
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis and antitubercular evaluation of benzothiazinones containing an oximido or amino nitrogen heterocycle moiety†
Rui Zhang‡
a,
Kai Lv‡a,
Bin Wangb,
Linhu Lia,
Bo Wanga,
Mingliang Liu *a,
Huiyuan Guoa,
Apeng Wanga and
Yu Lu*b
*a,
Huiyuan Guoa,
Apeng Wanga and
Yu Lu*b
aInstitute of Medicinal Biotechnology, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China. E-mail: lmllyx@126.com; Tel: +86-10-63030965
bBeijing Key Laboratory of Drug Resistance Tuberculosis Research, Department of Pharmacology, Beijing Tuberculosis and Thoracic Tumor Research Institute, Beijing Chest Hospital, Capital Medical University, Beijing 101149, China. E-mail: luyu4876@hotmail.com; Tel: +86-10-89509357
First published on 5th January 2017
Abstract
A series of 8-nitro-6-(trifluoromethyl)-1,3-benzothiazin-4-ones (BTZs) bearing an oximido or amino nitrogen heterocycle moiety through modifications at the C-2 position of BTZ043 and BPTZ169 were designed and synthesized as new antitubercular agents. Many of the target compounds demonstrate excellent in vitro activity (MIC: <0.016–0.088 μg mL−1) against the drug susceptive H37Rv strain and two clinically isolated multidrug-resistant Mycobacterium tuberculosis (MTB) strains. Compound 10a displays acceptable safety, aqueous solubility and pharmacokinetic properties, opening up a new possibility for further development.
Tuberculosis (TB), one of the world's major causes of illness and death, is caused mainly by Mycobacterium tuberculosis (MTB).1 The World Health Organization (WHO) estimated that approximately one third of the world population is infected with MTB, and 9.6 million people were infected and 1.5 million died from TB worldwide in 2014.2 In particular, the spread of multidrug-resistant (MDR) TB and the emergence of extensively drug-resistant (XDR) TB have revitalized drug discovery efforts in search of novel drugs recently.3–5 It is encouraging that bedaquiline and delamanid have been approved for the treatment of MDR-TB, after a huge gap of over 40 years.6,7 However, there are only two new chemical entities, Q203 and TBA-354, in phase 1 clinical trials at present. Therefore, it is urgent to develop new anti-TB drugs.8,9
8-Nitro-6-(trifluoromethyl)-1,3-benzothiazin-4-ones (BTZs), a novel class of TB agents targeting DprE1,10–12 were reported to have potent activity in multiple models.13 BTZ043 (Fig. 1) and PBTZ169 (Fig. 1) are being studied preclinically. Compared with BTZ043, PBTZ169 is slightly more potent and not being stereoselective which makes it easier and cheaper to synthesize.14 The structure–activity relationship (SAR) studies of BTZs show that –CF3 and –NO2 are the optimal groups at the C-6 and C-8 positions, respectively, so there is only one possible structural modifications at the C-2 position.
