
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium nanoparticles-modified reduced graphene prepared by a green method for high-performance supercapacitor application in neutral electrolyte†
Hagar K. Hassana,
Nada F. Attaa,
Maher M. Hameda,
Ahmed Galal *a and
Timo Jacob*bcd
*a and
Timo Jacob*bcd
aDepartment of Chemistry, Faculty of Science, Cairo University, 12613 Giza, Egypt. E-mail: galal@sci.cu.edu.eg
bInstitute of Electrochemistry, Ulm University, 89081 Ulm, Germany. E-mail: timo.jacob@uni-ulm.de
cHelmholtz-Institute-Ulm (HIU) [d], Helmholtzstr. 11, 89081 Ulm, Germany
dKarlsruhe Institute of Technology (KIT), P.O. Box 3640, 76021 Karlsruhe, Germany
First published on 13th February 2017
Abstract
Here, we report a one-pot preparation method of a ruthenium-based reduced graphene oxide hybrid (Runano-based RGO). The synthesis is based on a single step for the reduction of Ru3+ (RuCl3) into Ru nanoparticles, and graphene oxide (GO) into reduced graphene oxide (RGO) without the use of any reducing agent. The structure of the hybrid material was ascertained using X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy and high resolution transmission spectroscopy (HRTEM). The change in pH of the synthesis media resulted in different structures and morphologies. A mixture of Ru and RuO2 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was obtained at pH 8.0, while only Ru-nanoparticles were obtained at pH 4.0. The supercapacitor performance of the hybrid was investigated in a neutral electrolyte, 1.0 M NaNO3, in a three-electrode setup. Runano-based RGO prepared at pH 8 exhibited a maximum specific capacitance of 270 F g−1 (238.5 mF cm−2), and maximum energy and power densities of 15 W h kg−1 and 76.4 kW kg−1, respectively. The proposed supercapacitor has a stable potential window up to 1.1 V with very good cycling stability over 5000 cycles at 24 A g−1.
:2) was obtained at pH 8.0, while only Ru-nanoparticles were obtained at pH 4.0. The supercapacitor performance of the hybrid was investigated in a neutral electrolyte, 1.0 M NaNO3, in a three-electrode setup. Runano-based RGO prepared at pH 8 exhibited a maximum specific capacitance of 270 F g−1 (238.5 mF cm−2), and maximum energy and power densities of 15 W h kg−1 and 76.4 kW kg−1, respectively. The proposed supercapacitor has a stable potential window up to 1.1 V with very good cycling stability over 5000 cycles at 24 A g−1.
1. Introduction
Research and technology development for energy sources and storage alternatives are attracting considerable attention. This is because of the depletion of the energy supply from fossil fuels and increasing environmental pollution.1 Another challenge is electricity storage with high power and energy densities.2Electric double layer capacitors (EDLCs) are promising energy storage devices that store electric charges at the interface between the electrode surface and the electrolyte.3 Another class of capacitors is pseudocapacitors, in which the capacitance arises from a reversible reaction that takes place at the interface. Both EDLCs and pseudocapacitors belong to the class of supercapacitors or ultracapacitors due to their high specific capacitance compared to conventional capacitors. Moreover, supercapacitors provide several advantages over batteries such as high power density, long cycling stability, fast charging/discharging rate and lower environmental impact.4 Supercapacitors however have lower energy densities compared to batteries. Research work on the usage of energy storage devices is targeting the enhancement of the properties of supercapacitors for usage in many applications. Beside the low energy density of supercapacitors, pseudocapacitors have poor cycling stability.5 Construction of hybrid materials (for combination between EDLCs and pseudocapacitors) is one of the challenges to overcome the problem of both EDLCs and pseudocapacitors and to improve the properties of supercapacitors.6 This might allow for a maximum energy density at high power density without sacrificing the cycling stability.
In recent years, graphene attracted a significant interest as a promising electrode material for energy storage applications.7 Graphene-based supercapacitors showed high theoretical specific capacitance up to 550 F g−1.8 Since restacking or aggregation of its sheets due to van der Waals forces limits reaching its theoretical specific capacitance value in real applications, modifying the graphene sheets in order to avoid their aggregation is of prime importance.
Transition metals and their oxides are interesting candidates to act as spacers between graphene sheets to prevent their aggregation and hence, to increase the specific capacitance of graphene-based supercapacitors.5 Here, RuO2 is one of the most promising materials for supercapacitor applications due to its high specific capacitance from 1300 to 2200 F g−1,9,10 high rate capability, highly reversible faradic reaction and high thermal stability.11 However, Ru-based materials are usually suffering from their poor porosity, less cycling stability and rapid decrease of power density especially at high charge/discharge rates as well as the high price of Ru-based materials that limits their commercial use.5 The properties of Ru-based materials (i.e. Ru, RuO2 and Ru-based perovskites) can be strongly altered by changing the method of their preparation or changing the material they are grown over. There are several works related to the preparation of RuO2 over various carbon materials such as mesoporous carbon,12 activated carbon,5 graphene10,13–17 or carbon nanotubes.18 However, extra improvement is still required because almost all of the previous RuO2-based carbon supercapacitors are limited to operate in either highly acidic or basic electrolytes. This is in order to harvest the benefits of the high capacitance value of RuO2 in spite of the fact that such electrolytes are not targets for the next generation of supercapacitors due to environmental issues. Additionally, some of the previous methods require long preparation times, sophisticated preparation methods, the use of hazardous materials or further annealing steps. Therefore, it is preferred to construct a supercapacitor based on Ru-based materials, using a neutral electrolyte as well as a preparation method based on a “green” and simple (e.g. one-step) method. This can be achieved by tuning the structure and/or morphology of Ru-based materials through changing its preparation method, so that for instance nanoparticulate materials can be obtained. Wang et al.9 reported the preparation of Ru-based materials with smaller particle size decreasing the diffusion resistance, facilitating the proton transport in the bulk of RuO2 and increasing the specific capacitance as well as the storage efficiency.
In this work, we introduce the synthesis of Ru/RGO via a one-step, green and efficient method and its application as a supercapacitor electrode in a neutral electrolyte. The current method is based on the simultaneous reduction of a Ru precursor and GO in a single step and without using any reducing agent and under pH control. The mass percentage of the Ru precursor does not exceed 25% of the total weight of the material in order to minimize the cost of the capacitor. The proposed preparation method is accomplished without any annealing or calcination process. This is advantageous in reducing the energy consumption during the preparation step.
