Briais, P. Dumain, F. Belloc, J. Reiffers and P. Bernard, Blood, 1997, 90, 2895 Search PubMed.
- G. T. Stelzer, K. E. Shults and M. R. Loken, Ann. N. Y. Acad. Sci., 1993, 677, 265 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures with AFM, TEM, XPS and fluorescence images. Additional data characterizing the quantum dots. See DOI: 10.1039/c6ra27829a |
| This journal is © The Royal Society of Chemistry 2017 |