
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel self-assembled hybrid organogel of polypeptide-based block copolymers with inclusion of polypeptide-functionalized graphene†
Wei-Wei Lei,
Ling-Ying Shi*,
Hang Li,
Chen-Xi Li,
Yong-Fu Diao,
Yu-Lin Zhang and
Rong Ran
College of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, China. E-mail: shilingying@scu.edu.cn
First published on 5th January 2017
Abstract
Self-assembled hybrid organogels of polypeptides containing block copolymers with the inclusion of polypeptide-functionalized graphene were designed and elaborately prepared, and showed interesting microstructures as well as enhanced mechanical performances. Firstly, a series of peptide-based triblock copolymers (triBCPs), poly(γ-benzyl-L-glutamate)-b-poly-(dimethylsiloxane)-b-poly(γ-benzyl-L-glutamate) (PBLG-b-PDMS-b-PBLG, BDB), with different lengths of PBLG helices, were synthesized and characterized. As the length of the PBLG helices increased, the critical gelation concentration of the BDB triBCPs decreased while the gel–sol transition temperature increased. Moreover, the PBLG covalently modified graphene oxide (GO) sheets were successfully incorporated into the BDB organogels in toluene and hybrid organogels were prepared. In the presence of GO sheets, the minimum gelation concentration of the hybrid organogel was slightly lowered, and the hybrid organogels preserved thermoreversibility. In the hybrid gels, the triBCPs still self-assembled into nanoribbon structures and the functionalized GO sheets were well dispersed in the gel medium, which was obviously observed by transmission electron microscopy. In addition, the inclusion of the functionalized graphene greatly enhanced the mechanical performance of the hybrid gels, which was demonstrated by the significant increase of the moduli and the fracture stress of the hybrid gels compared with the corresponding native gels in the rheology experiments.
Introduction
Bioinspired polypeptides and polypeptides containing block copolymers have attracted significant scientific interest in polymer science over the last few decades due to their potential applications in biomedicine and biotechnology fields such as tissue-engineering scaffolds,1–3 drug-delivery carriers, biomedical imaging agents,4 and antibacterial agents.5–10 Poly(γ-benzyl-L-glutamate) (PBLG) is one of the most well studied polypeptides which could be easily synthesized by ring-opening polymerization (ROP) of N-carboxyanhydrides (NCA) of amino acids.11,12 The PBLG-based block copolymers can also be effectively synthesized through ROP method using amino-functionalized macroinitiator.13–15 Moreover, the polypeptide synthesized by ROP in some cases could maintain an amino group at the end of the chain, which provides the possibility of further functionalization.15,16 In addition, the PBLG chains with appropriate polymerization degrees can form the rigid rod-like conformation, i.e. α-helix conformation, stabilized by intramolecular hydrogen bonds,17 which is a vital precondition for the unique phase behavior of the PBLG and PBLG containing block copolymers such as liquid crystalline ordering and thermoreversible gelation.18–20 For instance, Manners and coworkers reported the discovery of thermoreversible gelation of diblock copolymers of PBLG in dilute solution and proposed a self-assembled nanoribbon mechanism.21 Wu and coworkers synthesized PE-b-PBLG diblick copolymer and prepared thermoreversible gels in toluene which represented the first example of polyethylene (PE) based material as a gelator.22The polymer organogels can be further extended to gel composites by the incorporation of the nanofillers which could provide materials with properties that are superior to the individual components or their noninteractive physical mixtures alone.23 The gel composites with advanced functions have widespread applications in materials and biological sciences. For the nanofillers used to generate gel composites, the graphene and their functionalized analogues are one kind of the most attractive candidates.23–25 Graphene oxide (GO) sheets as the derivate of graphene,26–29 a two-dimensional (2D) carbon nanostructure of one atom thickness with plenty of hydrophilic oxygenated groups, have good dispersing ability in water and many polar organic solvents. Moreover, the GO sheets can be easily functionalized through covalent grafting, which brings the opportunities for designing of composite materials based on graphene.30–32 The combination of polymer self-assembled gel and functionalized GO allows the fabrication of a series of novel materials. Many investigations devoted to the GO/polymer hybrid gels have been reported.25,33–35 As a chemical additive, GO can tailor the gelation behavior of other gelators to generate GO-containing hybrid gels. In addition, GO sheets often endow the hybrid supramolecular gel with some particular features from better thermal stability to strengthened mechanical strength. Typically, as a special two-dimensional nanofiller, the GO sheets with remarkable properties can be used to modulate the gelation behaviors as well as the mechanical properties of some polymer gels.23 For example, Jiang et al. reported a hybrid polymeric hydrogel comprised of block copolymer grafted graphene platelets prepared by noncovalent interaction between the cyclodextrins functionalized GO and PNIPAM block copolymers.24 The hybrid graphene inclusion complex showed rapid sol–gel transition at elevated temperature than the corresponding native block copolymers.24 Hao and coworkers used a urea-based gelator synthesized by long alkyl chain and chromophore to produce a hybrid gel with GO, and the hybrid gel was capable of stabilizing GO sheets in apolar solvents. In addition, the mechanical strength, fluorescent emissions as well as thermal stability of the gel were also tuned after capturing GO sheets.35 Recently, we reported a kind of poly(glutamate) covalently modified GO-containing hybrid organgels with enhanced mechanical performance, in which the modified graphene nanostructures acted as nanoscale skeletons and interfacial adhesives in the hybrid gels.16 However, as far as we know, the incorporation of the graphene sheets into the polypeptide containing rod–coil block copolymer self-assembled organogels as well as the influence of the graphene sheets on the phase behavior of the block copolymers in sol and gel state have not been reported.