2. Experimental
2.1 One-step preparation of Ru–RGO
Graphene oxide was prepared by a modification of Hummer's method, following the same procedure as reported previously.19 Briefly, 5 g of high purity graphite was cured with H2SO4, P2O5 and K2S2O8 in order to prepare the per-oxidized graphite. This is followed by stirring 5 g of dried per-oxidized graphite with 115 mL of concentrated H2SO4 in an ice bath for about 10 minutes, then 15 g of KMnO4 was gradually added and stirring was continued for two more hours. The mixture acquired a bright yellow color after its dilution by water and treatment with H2O2. The bright yellow suspension was filtered and washed with 1:10 HCl solution, afterwards it was dried overnight at 80 °C in an oven. Ru/RGO was prepared by adding 0.2 g of GO to 0.069 g of RuCl3 and dissolved in distilled water followed by ultrasonication of the mixture for two hours. Ammonia solution was used to adjust the pH of the suspension to (2.0, 4.0 and 8.0), then it was placed in a conventional microwave that was operated at 720 W using 30 s cycles (20 s: on and 10 s: off) until the ignition process takes place; the total reaction time is 30 minutes. During the irradiation, the suspension becomes viscous with time and dehydrated. The resulting powder was ignited and a strong firing takes place, resulting in a black powder that indicates the successful reduction of RGO. For comparison, the chemically reduced graphene oxide (RGO) was also prepared following the procedure mentioned in ref. 20.
2.2 Structural, spectral and surface analysis
The prepared materials were characterized using X-ray diffraction (XRD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), field-emission scanning electron microscope (FESEM) with energy-dispersive analysis by X-ray (EDX) and high-resolution transmission electron microscope (HR-TEM), the XRD was recorded by Panlytical X'Pert using Cu-Kα radiation (λ = 1.540 Å), the Raman spectra were obtained using Witec alpha 300R confocal Raman microscope with a 532 nm Nd:Yag laser. XPS measurements were performed with a Perkin Elmer 5300 XPS system with a non-monochromatised Mg-Kα X-ray source. Calibration was performed using the C 1s component (binding energy is 284.6 eV). An Mg-Kα X-ray was used with 300 W applied to the anode. The surface morphological analysis was performed using HR-TEM (Tecnai G20, FEI, Netherland, 200 kV, LaB6 Gun), FESEM and EDX (JEOL JSM-6360LA and Philips XL30).2.3 Electrode fabrication and electrochemical measurements
An amount of 20 mg of the prepared Runano-based RGO was sonicated with 1 mL of dimethyl formamide (DMF) for about 30 minutes in order to prepare a homogeneous suspension of 20 mg mL−1 Ru–RGO suspension. By the same way we also prepared 20 mg mL−1 of RGO (sonicated until a homogeneous suspension obtained). The electrodes used for the electrochemical capacitance measurements were prepared by adding 5 μL of 2% Nafion solution on the surface of well-polished mirror-like Ni electrodes of diameter 0.6 cm (surface area 0.283 cm2) and connected with a copper rod for electrical conductivity. Then the Ni/Nafion electrode was dried at 80 °C followed by casting the active material (Runano-based RGO or RGO) and left to dry in an oven in order to obtain Ni/N/Runano-based RGO and Ni/N/RGO. The masses of the electroactive materials were measured as 0.25 mg for both Runano-based RGO and RGO.The electrochemical measurements were performed in a conventional three-electrode system using Ag/AgCl (4.0 M KCl) as reference electrode and a Pt wire as counter electrode. Cyclic voltammetry and galvanostatic charging/discharging experiments were performed using a Voltalab PGZ301 potentiostat. Electrochemical impedance spectroscopy (EIS) was performed using the same instrument at OCP from 100 mHz to 100 kHz with an AC amplitude of 10 mV. Cyclic voltammetry curves were recorded from Ei = 0.0 V to Ef = 0.7, 0.8, 0.9, 1.0 and 1.1 V at scan rates ranging from 5.0 to 200 mV s−1. Galvanostatic charging/discharging curves were recorded using chronopotentiometry at various current densities ranging from 1.0 A g−1 to 26 A g−1 and limiting the potential between 0.0 V to 0.9 V. The specific capacitance was calculated from cyclic voltammetry using eqn (1) and from galvanostatic charging–discharging data using eqn (2). Eqn (3) and (4) are used to calculate the energy and the power densities, respectively.
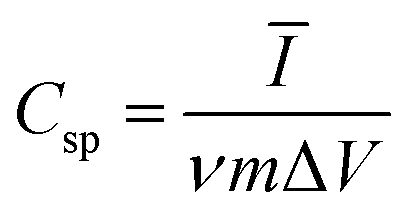 | (1) |
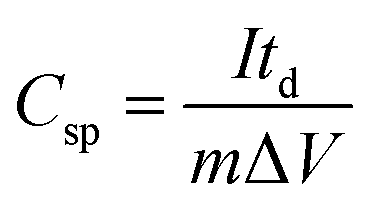 | (2) |
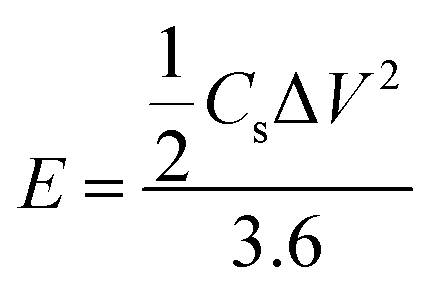 | (3) |
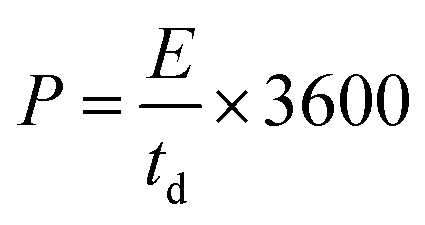 | (4) |
3. Results and discussion
By changing the preparation method one is able to form particles of different morphologies, sizes and of different structures as well. This should additionally alter the electric and electrochemical properties of the final hybrid. Hence, in the current study we investigated the effect of microwave irradiation on the precursors GO and RuCl3 in the absence of any reducing, stabilizing or precipitating agents. The microwave irradiation was used to provide a simultaneous reduction of both GO and Ru3+, and the structure of the obtained material was investigated. Microwave chemistry is the science that uses microwave irradiation to boost chemical reactions.21 There are two main heating mechanisms during the microwave irradiation; dipolar polarization and ionic conduction. Generally, microwave heats any polar molecules such as water or the conducting ions in a solvent or in a solid. Dipolar mechanism includes the reorientation of water molecules under the effect of alternating electric field. The frictions and collisions between molecules during the reorientation as well as the rotation are responsible for the heating effect.21 In the case of ions, the ionic conduction is based on the movement of ions in the solution under the effect of the orientation of the electric field. And since the fluctuation of the electric field is constant, the direction of ions movement is constant through the solution. This will result in an increase in the temperature due to frictions and collisions. So, the semiconducting or conducting samples heat up when their ions lose their electric energy that is generated during the irradiation. This loss in the electric energy is due to the electrical resistance of the materials.22 Microwave chemistry is therefore based on the efficient superheating of the material so that the product may acquire different properties. There is extensive work using microwave irradiation to reduce metal nanoparticles in the presence of a reducing agent such as hydrazine hydrate, urea, sodium borohydride, sugar, sodium citrate, citric acid, etc.22 The use of some of these compounds such as citric acid as a reducing agent is based upon the formation of a complex with metal salts. GO contains in its structure oxygenated functional groups that can form a complex with many metal salts. This complex facilitates the reduction of metal ions into metal nanoparticles as a result of the high density of electrons on the GO surface.3.1 Surface, structural and spectral characterization
:2 ratio.