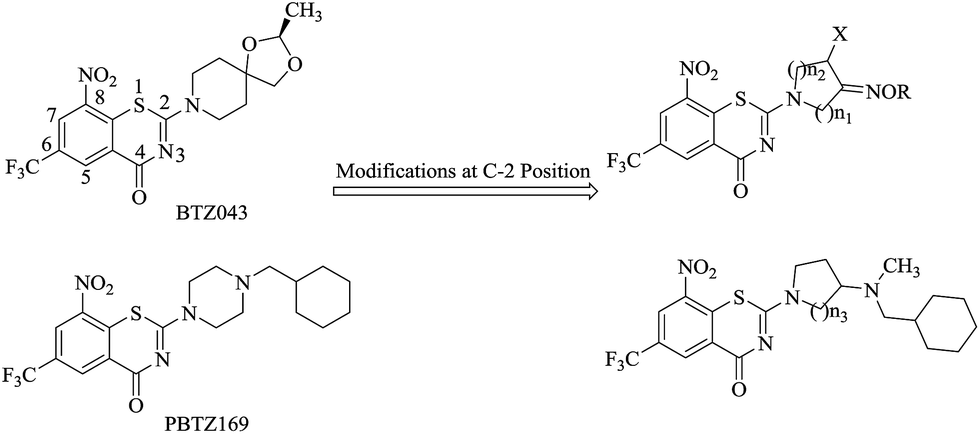 | ||
| Fig. 1 Design of the new molecules. | ||
On the other hand, some of fluoroquinolones (FQs), a class of important second-line anti-TB drugs, such as ciprofloxacin, ofloxacin and levofloxacin, are frequently used for management of TB including MDR-TB.15 Pyrrolidinyl, piperazinyl and piperidinyl are the most common groups at the C-7 position of FQs. It is interesting that recently the discovery of gemifloxacin and IMB-070593 highlights the importance of oxime-functionalized nitrogen heterocycles with respect to antibacterial activity.16–18
Inspired by the above research results, we intended to replace the spiroketal moiety of BTZ043 with nitrogen cycloketone oximes and shift the terminal nitrogen on the piperazine ring of BPTZ169 outside the ring (Fig. 1), which were expected to explore SAR of BTZs though variations on the sizes of the heterocycle and the alkyl group of the oxime. Thus, a series of novel BTZ derivatives containing an oximido or amino nitrogen heterocycle moiety were designed and synthesized in this study. Our primary objective was to identify alternative groups at the C-2 position of BTZs with potent antimycobacterial activity and facilitate the further development of BTZs.
Detailed synthetic pathways to novel BTZ derivatives 10a–i and 11a, 11b are outlined in Scheme 1. 4-7-Membered nitrogen cycloketones 1a–i were smoothly converted to the oximes 2a by reaction with O-alkylhydroxyamines in ethanol at room temperature. The N-Boc protecting group on 2a–i was removed with trifluoroacetic acid (TFA) in dichloromethane to afford the nitrogen cycloketone oximes 3a–i in 40–65% yield.