In this work, a series of rod–coil–rod triblock copolymers, PBLG-b-PDMS-b-PBLG, with different volume fractions of PBLG (fPBLG) was synthesized and the hybrid gels of PBLG-b-PDMS-b-PBLG copolymers with the inclusion of PBLG-functionalized GO sheets were prepared and investigated. The native gels of BDB triblock copolymers with different lengths of PBLG helices exhibited nanoribbon structures and the hybrid gels maintained the self-assembled nanoribbon structure with the 2D graphene sheets well dispersed in the gels were observed, and these hybrid organogels thereof showed enhanced mechanical performances compared to the native BDB triblock copolymer self-assembled gels.
Experimental
Materials
Triphosgene (Aladdin Industrial Inc.), γ-benzyl-L-glutamate (A.R., Tongsheng amino acids Ltd., China), bis(3-aminopropyl) terminated poly(dimethylsiloxane) (H2N–PDMS–NH2) (Sigma-Aldrich, >99.8%) were used as received. N,N-Dimethylformamide (DMF, A.R., Kelong Chemical Co., China), ethyl acetate (A.R., Bodi Chemical Co.) and n-hexane (A.R., Bodi Chemical Co.) were dried and distilled. GO-g-PBLG (Mn(PBLG) = 5800 g mol−1, DPPBLG = 21, and the grafting density of PBLG on GO platelets, wtPBLG/wtGO was 28 wt%) was prepared according to our previous work.16 Other commercially available reagents were purchased and used as received.Synthetic procedures
For the preparation of the GO-g-PBLG/PBLG-b-PDMS-b-PBLG hybrid organogel in toluene, a certain amount of GO-g-PBLG was first dispersed in toluene and then the triblock copolymer was added to the solution. Afterward, the sealed vial was heated in an oil bath until the hybrid complex became homogeneous, followed by taking out to the ambition condition. The sol–gel transition temperatures (Tgel) and gelation concentrations (Cgel) were determined by tube inversion technology.36,37 All Cgel and Tgel values were measured in triplicates.
Characterization
1H NMR spectra were obtained with Bruker 400 MHz spectrometers using CDCl3 (contains 0.1% TMS) and CDCl3 (without TMS) as solvents. Gel permeation chromatography (GPC) was conducted on a HLC-8380 instrument (TOSOH Corporation, Japan) with DMF as eluent (0.6 mL min−1) in presence of LiBr (1.0 g L−1). The calibration curve was obtained with linear polymethyl methacrylate as standards. Fourier transform infrared (FTIR) spectra was conducted on Nicolet 6700 spectrophotometer (Thermal Scientific, USA) in attenuated total reflection mode at resolution of 0.5 cm−1. The 1D wide-angle X-ray scattering (WAXS) experiments on the samples after annealing were carried out on a SAXSess instrument (Anton Paar) using Cu Kα radiation at a wavelength of 0.154 nm. The working voltage and current were 40 kV and 40 μA, respectively. The scattering profiles of both SAXS and WAXS were simultaneously recorded on an imaging plate (IP) with a pixel size of 42.3 × 42.3 μm2 which extended to the high-angle range (the q range covered by the IP was from 0.06 to 29 nm−1). The scattering peak positions were calibrated with silver behenate for the small-angle region and silicon powder for the wide-angle region, respectively. The scattering vector q is defined as q = 4π/λ[sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ], where the scattering angle is 2θ, and the d-spacing (d) is given by 2π/q. Differential scanning calorimetry (DSC) was carried out using a TA Q2000 DSC (TA Instruments, USA) at a heating rate of 10 °C min−1 under nitrogen flow. Rheological measurements were carried out on a Haake Mars III rheometer with a parallel plate (diameter 20 mm). The dynamic oscillatory stress sweep measurements were performed from 0.1 to 5000 Pa at a constant frequency of 1 Hz. Transmission electron microscopy (TEM) images were obtained on Tecnai G2 F20 transmission electron microscope (SEI, USA) at 200 kV. The TEM gel samples were prepared by inserting the carbon-coated copper grids into the gels and dried under air. Atomic force microscopy (AFM) samples were prepared by casting a drop of hot toluene solution of a block copolymer under spin coating. Then, the sample was conducted with AFM (Anasys Company, USA) using tapping mode to investigate the morphology and height of dry gel.