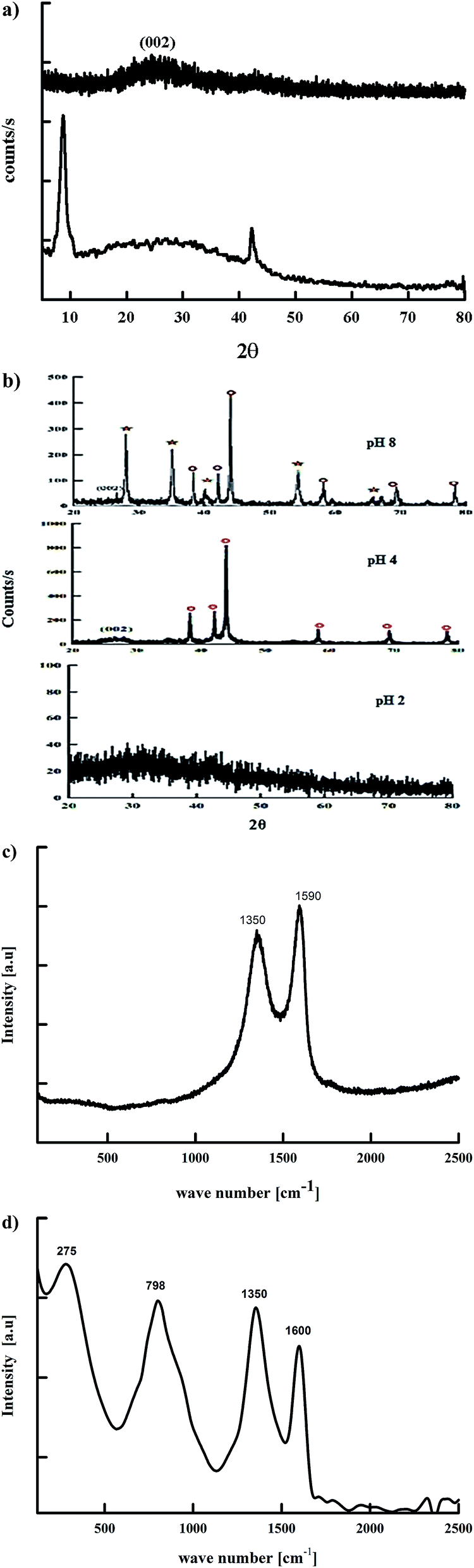 | ||
| Fig. 1 XRD of (a) GO and RGO, (b) Runano-based RGO (prepared at pH 2, 4 and 8) (circles are related to the phases of Ru and stars are related to RuO2), (c) Raman spectra of GO and (d) Raman spectra of Runano-based RGO (pH 8). | ||
It is worth mentioning that this is the first time Runano-based RGO with Ru-rich composition was prepared without any reducing agent. The microwave irradiation provided enough energy to successfully reduce both GO and Ru3+. RuCl3 dissolved in water forms hydroxyl-salts of Ru of the type [Ru(OH)x, Cl3−x] (see eqn (5)). In the presence of high concentrations of NH4(OH), [Ru(OH)x, Cl3−x] leads to the formation of Ru(OH)3. Further oxidation results in the formation of the corresponding oxide according to eqn (5)–(9).13 The major fraction of ruthenium ions are bonded to the surface of graphene and consequently reduced on the RGO surface, forming Ru nanoparticles.
| RuCl3·nH2O + H2O → Ru(OH)xCl3−x + xH+ + xCl− | (5) |
| NH4OH ↔ NH4+ + OH− | (6) |
| Ru(OH)xCl3−x + 3OH− → Ru(OH)3 + xOH− + (3 − x)Cl− | (7) |
| 2Ru(OH)3 + ½O2 + H2O → 2Ru(OH)4 | (8) |
| Ru(OH)4 + (n − 2)H2O → RuO2·nH2O | (9) |
As the pH of the precursors strongly affects the prepared material, we also investigated the influence of pH on the initial solution of hydrated RuCl3 and GO on the obtained composite structure. As shown in Fig. 1b the sample prepared at pH 4.0 contains solely Ru nanoparticles, where decorated RGO is the phase obtained without the formation of RuO2. Wang et al.9 reported that Ru nanoparticle formation is favored at pH values less than 4.9. At pH 4.0 Ru3+ ions are bound to GO sheets via complexation that helps reducing Ru3+ ions into Ru nanoparticles that are strongly attached to RGO sheets as a result of a high electron density on the GO surface. On the other hand, the sample prepared at pH 2.0 does not show any diffraction peaks, indicating an amorphous-like structure. This leads to the conclusion that pH 2.0 is not suitable for preparation of neither crystalline Ru nor RuO2. This is due to the strong acidic solution favoring “free” ionic formation and minimizing the possible reduction of Ru3+ ions to bind “in situ” onto RGO. However, the firing process that occurred during the microwave irradiation of the sample results in the reduction of GO into RGO as indicated by the absence of GO-related peaks. Hence, only samples prepared at pH 4.0 and 8.0 were electrochemically tested as supercapacitor electrodes.