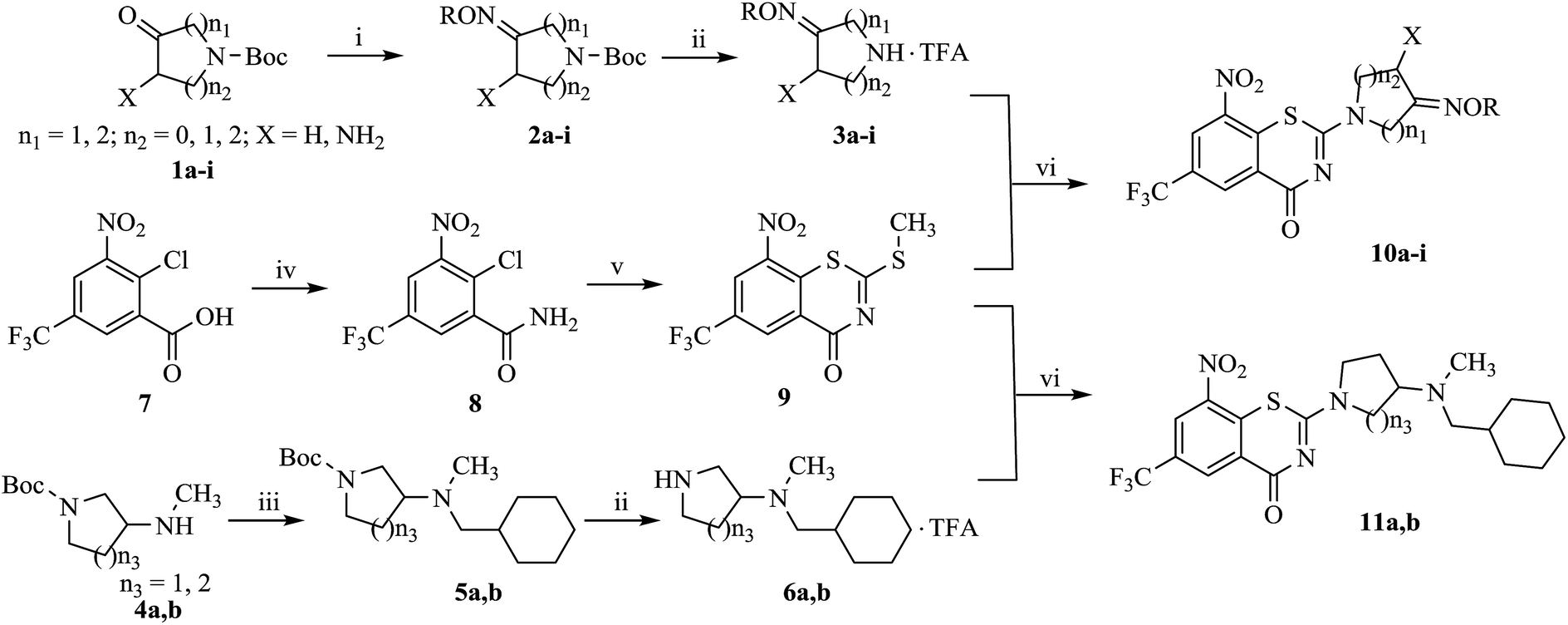 | ||
| Scheme 1 Synthesis of target compounds 10a–i and 11a, 11b. Reagents and conditions: (i) RONH2·HCl, K2CO3, C2H5OH, rt, 40–65%; (ii) TFA, CH2Cl2, rt, 50–52%; (iii) c-C6H11CH2Br, K2CO3, DMF, 80 °C, 50–52%; (iv) SOCl2, NH3·H2O, reflux, 76%; (v) CS2, CH3I, DMSO, rt, 41%; (vi) (C2H5)3N, C2H5OH, 60 °C, 45–59%. | ||
Compounds 6a, 6b were easily prepared from the corresponding tert-butyl 3-(methylamino) pyrrolidine/piperidine-1-carboxylate 4a, 4b via nucleophilic substitution reaction with (bromomethyl)cyclohexane in the presence of K2CO3 in dimethyl formamide (DMF) at 80 °C, and then the resulting cyclohexylmethylates 2a, 2b were treated with TFA.
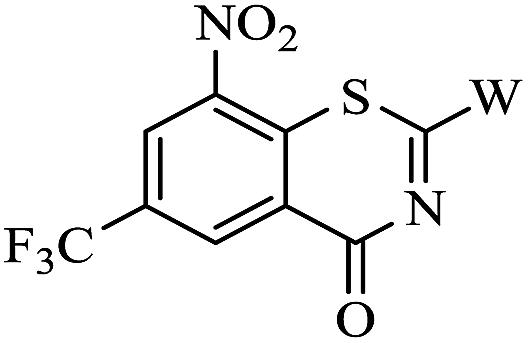
Amidation of 2-chloro-3-nitro-5-(trifluoromethyl) benzoic acid 7 gave amide 8, which was condensated with carbon disulfide and methyl iodide yielded BTZ core compound 9.10 Derivatives 10a–i and 11a, 11b were conveniently obtained from 9 by nucleophilic substitution with side chain compounds 3a–i and 6a, 6b, respectively.
The synthesized compounds 10a–i and 11a, 11b were preliminarily screened for in vitro activity against MTB H37Rv ATCC 27294 strain, using the Microplate Alamar Blue Assay (MABA).19 The minimum inhibitory concentration (MIC) is defined as the lowest concentration effecting a reduction in fluorescence of >90% relative to the mean of replicate bacterium-only controls. The minimum inhibitory concentration (MIC) values of the compounds along with PBTZ169, isoniazid (INH) and rifampicin (RFP) for comparison are presented in Table 1.