θ], where the scattering angle is 2θ, and the d-spacing (d) is given by 2π/q. Differential scanning calorimetry (DSC) was carried out using a TA Q2000 DSC (TA Instruments, USA) at a heating rate of 10 °C min−1 under nitrogen flow. Rheological measurements were carried out on a Haake Mars III rheometer with a parallel plate (diameter 20 mm). The dynamic oscillatory stress sweep measurements were performed from 0.1 to 5000 Pa at a constant frequency of 1 Hz. Transmission electron microscopy (TEM) images were obtained on Tecnai G2 F20 transmission electron microscope (SEI, USA) at 200 kV. The TEM gel samples were prepared by inserting the carbon-coated copper grids into the gels and dried under air. Atomic force microscopy (AFM) samples were prepared by casting a drop of hot toluene solution of a block copolymer under spin coating. Then, the sample was conducted with AFM (Anasys Company, USA) using tapping mode to investigate the morphology and height of dry gel.
Results and discussion
Synthesis and characterization of PBLG-b-PDMS-b-PBLG TriBCPs
As illustrated in Scheme 1, PBLG-b-PDMS-b-PBLG triblock copolymers with different molecular weights were prepared by the ring-opening polymerization (ROP) of the BLG-NCA monomer initiated by the corresponding H2N–PDMS–NH2 initiators (Mn = 3500 and 5100 g mol−1). First, the monomer, BLG-NCA, was synthesized from the commercially available γ-benzyl-L-glutamate using the method as reported.11 Dissolving the monomer and macroinitiator in DMF, different molecular weighted copolymers were synthesized through ROP by changing the ratio of initiator to monomer under N2 flow. Successful preparation of block copolymers could be demonstrated from the 1H NMR spectra and GPC results as illustrated in Fig. 1 and 2. As shown by the 1H NMR spectra in Fig. 1, signals related to both PDMS and PBLG segments were observed respectively and in accord with the expected chemical structure of the triblock copolymer. From the GPC results, the relative molecular weights and the polydispersity indexes (PDI) of all block copolymers were obtained as shown in Table 1. The absolute number-average molecular weights (Mn) and the polymerization degrees (DP) of PBLG-b-PDMS-b-PBLG copolymers