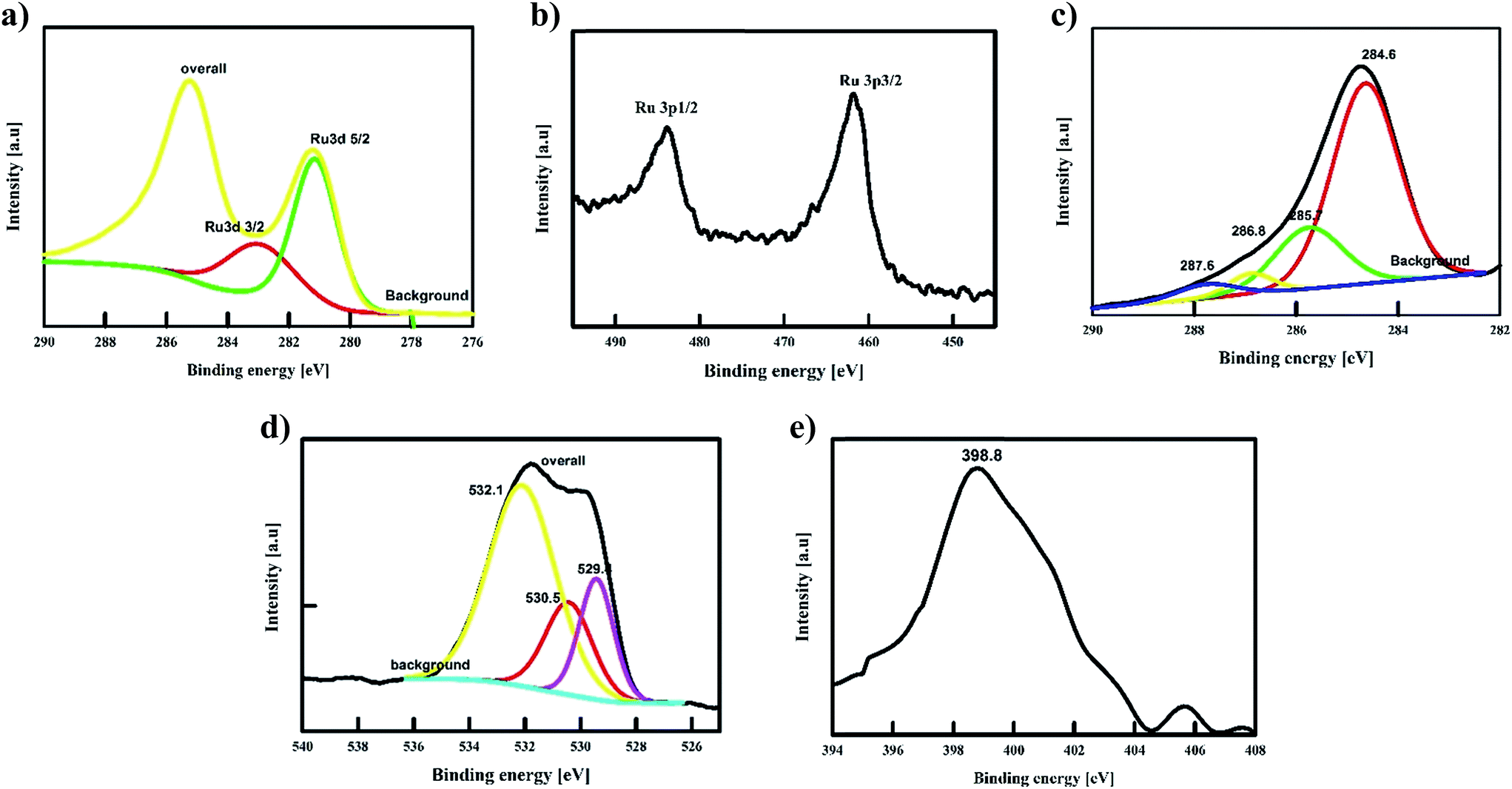 | ||
| Fig. 2 XPS spectra of Runano-based RGO (pH 8) shows the deconvoluted peaks of Ru 2p, Ru 3d, C 1s, O 1s and N 1s. | ||
The de-convolution of C-1s showed four peaks at 287.6, 286.8, 285.7 and 284.6 eV that can be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COO/COH, C–O and CC/C–H. The relative intensity of C–C/CC compared to the oxygenated peaks as well as the corresponding peak area are used to evidence the successful reduction of GO into RGO. As depicted in the C-1s spectra of GO before its reaction (Fig. S1†), the C–C/CC peak to the oxygenated peak ratio is 1:4. While in the case of Runano-based RGO (pH = 8.0), C–C/CC to the oxygenated peaks ratio is relatively higher compared to the case of GO as shown in Fig. 2c. In the latter case, the ratio is about 1.02:1.0 that confirms the successful reduction of GO into RGO. Moreover, the peaks of the O-1s spectra at 232.1 eV and 230.5 eV are assigned to the graphenic oxygen, while the peak at 229.4 eV corresponds to the oxygen of RuO2. Additionally, the presence of nitrogen-doped graphene is confirmed by the N-1s peak at 398.78 eV that corresponds to the N–C bond, indicating the formation of N-doped graphene, which in turn plays a crucial role in separating graphene sheets and preventing their restacking.
O, COO/COH, C–O and CC/C–H. The relative intensity of C–C/CC compared to the oxygenated peaks as well as the corresponding peak area are used to evidence the successful reduction of GO into RGO. As depicted in the C-1s spectra of GO before its reaction (Fig. S1†), the C–C/CC peak to the oxygenated peak ratio is 1:4. While in the case of Runano-based RGO (pH = 8.0), C–C/CC to the oxygenated peaks ratio is relatively higher compared to the case of GO as shown in Fig. 2c. In the latter case, the ratio is about 1.02:1.0 that confirms the successful reduction of GO into RGO. Moreover, the peaks of the O-1s spectra at 232.1 eV and 230.5 eV are assigned to the graphenic oxygen, while the peak at 229.4 eV corresponds to the oxygen of RuO2. Additionally, the presence of nitrogen-doped graphene is confirmed by the N-1s peak at 398.78 eV that corresponds to the N–C bond, indicating the formation of N-doped graphene, which in turn plays a crucial role in separating graphene sheets and preventing their restacking.
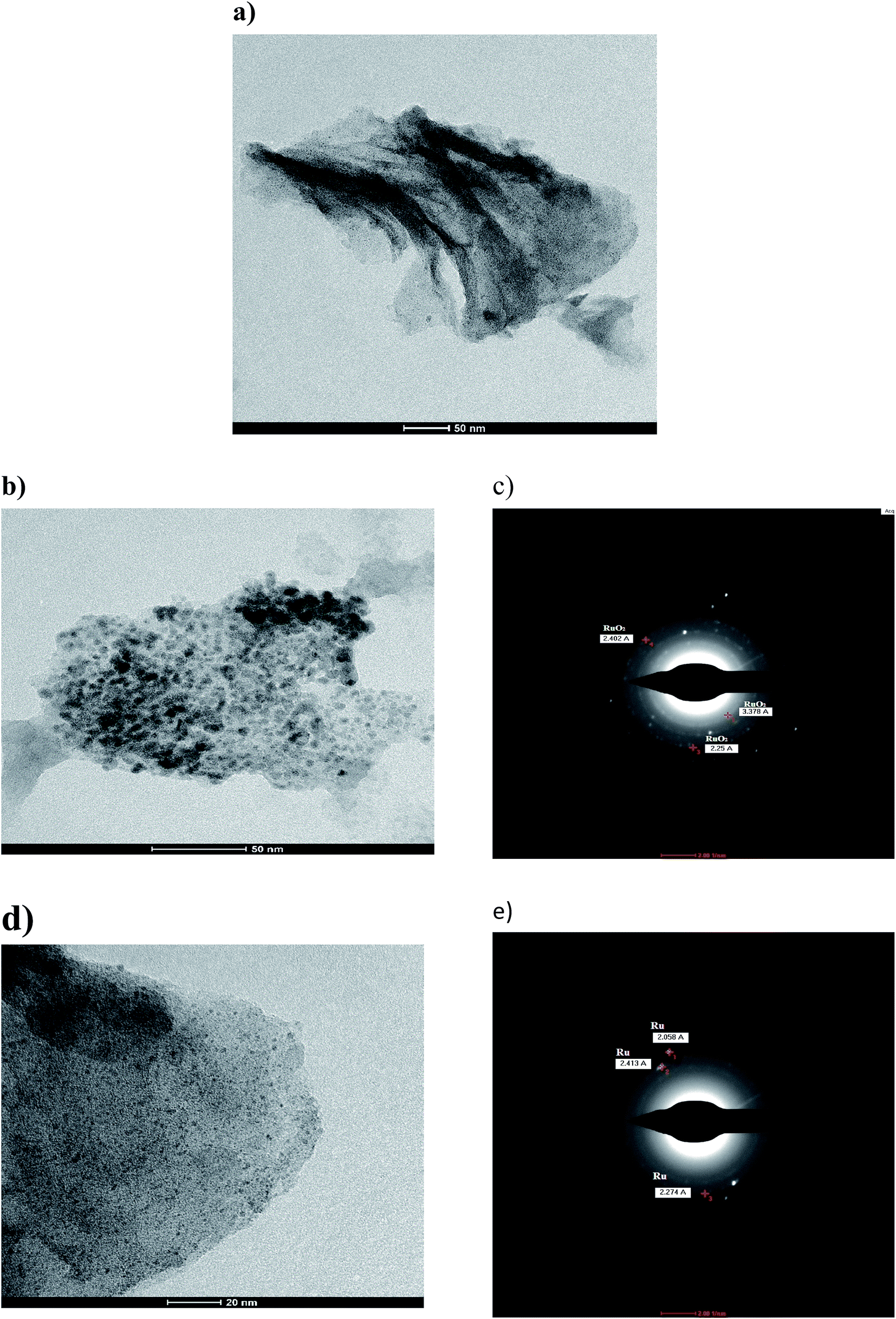 | ||
| Fig. 3 (a) TEM of Runano-based RGO (pH 8) (b) zoom in TEM of Runano-based RGO (pH 8) showing Ru nanoparticles and (c) its corresponding SEAD pattern, (d) zoom in TEM of Runano-based RGO (pH 8) showing RuO2 nanoparticles and (e) its corresponding SEAD pattern. | ||
3.2 Electrochemical characterization