|
||||
|---|---|---|---|---|
| Compd | W | MIC (μg mL−1) | CC50b (μg mL−1) | SIc |
| a MTB H37Rv ATCC 2729 was acquired from ATCC.b CC50: 50% cytotoxic concentration.c SI: selectivity index for MTB H37Rv ATCC 27294, CC50/MIC; INH: isoniazid: RFP: rifampicin. | ||||
| 10a | 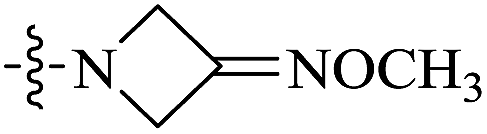 |
0.030 | 91.37 ± 35.18 | 3046 |
| 10b | 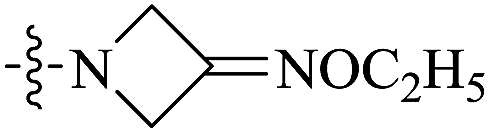 |
<0.016 | 707.33 ± 133.77 | >44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 208 208 |
| 10c | 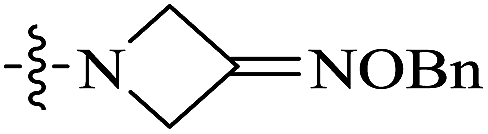 |
<0.016 | 157.74 ± 66.57 | >9858 |
| 10d | 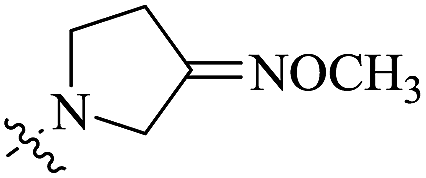 |
0.116 | 57.66 ± 11.34 | 497 |
| 10e | 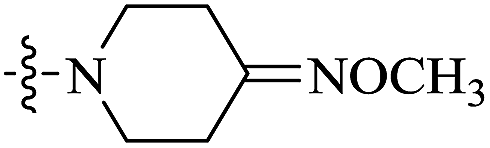 |
0.029 | 102.07 ± 7.13 | 3520 |
| 10f | 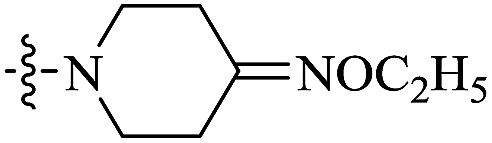 |
0.232 | 191.29 ± 33.94 | 825 |
| 10g | 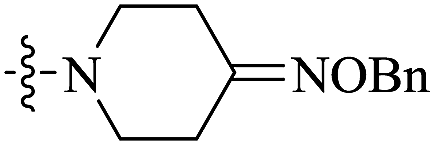 |
<0.016 | 440.69 ± 102.73 | >27543 |
| 10h | 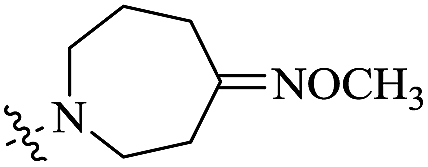 |
0.060 | 58.30 ± 3.78 | 972 |
| 10i |  |
0.436 | 241.49 ± 47.5 | 554 |
| 11a | 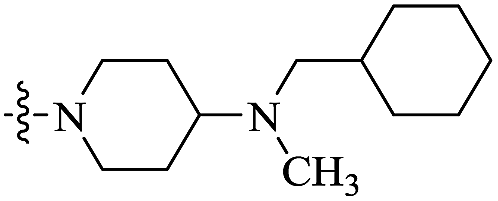 |
0.051 | 530.97 ± 180.70 | 10411 |
| 11b | 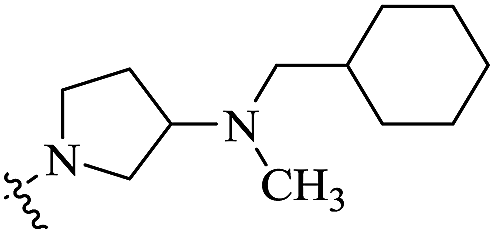 |
0.088 | 67.46 ± 19.96 | 7667 |
| PBTZ169 |  |
0.116 | 784.20 ± 185.8 | 6760 |
| INH | 0.050 | |||
| RFP | 0.049 | |||
The data reveal that with a few exceptions (10d, 10f, 10i), all of the BTZ derivatives have potent in vitro activity against this strain (MIC: <0.1 μg mL−1), which is better than BPTZ169 (MIC: 0.116 μg mL−1). In particular, the most active compounds 10b, 10c and 10g (MIC: <0.016 μg mL−1) were found to be >3–>7 fold more potent than BPTZ169, INH and RFP (MIC: 0.049–0.116 μg mL−1). The potency of the oxime BTZ derivatives (10a–h) in this study is related to both of the sizes of the heterocycle and alkyl group (R). Generally, 4- and 6-membered heterocycles show the best activity, followed by 7- and 5-membered ones with the same R group (CH3) successively (10a vs. 10d vs. 10e vs. 10h). Moreover, the contribution of the alkyl groups of the oxime moiety to the activity is relevant to the heterocycles. The activity of the R groups is as follows: benzyl = ethyl > methyl for azetidyl-based BTZs (10a vs. 10b vs. 10c), and but benzyl > methyl ![[right double angle bracket]](https://www.rsc.org/images/entities/char_27eb.gif) ethyl for piperidinyl-based ones (10e vs. 10f vs. 10g). Further investigation also suggests that introduction of an amino group on the heterocycles is detrimental. For instance, 3-amino-4-(methoxyimino)piperidin-1-yl derivative (10i) displays MIC value of 0.436 μg mL−1 which is 15-fold less potent than 10e (MIC: 0.029 μg mL−1).