in Table 1 were calculated from the absolute DP values of the PDMS blocks and the integral ratio between methylene proton b on benzyl group of PBLG and methyl proton f of PDMS in 1H NMR spectrum shown in Fig. 1. And the calculated DP values of the PBLG varied from 16 to 150. The glass transition temperatures of the PBLG blocks were among 5–20 °C determined by the DSC experiments shown in Fig. S1.† All structural information of the triblock copolymers were given in Table 1.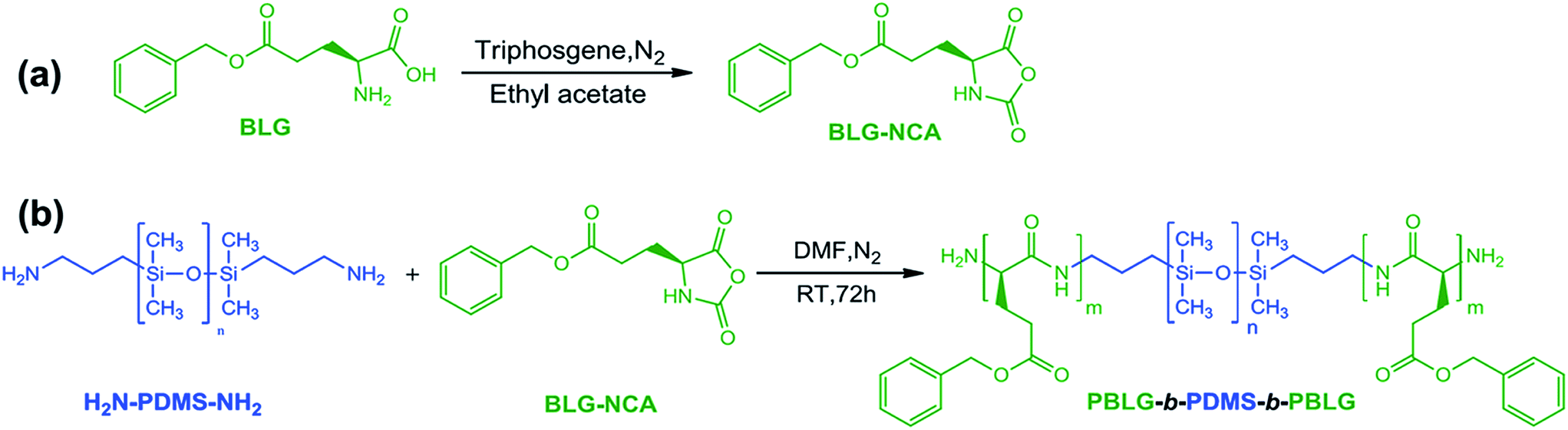 | ||
| Scheme 1 Schematic illustration of synthesis of (a) BLG-NCA compound and (b) PBLG-b-PDMS-b-PBLG triblock copolymers. | ||
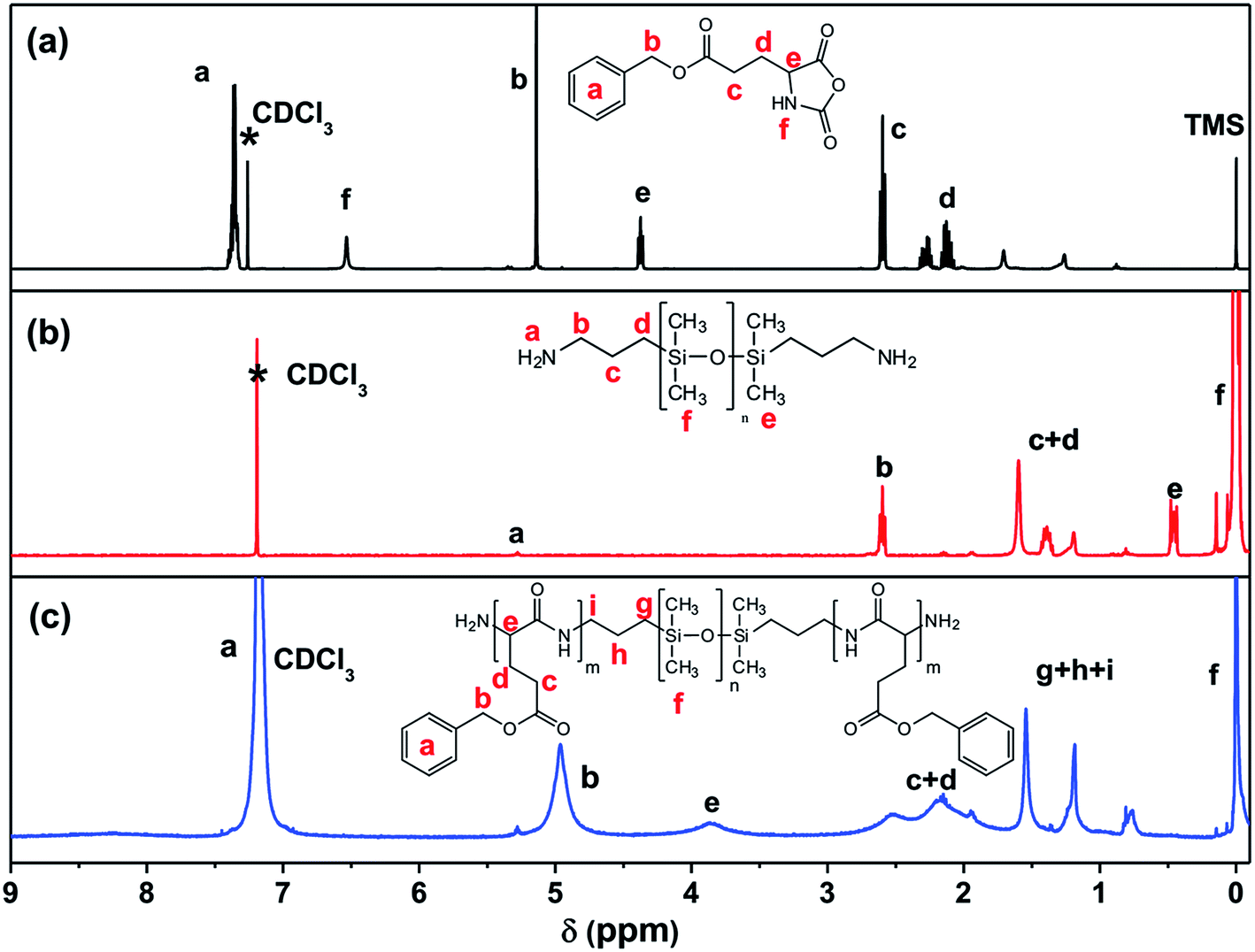 | ||
| Fig. 1 1H NMR spectra of (a) BLG-NCA (CDCl3, 0.1% TMS), (b) H2N–PDMS64–NH2 and (c) B90D45B90 (CDCl3 without TMS). | ||
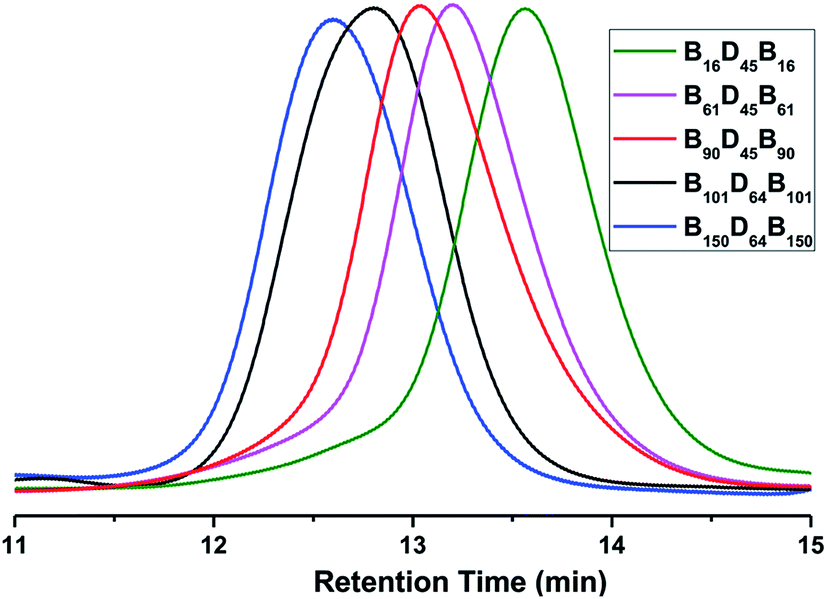 | ||
| Fig. 2 GPC curves of different BDB triblock copolymers in DMF. | ||
| Samples | Mna (g mol−1) | Mn,PBLGa (g mol−1) | PDI | fPBLGb (%) | Cgelc (wt%) | Tgeld (°C) | Lhelixe (nm) |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR.b Calculated from the following equation: fPBLG = Mn,PBLG × ρPBLG−1/(Mn,PBLG × ρPBLG−1 + Mn,PDMS × ρPDMS−1), where ρPBLG = 1.278 g cm−3 and ρPDMS = 0.970 g cm−3.c Apparent critical gelation concentration of the diblock copolymer in toluene.d Gel–sol phase transition temperature was measured at Cgel.e The width of the PBLG helix calculated by 1H NMR integration. Lhelix (nm) = DPPBLG × 0.15 nm (DPPBLG is the polymerization degree of the PBLG helix determined by 1H NMR, 0.15 is the distance between two amino acid residues).37 | |||||||
| B16D45B16 | 10500 |
7000 | 1.25 | 60 | 3.5 | 41 | 2.4 |
| B61D45B61 | 30200 |
26700 |
1.55 | 85 | 2.0 | 43 | 9.2 |
| B90D45B90 | 42900 |
39400 |
1.58 | 90 | 1.5 | 44 | 13.5 |
| B101D64B101 | 49300 |
44200 |
1.42 | 87 | 1.2 | 44 | 15.2 |
| B150D64B150 | 70800 |
65700 |
1.75 | 91 | 0.3 | 45 | 22.5 |