The electrochemical behavior of Ru-based graphene as supercapacitor electrode depends on the main phase within the structure.10,13–17 The main phase formed is RuO2 with the possible formation of some Ru nanostructures as minor phase.10,13–17 As it was anticipated in this work, Runano has the highest percentage in hybrid Runano-based RGO when being prepared at pH 8.0 and is the only phase at pH 4.0.Often H2SO and KOH are used as electrolytes in supercapacitors and pseudocapacitors measurements. Relatively high concentrations of these electrolytes are recommended in order to decrease the equivalent series resistance (ERS). While higher power densities are expected, the corrosive nature of the concentrated acid or base results in the deterioration of current collectors and their leakage will be detrimental, especially on decreasing the cycling stability of the capacitor. Limitation of the operating potential windows in acidic13,28 and basic11,29 electrolytes is another drawback.
RuO2-based supercapacitors have been reported to operate in highly concentrated H2SO4 30,31 and many researchers suggested that RuO2-based supercapacitors are suitable only in such electrolytes.13,28 A high pseudocapacitance was obtained due to a surface reaction between Ru ions and protons (H+) in acidic media according to eqn (10):
| RuOa(OH)b + σH+ + σe− ↔ RuOa−σ(OH)b+σ | (10) |
Using a neutral electrolyte not only solves the corrosion problem but also allows the supercapacitor to operate at a wider potential window.30
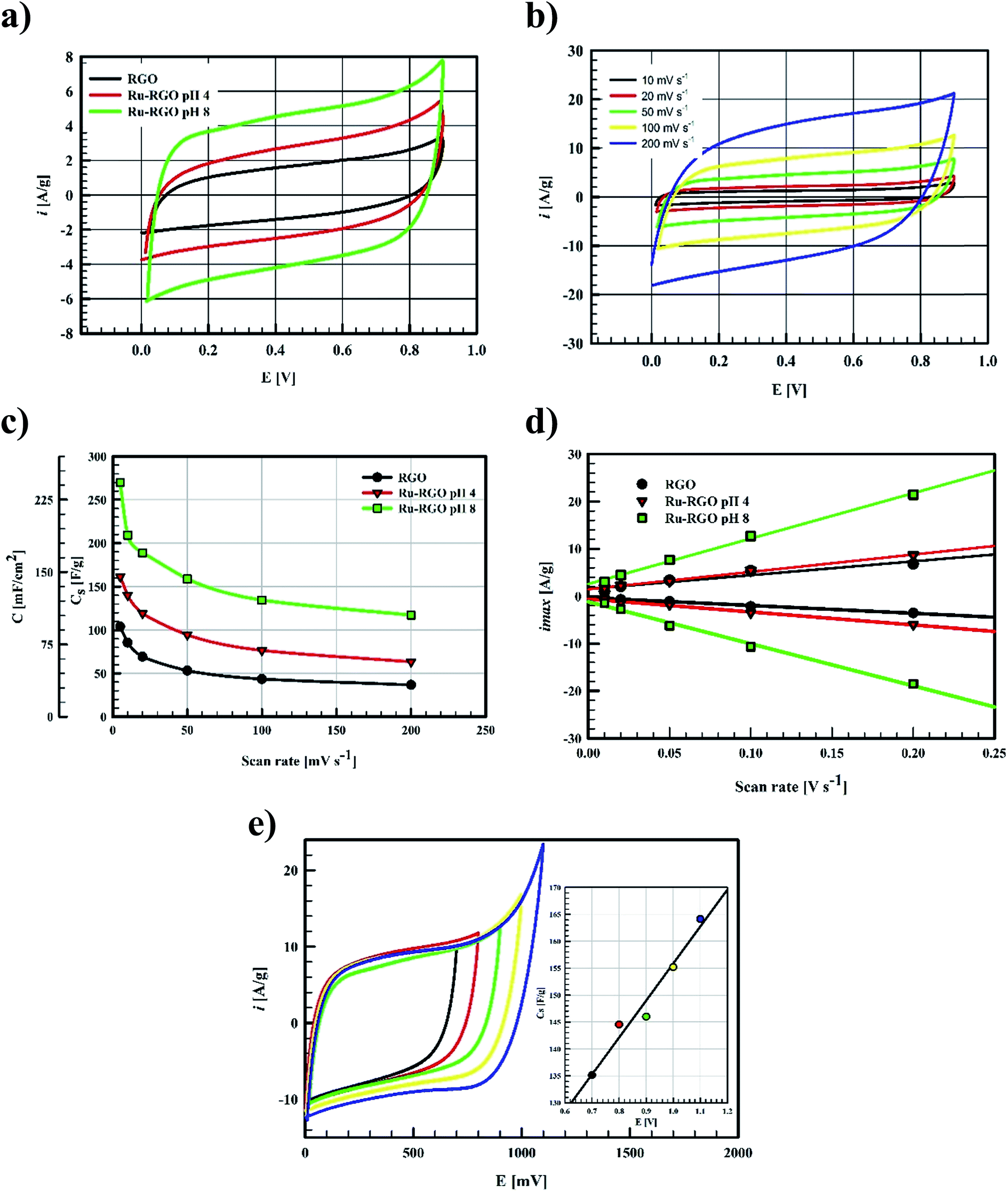 | ||
| Fig. 4 (a) CV of RGO (black line), Runano-based RGO (pH 4) (red line) and Runano-based RGO (pH 8) (green line) measured at scan rate 50 mV s−1 in 1 M NaNO3, (b) CVs of Ru–RGO (pH 8) at various SR from 5 mV s−1 to 200 mV s−1, (c) effect of scan rate on specific capacitance of RGO (black line), Runano-based RGO (pH 4) (red line) and Runano-based RGO (pH 8) (green line), (d) the effect of scan rate on the maximum current density obtained at charging and discharging process and (e) CVs of Runano-based RGO (pH 8) obtained at various upper potential and the relation between the upper potential and specific capacitance (inset). | ||
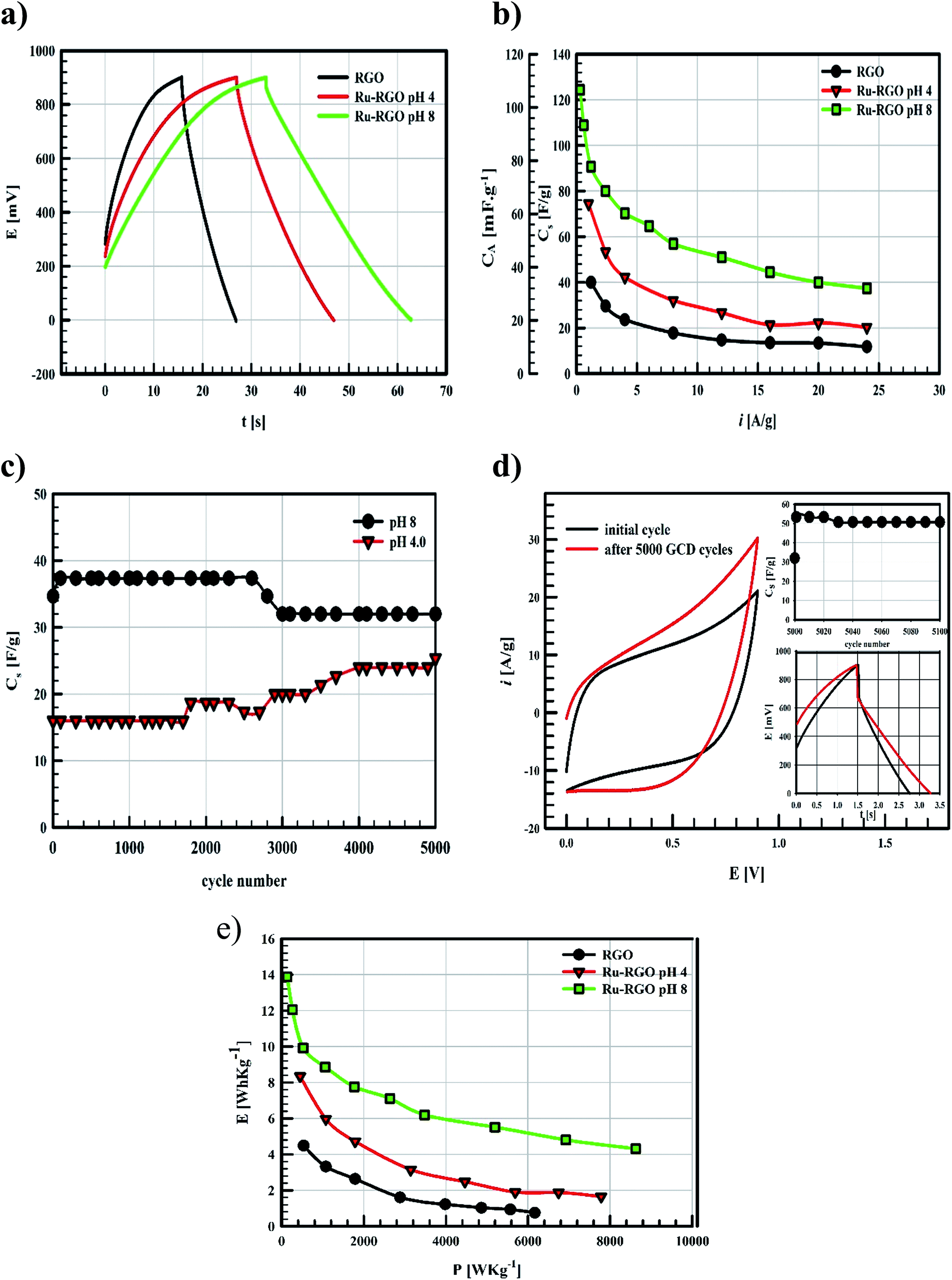 | ||
| Fig. 5 (a) GCD curves of RGO (black line), Runano-based RGO (pH 4) (red line) and Runano-based RGO (pH 8) (green line) measured at current density 24 A g−1 in 1.0 M NaNO3, (b) the relation between the applied current density and the measured specific capacitance, (c) cycling stability of and Runano-based RGO (pH 8 and 4) measured at current density 24 A g−1 for 5000 cycles (d) the initially recorded CV compared to after performing 5000 GCD cycles at 24 A g−1 followed by its storage overnight in the electrolyte solution. The inset is GCD at 24 A g−1 and the measured specific capacitance recorded for another 100 cycles after the storage process. And (e) Ragon plot of RGO (black line), Runano-based RGO (pH 4) (red line) and Runano-based RGO (pH 8) (green line). | ||