ethyl for piperidinyl-based ones (10e vs. 10f vs. 10g). Further investigation also suggests that introduction of an amino group on the heterocycles is detrimental. For instance, 3-amino-4-(methoxyimino)piperidin-1-yl derivative (10i) displays MIC value of 0.436 μg mL−1 which is 15-fold less potent than 10e (MIC: 0.029 μg mL−1).
On the other hand, when the nitrogen atom of the piperazine ring of PBTZ169 was converted to an exocyclic tertiary amino group, the resulting compound 11a demonstrates increased activity (MIC: 0.051 μg mL−1). It is clear that the piperidin-1-yl group (11a) could be displaced by a pyrrolidin-1-yl moiety (11b) without obviously affecting the potency. These results indicate that 4-aminopiperidine and 3-aminopyrrolidine rings are preferred over piperazine.
The compounds 10a–i and 11a, 11b were examined for toxicity (CC50) in human lung adenoma A549 cell lines by MTT assay and the results are reported in Table 1. All of them (CC50: 57.66–707.33 μg mL−1) are more cytotoxic than PBTZ169 (CC50: 784.20 μg mL−1), but the selectivity index (SI: 7667–>44208) of compounds 10b, 10c, 10g, 11a and 11b are bigger than PBTZ169 (SI: 6760) for MTB H37Rv ATCC 27294.
Encouraged by their strong potency against the drug-sensitive MTB H37Rv strain, these BTZ derivatives except compounds 10d, 10f and 10i were further evaluated against two clinical isolated MDR-MTB strains 16892 (resistant to both of INH and RFP) and 16802 (resistant to INH, RFP, streptomycin, ethambutol and levofloxacin) (Table 2). The results indicate that all of 10a–c, 10e, 10g, 10h and 11a, 11b have excellent activity against both of the two strains with similar MIC values (0.016–0.031 μg mL−1) to PBTZ169 (MIC: 0.016 μg mL−1), suggesting their promising potential for both drug-sensitive (MIC: <0.1 μg mL−1) and resistant MTB strains (Tables 1 and 2).
| Compd | MIC (μg mL−1) | Water Solubility (mg mL−1) | Metabolic stability t1/2 (min) | |
|---|---|---|---|---|
| MDR-MTB 16892b | MDR-MTB 16802b | |||
| a NT: not tested.b MDR-MTB 16892 and MDR-MTB 16802 were isolated from patients in Beijing Chest Hospital. | ||||
| 10a | 0.016 | 0.031 | 0.24 | >120 |
| 10b | 0.016 | 0.016 | 0.03 | NT |
| 10c | 0.016 | 0.016 | 0.05 | NT |
| 10e | 0.016 | 0.016 | 0.02 | NT |
| 10g | 0.016 | 0.016 | 0.05 | NT |
| 10h | 0.031 | 0.016 | 0.03 | NT |
| 11a | 0.016 | 0.016 | 4.05 | 49.1 |
| 11b | 0.016 | 0.016 | 3.11 | 24.3 |
| PBTZ169 | 0.016 | 0.016 | 0.30 | 19.5 |
| INH | >40 | 2.5 | NT | NT |
| RFP | >40 | 20 | NT | NT |
The BTZ derivatives were initially evaluated for their water solubility which was determined by HPLC measurement of the concentration of a micromembrane filtered saturated solution.18 Compared with other oxime derivatives (0.02–0.05 mg mL−1, clogP: 3.28–4.98), compound 10a (clogP: 2.83) has much greater solubility (0.24 mg mL−1) which is only slightly smaller than PBTZ169 (0.30 mg mL−1). As expected, exocyclic tertiary amino derivatives 11a, 11b possess excellent aqueous solubility (3.11–4.05 mg mL−1) which is more than ten times that of BPTZ169 (Table 2). Moreover, all of 10a, 11a and 11b show better metabolic stability (t1/2: 24.3–>120 min) in human liver microsomes compared to BPTZ169 (t1/2: 19.5 min).