The secondary structures of the PBLG blocks of all triblock copolymers were first determined by Fourier transform infrared spectroscopy (FTIR). The PBLG were known to form three different secondary structures: α-helixes, β-sheets and random coil conformation.38 The locations of the amide-I and amide-II bands in the FTIR spectra can be used to determine the conformation of the polypeptide. As shown in Fig. 3a, the signal at 1735 cm−1 was related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the benzyl ester protecting group of the PBLG block.38–42 In the high resolution FTIR spectra (Fig. 3b) with the wavenumber from 1800 to 1600 cm−1, all of the triblock polymers exhibited amide I and amide II bands at 1653 cm−1 and 1547 cm−1 that were characteristic of α-helical conformations. To be more specific, the amide I band at 1650–1660 cm−1 and amide II band at 1540–1550 cm−1 are associated with the α-helical conformations. The amide I band shifted to 1620–1640 cm−1 is characteristic of β-sheets conformations, and shifted to 1660–1670 cm−1 for random-coil conformations.17 Furthermore, it was clearly observed that the shifted amide I band at 1627 cm−1 in B16D45B16 (indicated with the arrow) was quite obvious, compared to other copolymers. Thus, both α-helix and β-sheet structures were present in B16D45B16, while PBGL chains of the other copolymers mainly assumed α-helix conformation.38–42 The results were consistent with previous research results.17,37 As reported in the literature, both α-helixes and β-sheets secondary structures are present in the polypeptide segments with shorter chain lengths (n < 18), whereas the β-sheets secondary structure becomes less stable in the longer polypeptides (n > 18) and the polypeptide segment almost exclusively adopts an α-helical secondary structures.17
O stretching of the benzyl ester protecting group of the PBLG block.38–42 In the high resolution FTIR spectra (Fig. 3b) with the wavenumber from 1800 to 1600 cm−1, all of the triblock polymers exhibited amide I and amide II bands at 1653 cm−1 and 1547 cm−1 that were characteristic of α-helical conformations. To be more specific, the amide I band at 1650–1660 cm−1 and amide II band at 1540–1550 cm−1 are associated with the α-helical conformations. The amide I band shifted to 1620–1640 cm−1 is characteristic of β-sheets conformations, and shifted to 1660–1670 cm−1 for random-coil conformations.17 Furthermore, it was clearly observed that the shifted amide I band at 1627 cm−1 in B16D45B16 (indicated