Ragon plots in Fig. 5e are used to compare the performances of the current surfaces, showing the relation between power density in W kg−1 and the corresponding energy density in W h kg−1. As shown in Fig. 5e, for a given power density the corresponding energy density is highest when using Runano-based RGO (prepared at pH 8.0). This also shows an energy density of 7.7 W h kg−1 at 1766 W kg−1, holding an energy density of 4.3 W h kg−1 at a higher power density (8624 W kg−1). The energy density decreases at high power densities due to the fast voltage decay at high power densities.31
The current supercapacitor shows higher specific capacitance and cyclic stability compared to that reported by Antiohos et al.32 at 90% MnO-10% GO (42.5 ± 9.2 mF cm−2) at scan rate 50 mV s−1, in 1.0 M NaNO3. Li et al.6 reported 211.5 F g−1 at 2.0 mV s−1 in 1.0 M Na2SO4. Further, He et al. used Ru (20%)/mesoporous carbon and reported 269 F g−1 at 2.0 mV s−1 in 6.0 M KOH,11 while Xia et al. reported 238.6 F g−1 at scan rate 50 mV s−1 in 6.0 M KOH over RGO/CoMoO4 material.33 Lou et al.34 synthesized Ru/porous activated carbon (derived from Moringa Oleifera fruit shells) composites using the bio-waste carbon source and ruthenium acetylacetonate as the co-feeding metal precursors (Ru/MOC). Ru/MOC provided a specific capacitance of 291 F g−1 (the area of the electrode is 1.0 cm2) at scan rate 10 mV s−1 in a potential range 0.0 to 800 mV in 1.0 M H2SO4 and showed 90% of its original Cs after only 2000 cycles. Finally, Takikawa and coworkers reported 153 F g−1 at 0.125 A g−1 using RuO2/activated carbon.12 The values reported in this work for Csp are comparable to those measured earlier,18 using RuO2 (30.4%)/p-MWCNT in 1.0 M H2SO4 (231 F g−1 at a scan rate of 20 mV s−1). Others reported the use of RGO/SnO2-CNT, resulting in 218 F g−1 at scan rate 200 mV s−1 in 30 wt% KOH (81% of initial capacitance remains after 6000 cycles).35 Although using RuO2–RGO as electrode material14 shows higher specific capacitance (that may be due to the use of 1.0 M H2SO4 as electrolyte), the electrode material used in the present study (Runano-based RGO) shows higher cycling stability. And by taking into account the very small area of the used electrode (0.283 cm2), its use in capacitor applications can be considered as a good candidate for a commercial, green and easily-prepared supercapacitor.