Based on the measured activity levels against the tested strains, solubility and metabolic stability, 10a, 11a and 11b were further tested for in vivo pharmacokinetic (PK) profiles in mice after a single oral administration of 50 mg kg−1. As shown in Table 3, compound 10a, with a 3-(methoxyimino) azetidine moiety, displays good PK properties, with Cmax of 5767 ng mL−1, AUC0–∞ of 7678 h ng mL−1 and t1/2 of 1.15 h. Compounds 11a and 11b have a relatively longer t1/2 of 1.69–1.76 h and MRT of 2.42–2.92 h, but a poor Cmax of 173–327 ng mL−1 and AUC0–∞ of 627–646 h ng mL−1. Unexpectedly, all of the three compounds have a shorter t1/2 of 1.15–1.76 h compared to BPTZ169 (t1/2: 3.31 h). The in vitro and in vivo half-times of inconsistency may be due to differences in species between human liver microsomes and mice.
| a Male CD-1 mice was acquired from WuXi AppTec (Shanghai) CO., Ltd; ND: not determined (parameters not determined due to inadequately defined terminal elimination phase). | ||||
|---|---|---|---|---|
| Compd | 10a | 11a | 11b | PBTZ169 |
| Cmax (ng mL−1) | 5767 ± 3190 | 173 ± 76.8 | 327 ± 137 | 1512 ± 696 |
| Tmax (h) | 0.333 ± 0.144 | 1.000 ± 0.866 | 0.250 ± 0 | 0.583 ± 0.382 |
| AUC0–∞ (h ng mL−1) | 7678 ± 4395 | 627 ± ND | 646 ± 460 | 5681 ± 1756 |
| t1/2 (h) | 1.15 ± 0.540 | 1.76 ± ND | 1.69 ± 0.483 | 3.31 ± 0.187 |
| MRT (h) | 1.85 ± 0.634 | 2.92 ± ND | 2.42 ± 0.171 | 4.48 ± 0.484 |
Conclusions
A series of novel BTZ derivatives bearing an oximido or amino nitrogen heterocycle moiety were designed as new antitubercular agents through modifications at the C-2 position of BTZ043 and BPTZ169. Many of them exhibit excellent in vitro inhibitory activity against both drug-sensitive MTB strain H37Rv and drug-resistant clinical isolates. Compound 10a displays acceptable safety, aqueous solubility and PK properties, and it may serve as a new and promising lead compound for further antitubercular drug discovery. Studies to determine the in vivo efficacy of 10a are currently underway.Ethical statement
Animals were maintained in accordance with the guidelines of the Chinese Association for Laboratory Animal Sciences, Beijing, China, and approved by the Institutional Ethical Committee (IEC) of Peking Union Medical College.Acknowledgements
This work is supported by the National S&T Major Special Project on Major New Drug Innovations (2014ZX09507009-003, 2015ZX09102007-008), NSFC (81502923, 81373267, 21502237) and Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201304).Notes and references
- A. Nusrath Unissa, L. E. Hanna and S. Swaminathan, Chem. Biol. Drug Des., 2016, 87, 537–550 CAS.
- L. Anderson, A. Dean and D. Falzon, et al., Global tuberculosis report, World Health Organization, Geneva, 20th edn, 2015 Search PubMed.
- D. Schraufnagel and J. Abubaker, JAMA, J. Am. Med. Assoc., 2000, 283, 54–55 CrossRef CAS PubMed.
- D. Jones, Nat. Rev. Drug Discovery, 2013, 12, 175–176 CrossRef CAS PubMed.
- D. T. Hoagland, J. Liu, R. B. Lee and R. E. Lee, Adv. Drug Delivery Rev., 2016, 102, 55–72 CrossRef CAS PubMed.
- J. Cohen, Science, 2013, 339, 130 CrossRef CAS PubMed.