with the arrow) was quite obvious, compared to other copolymers. Thus, both α-helix and β-sheet structures were present in B16D45B16, while PBGL chains of the other copolymers mainly assumed α-helix conformation.38–42 The results were consistent with previous research results.17,37 As reported in the literature, both α-helixes and β-sheets secondary structures are present in the polypeptide segments with shorter chain lengths (n < 18), whereas the β-sheets secondary structure becomes less stable in the longer polypeptides (n > 18) and the polypeptide segment almost exclusively adopts an α-helical secondary structures.17
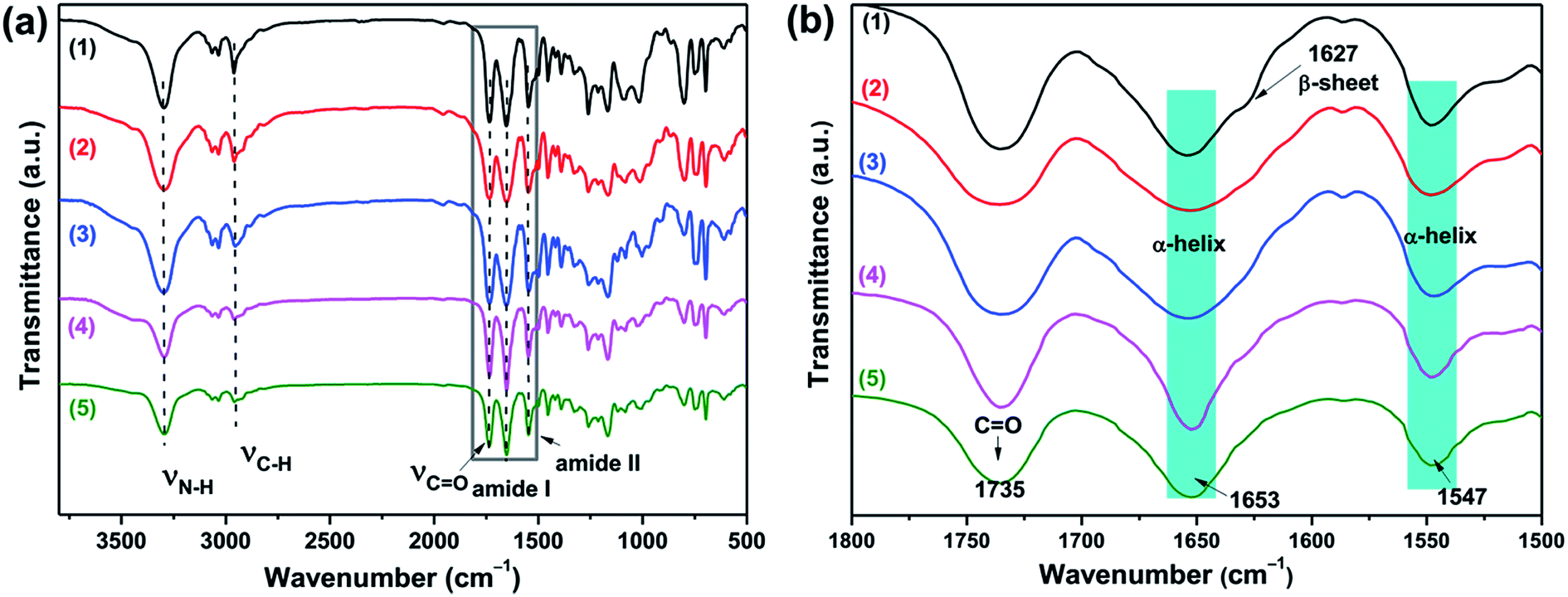 | ||
| Fig. 3 (a) FTIR spectra and (b) the corresponding high resolution FTIR spectra with the wavenumber from 1800 to 1500 cm−1 of the triblock copolymers: (1) B16D45B16, (2) B61D45B61, (3) B90D45B90, (4) B101D64B101, and (5) B150D64B150. | ||
1D WAXS experiments can also be used to identify the type of secondary structures. As shown in Fig. 4, the scattering intensity distributions of different BCPs revealed the effect of chain length on the secondary structure. In each profile, three reflection peaks with a scattering vector ratio of 1:√3:√4 were attributed to a hexagonal packing of cylinders composed of 18/5 α-helixes with a d-spacing of 1.37 nm, in agreement with the FTIR results. However, the primary reflection peak of B16D45B16 was asymmetrical and a small peak at q* = 4.10 nm−1 was observed characteristic of the β-sheets structure.14,38 In addition, the broad “amorphous halo” at about 14.5 nm−1 originated mainly from the long amorphous side-chains comprising a large part of the monomer unit.17 Therefore, FTIR and WAXS results allowed to conclude that the secondary structures of PBLG in the B16D45B16 were the mixture of α-helix and β-sheet, whereas the conformations of PBLG blocks of other BCPs were mostly α-helix with small part of the β-sheet.
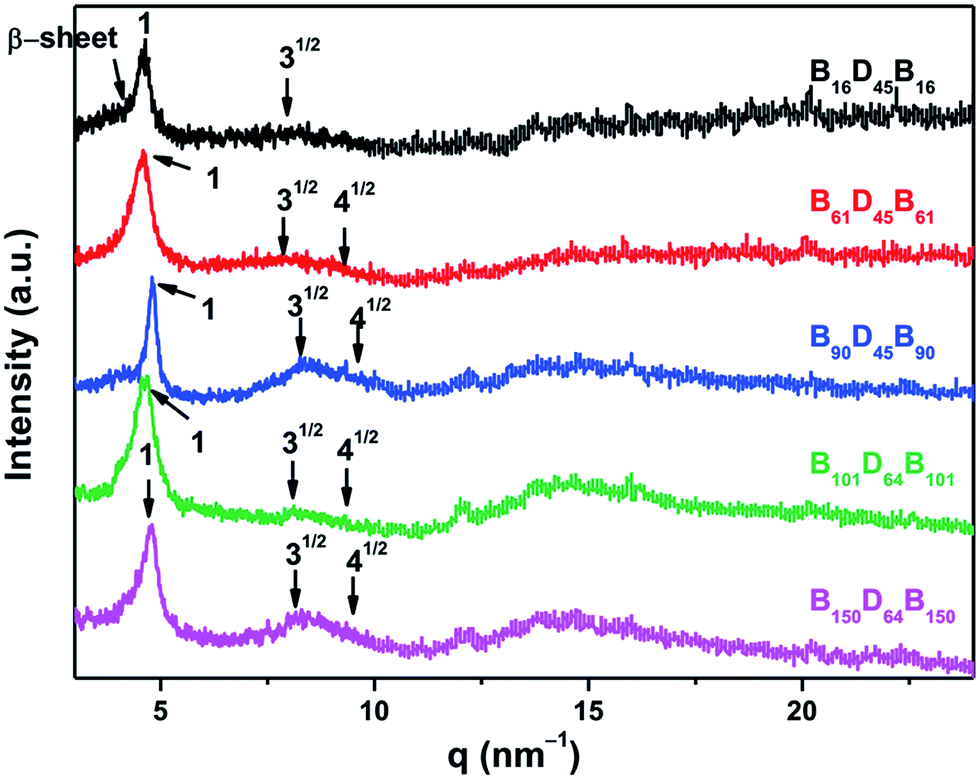 | ||
| Fig. 4 1D WAXD profiles of different BDB triblock copolymers. | ||
The gelation behaviors of BDB TriBCPs with the incorporation of PBLG functionalized graphene
The native gels of the PBLG-b-PDMS-b-PBLG triblock copolymers were first prepared and investigated. With the appropriate concentration of the BDB block copolymers in toluene, the BDB samples were soluble in hot toluene and transformed to transparent thermoreversible gels when temperature decreased. The gels of each triblock copolymer with different concentrations were prepared according to the method described above. The critical gelation concentrations (Cgel) and critical gel–sol transition temperatures (Tgel) were determined through tube inversion technology.36 As shown in Table 1, the Cgel of the synthesized BDB triblock copolymers varied from 3.50 wt% to 0.30 wt% and the Tgel varied from 41 to 45 °C. And the value of Cgel decreased with the increase of PBLG length, while the Tgel increased as the PBLG length increased. Thus, the degree of polymerization of the PBLG block had an important effect on the gelation of PBLG-b-PDMS-b-PBLG triblock copolymers since the stacking between the PBLG helices in combination with the π–π interaction were the main driving force of the self-assembly.21 And relative higher gelation concentration of the BDB block copolymers with shorter PBLG lengths, e.g. B16D45B16, should be ascribed to the weaker driving force of the stacking between the PBLG helices because of the lower α-helical content and the weaker π–π interaction of phenyl groups between the PBLG helices due to the shorter PBLG length.43–45To obtain the hybrid organogels of the BCPs with the incorporation of graphene, a certain amount (0.05 or 0.10 wt%) of PBLG covalently functionalized graphene sheets (GO-g-PBLG, Mn(PBLG) = 5800 g mol−1, DPPBLG = 21, grafting density wtPBLG/wtGO = 28 wt%) that could be well dispersed in toluene, was dispersed in toluene and then the BCP was dissolved in the solution with heating. Afterward, the homogeneous hybrid complex was cooled down to the room temperature and a blackish-colored hybrid organogel was produced. The gelation ability of the hybrid complex with a concentration of the BDB triblock copolymers near Cgel of the native triblock copolymers was investigated. Interestingly, the gelation concentration of the triblock copolymer in the hybrid complex slightly deceased which indicated that the addition of GO-g-PBLG did not influence the