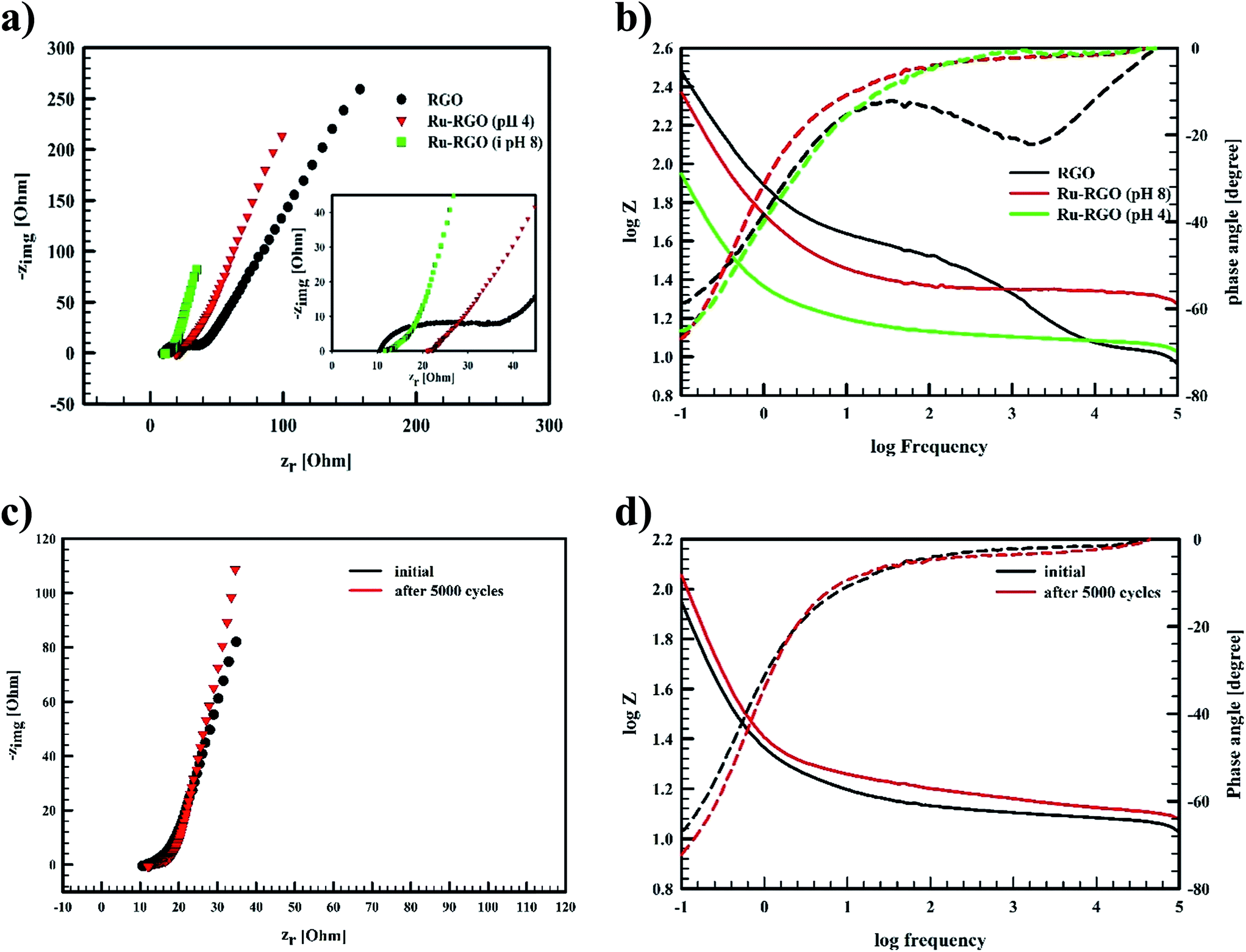 | ||
| Fig. 6 (a) Nyquist plot of RGO (black line), Runano-based RGO (pH 4) (red line) and Runano-based RGO (pH 8) (green line), (b) Bode plot of RGO (black line), Runano-based RGO (pH 4) (red line) and Runano-based RGO (pH 8) (green line), (c) the initial Nyquist plot of Runano-based RGO (pH 8) (black) and after 5000 cycles (red) at 24 A g−1, and (d) the corresponding Bode plot. | ||
The Bode plot in Fig. 6b reveals that Runano-based RGO (prepared at pH 8.0) has a phase angle φ of 70° at a frequency of 100 mHz, compared to 65° for Runano-based RGO (prepared at pH 4) and 54° for RGO. The value of φ = 70° is close to the ideal capacitor at this frequency. Additionally, the time constants (τ) for Runano-based RGO (prepared at pH 8.0) can be calculated from the Bode plot, which is the time necessary to discharge the capacitor to 36.8% and it can be directly calculated by:
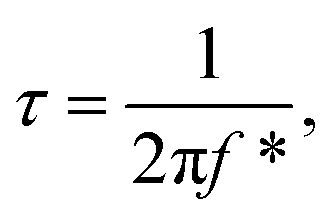 | (11) |
It is established that the lower the time constant, the faster the charge/discharge characteristics. The calculated value of τ for Runano-based RGO (prepared at pH 8) is 0.357 s. This small value indicates fast charge/discharge properties of the current material. From the calculated time constant, the value of the maximum energy density can be deduced from the following relations:
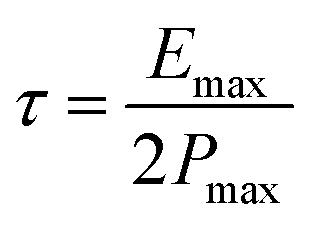 | (12) |
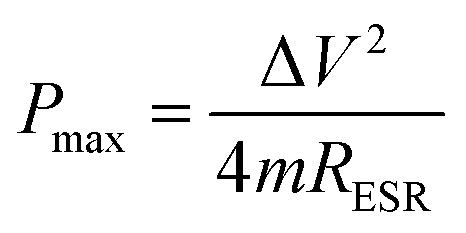 | (13) |
The calculated Emax and Pmax of Runano-based RGO (prepared at pH 8) are 15 W h kg−1 and 76.4 kW kg−1, respectively.