- N. J. Ryan and J. H. Lo, Drugs, 2014, 74, 1041–1045 CrossRef CAS PubMed.
- Z. Ma, C. Lienhardt, H. McIlleron, A. J. Nunn and X. Wang, Lancet, 2010, 375, 2100–2109 CrossRef.
- B. Villemagne, C. Crauste, M. Flipo, A. R. Baulard, B. Deprez and N. Willand, Eur. J. Med. Chem., 2012, 51, 1–16 CrossRef CAS PubMed.
- J. Neres, F. Pojer, E. Molteni, L. R. Chiarelli, N. Dhar, S. Boy-Rottger, S. Buroni, E. Fullam, G. Degiacomi, A. P. Lucarelli, R. J. Read, G. Zanoni, D. E. Edmondson, E. De Rossi, M. R. Pasca, J. D. McKinney, P. J. Dyson, G. Riccardi, A. Mattevi, S. T. Cole and C. Binda, Sci. Transl. Med., 2012, 4, 150ra121 Search PubMed.
- C. Trefzer, M. Rengifo-Gonzalez, M. J. Hinner, P. Schneider, V. Makarov, S. T. Cole and K. Johnsson, J. Am. Chem. Soc., 2010, 132, 13663–13665 CrossRef CAS PubMed.
- A. L. Ribeiro, G. Degiacomi, F. Ewann, S. Buroni, M. L. Incandela, L. R. Chiarelli, G. Mori, J. Kim, M. Contreras-Dominguez, Y. S. Park, S. J. Han, P. Brodin, G. Valentini, M. Rizzi, G. Riccardi and M. R. Pasca, PLoS One, 2011, 6, e26675 Search PubMed.
- V. Makarov, G. Manina, K. Mikusova, U. Mollmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, S. Buroni, A. P. Lucarelli, A. Milano, E. De Rossi, M. Belanova, A. Bobovska, P. Dianiskova, J. Kordulakova, C. Sala, E. Fullam, P. Schneider, J. D. McKinney, P. Brodin, T. Christophe, S. Waddell, P. Butcher, J. Albrethsen, I. Rosenkrands, R. Brosch, V. Nandi, S. Bharath, S. Gaonkar, R. K. Shandil, V. Balasubramanian, T. Balganesh, S. Tyagi, J. Grosset, G. Riccardi and S. T. Cole, Science, 2009, 324, 801–804 CrossRef CAS PubMed.
- V. Makarov, B. Lechartier, M. Zhang, J. Neres, A. M. van der Sar, S. A. Raadsen, R. C. Hartkoorn, O. B. Ryabova, A. Vocat, L. A. Decosterd, N. Widmer, T. Buclin, W. Bitter, K. Andries, F. Pojer, P. J. Dyson and S. T. Cole, EMBO Mol. Med., 2014, 6, 372–383 CrossRef CAS PubMed.
- J. Huang, M. Wang, B. Wang, Z. Wu, M. Liu, L. Feng, J. Zhang, X. Li, Y. Yang and Y. Lu, Bioorg. Med. Chem. Lett., 2016, 26, 2262–2267 CrossRef CAS PubMed.
- K. Lv, M. L. Liu, L. S. Feng, L. Y. Sun, Y. X. Sun, Z. Q. Wei and H. Q. Guo, Eur. J. Med. Chem., 2012, 47, 619–625 CrossRef CAS PubMed.
- C. Y. Hong, Y. K. Kim, J. H. Chang, S. H. Kim, H. Choi, D. H. Nam, Y. Z. Kim and J. H. Kwak, J. Med. Chem., 1997, 40, 3584–3593 CrossRef CAS PubMed.
- T. Zhang, W. Shen, M. Liu, R. Zhang, M. Wang, L. Li, B. Wang, H. Guo and Y. Lu, Eur. J. Med. Chem., 2015, 104, 73–85 CrossRef CAS PubMed.
- Y. Lu, M. Zheng, B. Wang, L. Fu, W. Zhao, P. Li, J. Xu, H. Zhu, H. Jin, D. Yin, H. Huang, A. M. Upton and Z. Ma, Antimicrob. Agents Chemother., 2011, 55, 5185–5193 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25712g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |