
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A practical iodine-catalyzed oxidative conversion of aldehydes to nitriles†
Chaojie Fang,
Meichao Li*,
Xinquan Hu,
Weimin Mo,
Baoxiang Hu,
Nan Sun,
Liqun Jin and
Zhenlu Shen *
*
College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China. E-mail: zhenlushen@zjut.edu.cn; limc@zjut.edu.cn
First published on 5th January 2017
Abstract
A simple and efficient method for the direct synthesis of nitriles from aldehydes using ammonium acetate as the nitrogen source has been developed. The reactions were performed with iodine as the catalyst and tert-butyl hydroperoxide (TBHP) as the oxidant under mild conditions. A variety of aromatic, heteroaromatic, aliphatic and allylic aldehydes could be converted into their corresponding nitriles in good to excellent yields.
Introduction
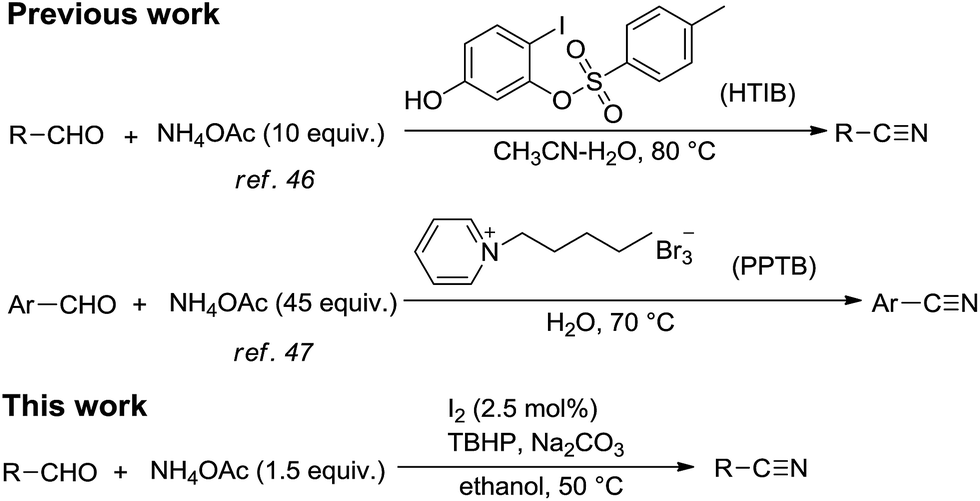
Nitriles are important starting materials for the synthesis of amides,1–3 amines,4,5 ketones,6,7 carbonic acids8,9 and esters.10,11 Furthermore, the cyano group is a key motif in the structure of numerous useful compounds, such as bioactive natural products, pharmaceuticals12,13 and functional materials.14 Traditionally, the Sandmeyer15 and Rosenmund–von Braun reaction16 were widely used in synthesizing aryl nitriles, while highly toxic cyanide salts, were always required and the reactions produced large amounts of inorganic salts as waste. Therefore, the development of new pathways for the synthesis of nitriles has gained increasing attention. In the past decades, a large number of new methods utilizing different kinds of compounds, like alcohols,17–21 amines,22–25 amides26–28 and aldoximes29,30 as starting materials have been reported. These methods offer great improvements, but always need high temperature or pressure, stoichiometric or excess amount of highly reactive oxidants, transition metal catalysts or metal complex catalysts.Aldehydes have been utilized as attractive starting materials for the synthesis of nitriles due to their ready availability, and several methods have been reported with different compounds as nitrogen sources.31–42 Inevitably, there were some drawbacks existing in these methods: (i) high reaction temperature was still needed; (ii) stoichiometric halogen-containing or metal oxidants were always required; (iii) limited substrate scopes; (iv) transition metal catalysts were essential in most cases.
Metal-free catalysis in modern organic synthesis has received much attention because of its economy and low toxicity. The I2/tert-butyl hydroperoxide (TBHP) catalytic oxidation system is one of the most popular examples. TBHP is a kind of mild, efficient and manageable oxidant, giving tert-butanol as the benign by-product in most cases. Recently, the I2/TBHP system has been widely used in oxidative reactions, such as C–C bond cleavage,43 oxidation of indoles,44 oxidative coupling reaction45 and oxidative cyclization,46 etc. Reddy and co-workers47 reported a KI/I2–TBHP system for the conversion of alcohols, aldehydes and amines into nitriles. Under this catalytic system, they prepared a series of aromatic nitriles from aromatic aldehydes by using ammonia as the nitrogen source in water. However, the yields of products were general low and the nitro-substituted substrate cannot be converted into the desired product. What's more, the large excess of ammonia may cause environmental problems as a result of its easy volatility and would be an undesirable contaminant in waste water.