EIS of Runano-based RGO (pH 8.0) was also measured after applying 5000 cycles at 24 A g−1 and the results were compared with the initial EIS measurement shown in Fig. 6c and d. We observe no significant change in the solution resistance after 5000 GCD cycles. However, the line at low frequency region became slightly more vertical after 5000 GCD cycles compared to the initial one. Moreover, there was no significant change in the phase angle as shown in the Bode plot (Fig. 6d).
4. Conclusion
Runano-based material supported on RGO was successfully prepared by a one-pot, fast, green and pH-controlled synthesis method with the aid of microwave irradiation and in the absence of any other reducing or stabilizing agent. No calcination steps were necessary to obtain crystalline Ru-nanoparticles. The results show that Runano-based RGO (prepared at pH 8.0) possesses the highest electrochemical capacitance compared to the one prepared at pH 4.0. Synthesis at pH 2.0 was not suitable to reduce Ru3+ ions, and consequently did not show a good capacitive behavior. All measurements were carried out in a neutral electrolyte (1.0 M NaNO3). Runano-based RGO (prepared at pH 8.0) showed a specific capacitance of 270 F g−1 at scan rate of 5.0 mV s−1 (238.5 mF cm−2, surface area 0.283 cm2). Runano–RGO systems prepared at pH 8 reveal a very good cycle stability even after 5000 cycles, where it showed a Csp retention 92.5% that increased to 146% when the electrode stored in the electrolyte solution overnight. Runano–RGO (prepared at pH 8.0) showed a maximum power density of 76.4 kW kg−1 with a maximum energy density of 15.0 W h kg−1.Acknowledgements
The authors would like to acknowledge the partial financial support from Cairo University through the Vice President Office for Research Funds.References
- Y. Cao, B. Lin, Y. Sun, H. Yang and X. Zhang, J. Alloys Compd., 2015, 638, 204–213 CrossRef CAS.
- Z. B. Zhou, M. Benbouzid, J. F. Charpentier, F. Scuiller and T. H. Tang, Renewable Sustainable Energy Rev., 2013, 18, 390–400 CrossRef CAS.
- J. W. Leea, J. M. Kob and J.-D. Kima, Electrochim. Acta, 2012, 85, 459–466 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- N. Lin, J. Tian, Z. Shan, K. Chen and W. Liao, Electrochim. Acta, 2013, 99, 219–224 CrossRef CAS.
- Z. Li, J. Wanga, S. Liu, X. Liu and S. Yang, J. Power Sources, 2011, 196, 8160–8165 CrossRef CAS.
- C. G. Liu, Z. N. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS PubMed.
- P. Wang, H. Liu, Y. Xu, Y. Chen, J. Yang and Q. Tan, Electrochim. Acta, 2016, 194, 211–218 CrossRef CAS.
- J. Y. Hwang, M. F. El-Kady, Y. Wang, L. Wang, Y. Shao, K. Marsh, J. M. Ko and R. B. Kaner, Nano Energy, 2015, 18, 57–70 CrossRef CAS.
- X. He, K. Xie, R. Li and M. Wu, Mater. Lett., 2014, 115, 96–99 CrossRef CAS.
- X. J. He, Y. J. Geng, S. Oke, K. Higashi, M. Yamamoto and H. Takikawa, Synth. Met., 2009, 159, 7–12 CrossRef CAS.
- X. Leng, J. Zou, X. Xiong and H. He, J. Alloys Compd., 2015, 653, 577–584 CrossRef CAS.
- L. Na, J. Tian, Z. Shan, C. Kuan and W. Liao, Electrochim. Acta, 2013, 99, 219–224 CrossRef.
- R. Rakhi, W. Chen, D. Cha and H. Alshareef, J. Mater. Chem., 2011, 21, 16197–16204 RSC.
- N. Soin, S. S. Roy, S. K. Mitra, T. Thundat and J. A. McLaughlin, J. Mater. Chem., 2012, 22, 14944–14950 RSC.
- L. Deng, J. Wang, G. Zhu, L. Kang, Z. Hao, Z. Lei, Z. Yang and Z.-H. Liu, J. Power Sources, 2014, 248, 407–415 CrossRef CAS.
- X. Liu, T. A. Huber, M. C. Kopac and P. G. Pickup, Electrochim. Acta, 2009, 54, 7141–7147 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- N. F Atta, H. K. Hassan and A. Galal, Anal. Bioanal. Chem., 2014, 406, 6933–6942 CrossRef PubMed.
- M. J. Collins Jr, Future Med. Chem., 2010, 2, 151–155 CrossRef PubMed.
- Y.-J. Zhu and F. Chen, Chem. Rev., 2014, 114, 6462–6555 CrossRef CAS PubMed.
- R. C. Rocha, M. G. Brown, C. H. Londergan, J. C. Salsman, C. P. Kubiak and A. P. Shreve, J. Phys. Chem. A, 2005, 109, 9006–9012 CrossRef CAS PubMed.
- L. M. M. Tejada, A. Muñoz, M. A. Centeno and J. A. Odriozola, J. Raman Spectrosc., 2016, 47, 189–197 CrossRef.
- R.-R. Bi, X.-L. Wu, F.-F. Cao, L.-Y. Jiang, Y.-G. Guo and L.-J. Wan, J. Phys. Chem. C, 2010, 114, 2448–2451 CAS.
- J. Y. Shen, A. Adnot and S. Kaliaguine, Appl. Surf. Sci., 1991, 51, 47–60 CrossRef CAS.
- J. C. Fuggle, T. E. Madey, M. Steinkilberg and D. Menzel, Surf. Sci., 1975, 52, 521–541 CrossRef CAS.
- M. Y. Ho, P. S. Khiew, D. Isa, T. K. Tan, W. S. Chiu and C. H. Chia, Nano, 2014, 9(6), 1430002–1430027 CrossRef CAS.
- K. Seung and Y. B.-H. Kim, Electrochim. Acta, 2015, 186, 337–344 CrossRef.
- P. K. Shen, C.-Y. Wang, S. P. Jiang, X. Sun and J. Zhan, Electrochemical Energy, advanced materials and technology, CRC Press, 2016 Search PubMed.
- P. Tamilarasan and S. Ramaprabhu, Energy, 2013, 51, 374–381 CrossRef CAS.
- D. Antiohos, K. Pingmuang, M. S. Romano, S. Beirne, T. Romeoa, P. Aitchison, A. Minett, G. Wallace, S. Phanichphant and J. Chen, Electrochim. Acta, 2013, 101, 99–108 CrossRef CAS.
- X. Xia, W. Lei, Q. Hao, W. Wang and X. Wang, Electrochim. Acta, 2013, 99, 253–261 CrossRef CAS.
- B.-S. Lou, P. Veerakumar, S.-M. Chen, V. Veeramani, R. Madhu and S.-B. Liu, Sci. Rep., 2016, 6, 19949, DOI:10.1038/srep19949 . PMID: 26818461.
- R. B. Rakhi and H. N. Alshareef, J. Power Sources, 2011, 196, 8858–8865 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27415c |
| This journal is © The Royal Society of Chemistry 2017 |