Ammonium acetate (NH4OAc), which has many remarkable advantages, including good stability and low toxicity, is an idea low-cost alternative nitrogen source instead of ammonia. Previously, ammonium acetate as the cyanide source has already been developed in the direct synthesis of nitriles from aldehydes, like NH4OAc/HTIB48 and NH4OAc/PPTB49 systems. One of limitations of these examples is the use of expensive halogen-containing oxidants, which could generate hazardous wastes during work-up. On the other hand, about decade times of NH4OAc had to be employed to ensure the full conversion of aldehydes, which is extremely wasteful.
Inspired by hereinbefore work, as a part of our research program aimed at synthesis of nitriles and I2-catalyzed reaction,50–52 we attempted to develop a simple, efficient, economical and environmentally friendly method for the direct synthesis of nitriles from aldehydes using NH4OAc as the nitrogen source under I2/TBHP system (Scheme 1).
 | ||
| Scheme 1 The improved method for the synthesis of nitriles from aldehydes with NH4OAc as a nitrogen source. | ||
Results and discussion
At the outset, benzaldehyde (1a) was chosen as the model substrate to examine a variety of reaction condition parameters and the results are summarized in Table 1. Firstly, a series of solvents were screened and the results indicated 1a could be transformed to benzonitrile (1b) in 86% yield in ethanol within 8 h at 60 °C (entry 4). As is known to all, water is an ideal solvent. We have also tried to verify the feasibility of using water as the solvent. Unfortunately, the easy formation of amide in aqueous medium resulted in a low yield of nitrile (35%) (entry 8). Then 1.5 equiv. of K2CO3 as acid-binding agent was tentatively added into the reaction, and 1a was almost completely converted into 1b in 2.5 h (entry 9). Some other acid-binding agents, such as NaHCO3, NaOAc, Na2CO3 (entries 10–12), were also tested. Gratifyingly, the reaction could be performed more smoothly in the presence of Na2CO3 (entry 12).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | NH4OAc (equiv.) | I2 (mol%) | Solvent | Addictive (equiv.) | T (°C) | t (h) | Yieldb (%) |
| a Reaction conditions: 1a (4 mmol, 0.424 g), 5 mL of solvents, 1.1 equiv. of TBHP.b Yields were determined by GC using an internal standard method. | |||||||
| 1 | 2.0 | 5.0 | CH3CN | — | 60 | 8.0 | 72 |
| 2 | 2.0 | 5.0 | PhCH3 | — | 60 | 8.0 | 14 |
| 3 | 2.0 | 5.0 | PhCl | — | 60 | 8.0 | 25 |
| 4 | 2.0 | 5.0 | C2H5OH | — | 60 | 8.0 | 86 |
| 5 | 2.0 | 5.0 | THF | — | 60 | 8.0 | 25 |
| 6 | 2.0 | 5.0 | DMF | — | 60 | 8.0 | 70 |
| 7 | 2.0 | 5.0 | EtOAc | — | 60 | 8.0 | 37 |
| 8 | 2.0 | 5.0 | H2O | — | 60 | 8.0 | 35 |
| 9 | 2.0 | 5.0 | C2H5OH | K2CO3 (1.5) | 60 | 2.5 | 98 |
| 10 | 2.0 | 5.0 | C2H5OH | NaOAc (1.5) | 60 | 7.0 | 96 |
| 11 | 2.0 | 5.0 | C2H5OH | NaHCO3 (1.5) | 60 | 4.0 | 99 |
| 12 | 2.0 | 5.0 | C2H5OH | Na2CO3 (1.5) | 60 | 1.5 | 99 |
| 13 | 2.0 | 5.0 | C2H5OH | Na2CO3 (1.0) | 60 | 2.5 | 99 |
| 14 | 2.0 | 5.0 | C2H5OH | Na2CO3 (0.5) | 60 | 8.0 | 94 |
| 15 | 2.0 | 2.5 | C2H5OH | Na2CO3 (1.0) | 60 | 5.0 | 99 |
| 16 | 2.0 | 1.0 | C2H5OH | Na2CO3 (1.0) | 60 | 12.0 | 94 |
| 17 | 2.0 | 2.5 | C2H5OH | Na2CO3 (1.0) | 50 | 5.5 | 99 |
| 18 | 2.0 | 2.5 | C2H5OH | Na2CO3 (1.0) | 40 | 11.0 | 94 |
| 19 | 1.5 | 2.5 | C2H5OH | Na2CO3 (1.0) | 50 | 6.0 | 98 |
| 20 | 1.0 | 2.5 | C2H5OH | Na2CO3 (1.0) | 50 | 12.0 | 90 |
When the loading of Na2CO3 was reduced to 1.0 equiv. and 0.5 equiv., the reaction time had to be prolonged to 2.5 h and 8 h, respectively (entries 13 and 14). Thus, we chose to add 1.0 equiv. of Na2CO3 into the reaction system. When the loading of I2 was reduced to 2.5 mol%, 1b could be obtained in 99% yield in 5 h (entry 15). After that, the reaction temperature was decreased and we found that 1b could be obtained in 99% yield at 50 °C in 5.5 h (entries 17 and 18). At last, when the loading of NH4OAc was reduced to 1.5 equiv., 1a could also be converted into 1b successfully with high yield in 6 h (entry 19).
Having defined the optimum conditions for the reaction, the substrate scope of this methodology was investigated and the results are summarized in Scheme 2. Initially, the catalytic system was examined to tolerate a wide range of aromatic aldehydes which bearing electron-withdrawing or electron-donating groups on benzene ring. It was observed that substrates carrying electron-withdrawing groups could be smoothly converted into the desired products in 5 h (2b–9b). Benzaldehydes carrying electron-donating groups, need much more time to complete their transformation (10b–17b). The I2–TBHP catalytic system was also tolerant of substrates containing heteroatoms such as oxygen, sulfur and nitrogen. In the case of 20a and 22a, the conversions could perform smoothly in 5 h (20b and 22b). However, in the case of electron-rich heteroaromatic aldehydes containing N and S atoms, prolonged time was needed for full conversions (19b and 21b). In addition, the representative polycyclic substrate, 1-naphthaldehyde (18a) tolerated our catalytic system to give the product 18b in excellent yield.
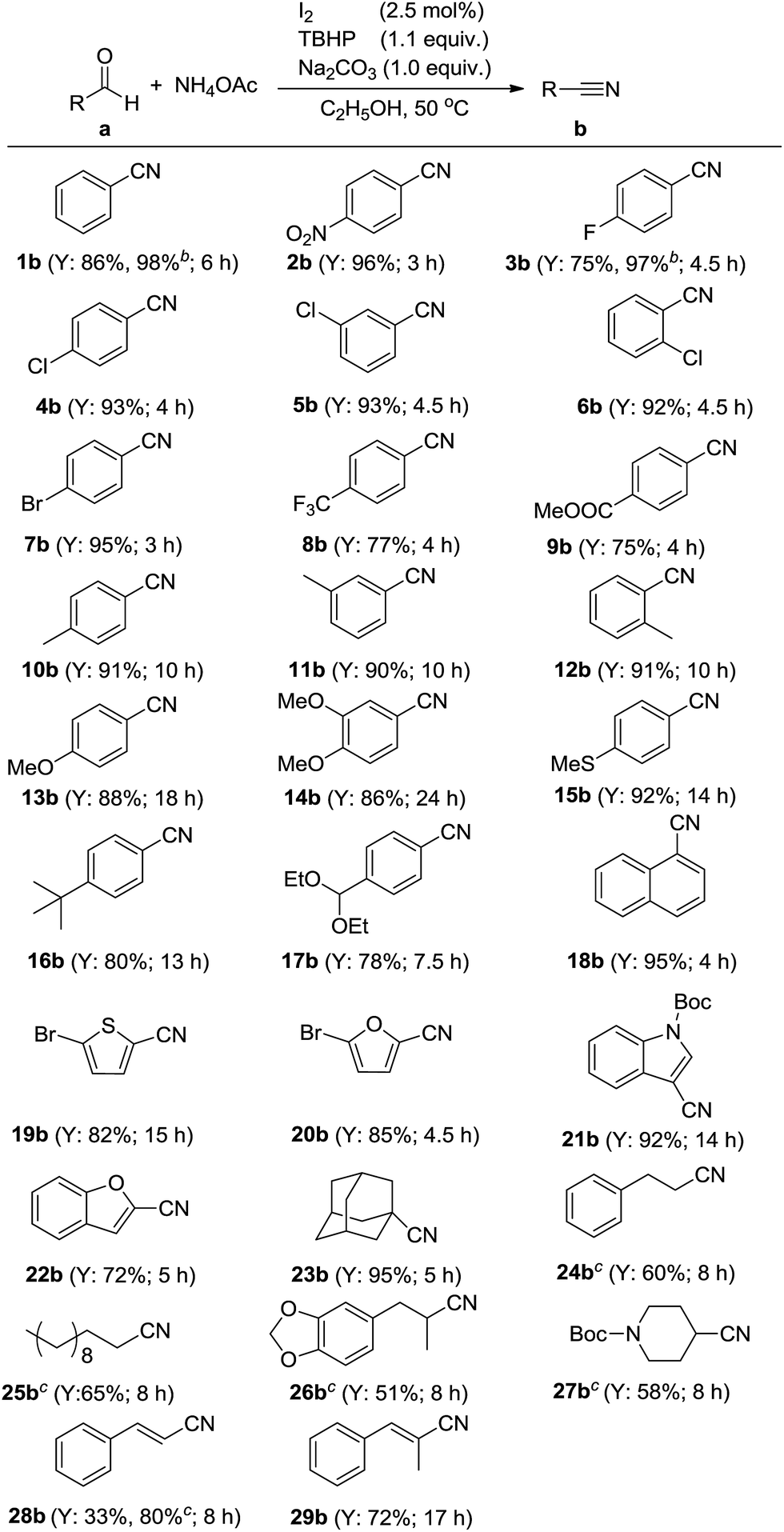 | ||
| Scheme 2 Oxidative conversion of aldehydes to nitrilesa. a Reaction conditions: aldehyde (4 mmol), NH4OAc (1.5 equiv.), I2 (2.5 mol%), TBHP (1.1 equiv.), 5 mL of ethanol, 50 °C. Y = yield of the isolated product. b Yields were determined by GC using an internal standard method. cAldehyde was slowly added into reaction solution. | ||
Adamantane-1-carbaldehyde (23a), an α-saturated aliphatic aldehyde, gave the desired nitrile 23b in 94% yield. As is known to all, aldehydes with α-H atoms can be halogenated at their α positions by reaction with halogens, like Cl2, Br2 and I2. This conclusion was proved by the representative aliphatic aldehyde 3-phenylpropanal (24a). When it was used as the substrate, the yield of 3-phenylpropanenitrile (24b) was very low. However, when aldehyde 24a was slowly added into the reaction solution, 60% yield of 24b could be obtained. This consequence suggested there was a competitive relationship between the halogenated reaction and the desired reaction.
With this encouraging finding in hand, some other α-unsaturated aldehydes, such as undecanal (25a), helional (26a) and N-Boc-piperidin-4-ylformaldehyde (27a) were also examined in this modified method. To our delight, all of them could be converted into the desired nitriles with satisfying yields. At last, two allylic aldehydes, cinnamaldehyde (28a) and (E)-2-methyl-3-phenylacrylaldehyde (29a) also tolerated our catalytic system. Cinnamonitrile (28b) could be obtained in 80% yield under optimized and modified conditions.
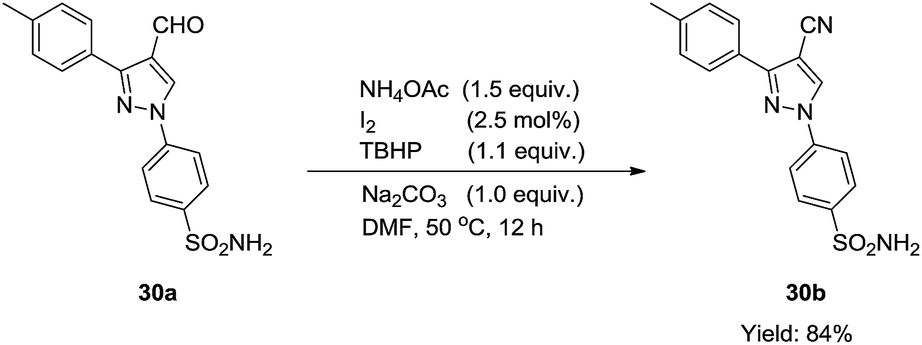
In addition, this catalytic system could also be applied in the synthesis of a complex compound (30b) (eqn (1)), which displays appreciable anti-inflammatory and selective COX-2 inhibitory activity.51 Because of the poor solubility of 30a in ethanol, equal volume of DMF was chosen as the solvent in the transformation of 30a to 30b. To our delight, the reaction was smoothly performed and 84% yield of 30b was obtained.
 | (1) |
In order to examine the practicability of this catalytic system, gram-scale reaction was also performed under the optimized conditions (14.8 g of 4-bromobenzaldehyde (7a)). It was gratifying that the isolated product 13.5 g of 4-bromobenzonitrile (7b) could be obtained (93% yield).
Next, we envisioned oxidative conversion of primary alcohol to nitrile using our method (eqn (2)). However, the oxidative conversion of benzyl alcohol under the optimized conditions gave only 1b in 58% yield after 24 h although the loading of TBHP was increased to 2.5 equiv. A large amount of benzyl alcohol was remained, which cannot be converted into the desired product.
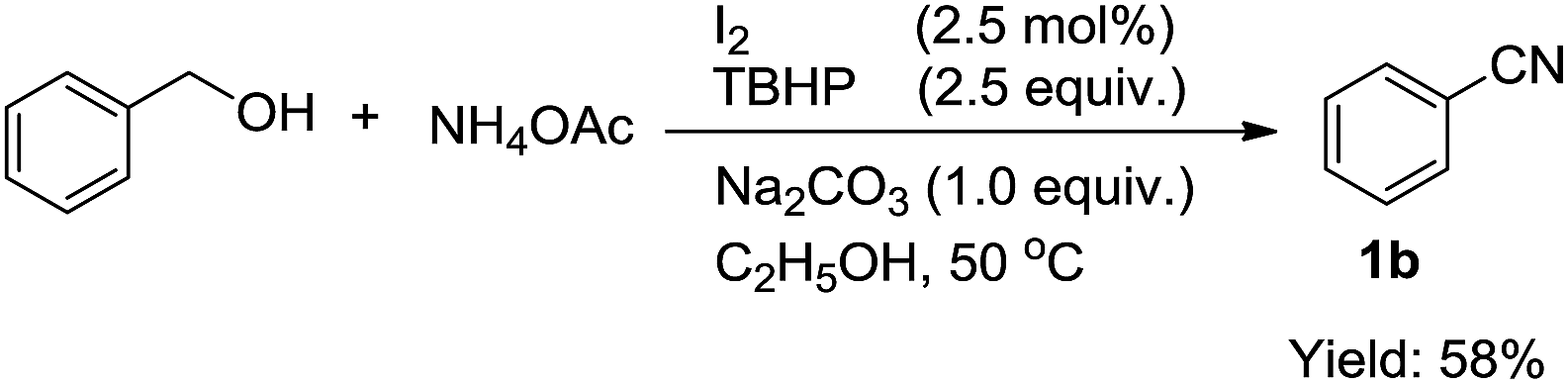 | (2) |
With these results in hand, we tried to explore the plausible reaction mechanism. When 4 mmol of benzaldehyde (1a), 1.5 equiv. of NH4OAc, 1.0 equiv. of Na2CO3 and 5 mL of ethanol were mixed up and stirred under 50 °C for 8 h, phenylmethanimine was achieved in 13% yield determined by GC with the normalization method. Then 2.5 mol% of I2 and 1.1 equiv. of TBHP were added into the reaction mixture, it was founded that benzonitrile (1b) was the sole product after 6 h. These results implied that: (i) phenylmethanimine was the intermediate product; (ii) the process of transformation phenylmethanimine into 1b could promote the generation of phenylmethanimine from 1a.
According to literatures and our observations, a plausible reaction mechanism of this I2/TBHP catalytic system has been proposed in Scheme 3. First of all, aldehyde reacted with ammonium acetate to form aldimine 3 and sodium acetate in the presence of Na2CO3. Then aldimine 3 was oxidized by I2, affording N-iodo aldimine 4 as an intermediate, which was transformed into nitrile 2 by β-elimination of HI under alkaline condition. At the same time, I− was oxidized to I2 by TBHP, which was reduced to tert-butanol immediately.
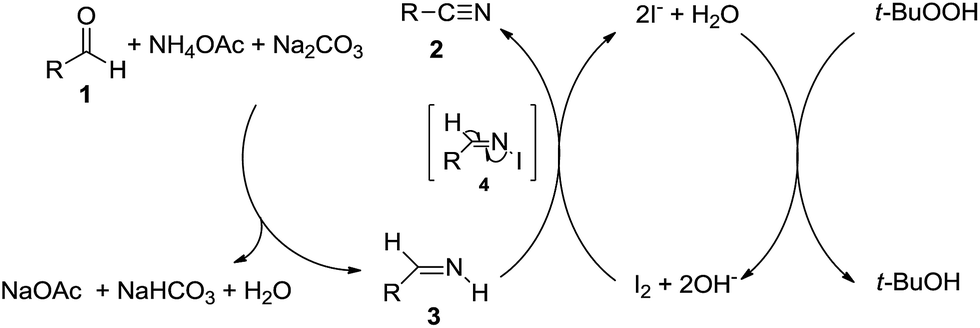 | ||
| Scheme 3 A plausible reaction mechanism. | ||
Conclusion
In conclusion, we have successfully developed a simple and efficient method for the direct synthesis of nitriles from aldehydes with ammonium acetate as the nitrogen source, using I2 as the catalyst and TBHP as the oxidant. This catalytic system is practical, environmentally friendly as it utilizes ethanol as solvent and TBHP as oxidant. A broad range of substrates including aromatic, heteroaromatic, aliphatic and allylic nitriles could be obtained in good to excellent yields.Experimental section
General procedure for the synthesis of 1b–23b, 29b and 30b
To a 20 mL, two-necked, round-bottom flask equipped with a magnetic stirring bar and a thermometer, were added 4 mmol of aldehyde, 0.462 g (6 mmol) of ammonium acetate, 0.424 g (4 mmol) of Na2CO3, 25.4 mg (0.1 mmol) of I2, 0.61 g of TBHP (wt% = 65, 1.1 equiv.) and 5 mL of ethanol (replaced by 5 mL of DMF for 30a). Then the flask was sealed by a bubbler and placed into a preheated oil bath (50 °C). After the reaction was finished (as determined by GC, except 30a which was determined by HPLC), to the reaction mixture were added 10 mL of 5% of Na2S2O3 solution and stirred for 15 min. Then the mixture was transferred into a separation funnel, and 100 mL of diethyl ether (replaced by 100 mL of dichloromethane for 30b) were also added to the separation funnel. The two-phase mixture was washed with saturated sodium bicarbonate solution (25 mL × 2). Then the organic layer was dried with Na2SO4, concentrated on a rotary evaporator and the residue was purified by column chromatography on silica gel using hexanes/EtOAc as eluent to afford the pure product.General procedure for the synthesis of 24b–28b
To a 20 mL, two-necked, round-bottom flask equipped with a magnetic stirring bar and a thermometer, were added 0.462 g (6 mmol) of ammonium acetate, 0.424 g (4 mmol) of Na2CO3, 25.4 mg (0.1 mmol) of I2, 0.61 g of TBHP (wt% = 65%, 1.1 equiv.) and 5 mL of ethanol. Then the flask was sealed by a rubber plug and placed into a preheated oil bath (50 °C). After that, 4 mmol of aldehyde, which was distilled to 1 mL with ethanol, was injected into the solution slowly (4 h). The work-up procedure was as same as that for the synthesis of 1b–24b.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21376224, 21206147), the Natural Science Foundation of Zhejiang Province (LY17B060007), and Hangzhou Qianjiang Distinguished Experts Project.Notes and references
- A. R. Katritzky, B. Pilarski and L. Urogdi, Synthesis, 1989, 949–950 CrossRef CAS.
- J. Borau-Garcia, D. V. Gutsulyak, R. J. Burford and W. E. Piers, Dalton Trans., 2015, 44, 12082–12085 RSC.
- C. Battilocchio, J. M. Hawkins and S. V. Ley, Org. Lett., 2014, 16, 1060–1063 CrossRef CAS PubMed.
- F. Gould, G. Johnson and A. Ferris, J. Org. Chem., 1960, 25, 1658–1660 CrossRef CAS.
- H. Adkins and H. R. Billica, J. Am. Chem. Soc., 2002, 1948(70), 695–698 Search PubMed.
- W. B. Jang, W. S. Shin, J. E. Hong, S. Y. Lee and D. Y. Oh, Synth. Commun., 2006, 27, 3333–3339 CrossRef.
- M. Ortiz-Marciales, L. M. Tirado, R. Colon, M. L. Ufret, R. Figueroa, M. Lebron, M. DeJesus, J. Martinez and T. Malave, Synth. Commun., 1998, 28, 4067–4075 CrossRef CAS.
- D. Brady, N. Dube and R. Petersen, S. Afr. J. Sci., 2006, 102, 339–344 CAS.
- J. Raj, N. Singh, S. Prasad, A. Seth and T. C. Bhalla, Acta Microbiol. Immunol. Hung., 2007, 54, 79–88 CrossRef CAS PubMed.
- T. Kamitanaka, K. Yamamoto, T. Matsuda and T. Harada, Tetrahedron, 2008, 64, 5699–5702 CrossRef CAS.
- M. Noe, A. Perosa and M. Selva, Green Chem., 2013, 15, 2252–2260 RSC.
- D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, K. Ashish, J. G. Robertson, W. Aiying, L. M. Simpkins, T. Prakash and H. Qi, J. Med. Chem., 2005, 48, 5025–5037 CrossRef CAS PubMed.
- E. Pascual, F. Sivera, U. Yasothan and P. Kirkpatrick, Nat. Rev. Drug Discov., 2009, 8, 191–192 CrossRef CAS PubMed.
- H. Jiang, X. Yang, Z. Cui, Y. Liu, H. Li, W. Hu, Y. Liu and D. Zhu, Appl. Phys. Lett., 2007, 91, 123505, DOI:10.1063/1.2784970.
- D. T. Mowry, Chem. Rev., 1948, 42, 189–283 CrossRef CAS PubMed.
- G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779–794 CrossRef CAS.
- S. U. Dighe, D. Chowdhury and S. Batra, Adv. Synth. Catal., 2014, 356, 3892–3896 CrossRef CAS.
- R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123, DOI:10.1038/ncomms5123.
- W. Yin, C. Wang and Y. Huang, Org. Lett., 2013, 15, 1850–1853 CrossRef CAS PubMed.
- D.-W. Tan, J.-B. Xie, Q. Li, H.-X. Li, J.-C. Li, H.-Y. Li and J.-P. Lang, Dalton Trans., 2014, 43, 14061–14071 RSC.
- T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286–6288 CrossRef CAS PubMed.
- L. Gao, H. Tang and Z. Wang, Chem. Commun., 2014, 50, 4085–4088 RSC.
- K. M. Lambert, J. M. Bobbitt, S. A. Eldirany, K. B. Wiberg and W. F. Bailey, Org. Lett., 2014, 16, 6484–6487 CrossRef CAS PubMed.
- A. F. Shoair and A. A. El-Bindary, Spectrochim. Acta, Part A, 2014, 131, 490–496 CrossRef CAS PubMed.
- D. Damodara, R. Arundhathi and P. R. Likhar, Adv. Synth. Catal., 2014, 356, 189–198 CrossRef CAS.
- S. Zhou, D. Addis, S. Das, K. Junge and M. Beller, Chem. Commun., 2009, 4883–4885 RSC.
- S. Elangovan, S. Quintero-Duque, V. Dorcet, T. Roisnel, L. Norel, C. Darcel and J.-B. Sortais, Organometallics, 2015, 34, 4521–4528 CrossRef CAS.
- S. Itagaki, K. Kamata, K. Yamaguchi and N. Mizuno, ChemCatChem, 2013, 5, 1725–1728 CrossRef CAS.
- X. Zhang, J. Sun, Y. Ding and L. Yu, Org. Lett., 2015, 17, 5840–5842 CrossRef CAS PubMed.
- L. Yu, H. Li, X. Zhang, J. Ye, J. Liu, Q. Xu and M. Lautens, Org. Lett., 2014, 16, 1346–1349 CrossRef CAS PubMed.
- M. B. Erman, J. W. Snow and M. J. Williams, Tetrahedron Lett., 2000, 41, 6749–6752 CrossRef CAS.
- S. Talukdar, J. L. Hsu, T. C. Chou and J. M. Fang, Tetrahedron Lett., 2001, 42, 1103–1105 CrossRef CAS.
- H. Sharghi and M. H. Sarvari, ChemInform, 2003, 34, 10323–10328 Search PubMed.
- B. Movassagh and S. Shokri, Tetrahedron Lett., 2005, 46, 6923–6925 CrossRef CAS.
- S. Laulhe, S. S. Gori and M. H. Nantz, J. Org. Chem., 2012, 77, 9334–9337 CrossRef CAS PubMed.
- M. Sridhar, M. K. K. Reddy, V. V. Sairam, J. Raveendra, K. R. Godala, C. Narsaiah, B. C. Ramanaiah and C. S. Reddy, Tetrahedron Lett., 2012, 53, 3421–3424 CrossRef CAS.
- B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2012, 77, 5364–5370 CrossRef CAS PubMed.
- J. Nagarkar, U. Patil and S. Shendage, Synthesis, 2013, 45, 3295–3299 CrossRef.
- H. Veisi, Synthesis, 2010, 2631–2635 CrossRef CAS.
- C. Tao, F. Liu, Y. Zhu, W. Liu and Z. Cao, Org. Biomol. Chem., 2013, 11, 3349–3354 CAS.
- J. N. Noh and J. Kim, J. Org. Chem., 2015, 80, 11624–11628 CrossRef CAS PubMed.
- C. B. Kelly, K. M. Lambert, M. A. Mercadante, J. M. Ovian, W. F. Bailey and N. E. Leadbeater, Angew. Chem., Int. Ed., 2015, 54, 4241–4245 CrossRef CAS PubMed.
- M. Waterman, J. Knight, J. M. Stempak, K. Croitoru, G. C. Nguyen, Z. Cohen, R. S. Mcleod, G. R. Greenberg, A. H. Steinhart and M. S. Silverberg, RSC Adv., 2013, 3, 1311–1316 RSC.
- Y. Zi, Z. J. Cai, S. Y. Wang and S. J. Ji, Org. Lett., 2014, 16, 3094–3097 CrossRef CAS PubMed.
- X. Zhang and L. Wang, Green Chem., 2012, 14, 2141–2145 RSC.
- H. Jiang, H. Huang, H. Cao and C. Qi, Org. Lett., 2010, 12, 5561–5563 CrossRef CAS PubMed.
- K. Rajender Reddy, C. Uma Maheswari, M. Venkateshwar, S. Prashanthi and M. Lakshmi Kantam, Tetrahedron Lett., 2009, 50, 2050–2053 CrossRef CAS.
- Y. Wei, C. Zhu and C. Sun, Synthesis, 2010, 4235–4241 CrossRef.
- G. Bagherzade, A. Zali and A. Shokrolahi, Chin. Chem. Lett., 2015, 26, 603–606 CrossRef CAS.
- C. Fang, M. Li, X. Hu, W. Mo, B. Hu, S. Nan, L. Jin and Z. Shen, Adv. Synth. Catal., 2016, 358, 1157–1163 CrossRef CAS.
- Q. Chen, C. Fang, Z. Shen and M. Li, Electrochem. Commun., 2016, 64, 51–55 CrossRef CAS.
- S. Yi, M. Li, W. Mo, X. Hu, B. Hu, N. Sun, L. Jin and Z. Shen, Tetrahedron Lett., 2016, 57, 1912–1916 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and copies of 1H NMR spectra. See DOI: 10.1039/c6ra26435b |
| This journal is © The Royal Society of Chemistry 2017 |