gel-formation ability of the triblock copolymers in toluene. For example, it was found that the hybrid complex containing 0.05 wt% of GO-g-PBLG with 0.25 wt% B150D64B150 transformed from solution to gel during cooling process, while the Cgel of B150D64B150 was 0.30 wt%. When the overall weight fraction of the hybrid complex was almost equal to the corresponding weight fraction of the native BCP gel at the Cgel, the hybrid complex could form hybrid gels. The results demonstrated that addition of small amount of GO platelets did not hamper the gelation ability but slightly trigger the gelation of BDB. Moreover, the GO-containing hybrid organogels were still thermoreversible.
In order to investigate the gelation mechanism and the morphology of the native BDB gel and that of the hybrid complex, the TEM and AFM experiments were carried out. As shown in Fig. 5a and b, the gel of the B61D45B61 sample exhibited nanofibril-like morphology, and it was observed that average widths of the nanofiber was about 22 nm and lengths was from 0.5 to 3 μm in the TEM micrographs with higher magnifications. The TEM image of B150D64B150 in Fig. 5c also revealed the entangled nanofiber network structures. In the TEM micrographs with higher magnifications (Fig. 5d), it was observed that some significantly widened nanofiber should be ascribed the parallel alignment and overlapping of two or more fibers. Moreover, the width of different fibers and the width of a single fiber might be slightly fluctuating due to polydispersity of the block copolymers.46 However, the average width of the single fibers was about 26 nm and each fiber was a few micrometers in length. Moreover, through careful observation of the nanofibril in the TEM experiment with higher magnification, it was obviously found that the edge section on both sides of the fiber was darker while the center section was brighter. The darker region should be PDMS segments and the brighter part should be the PBLG helices due to the relatively higher electron density of PDMS than that of PBLG because of the Si elements in the PDMS.47 Furthermore, AFM experiments were carried out to determine the morphology of B150D64B150 (0.35 wt%) organogel. In the AFM image (Fig. S2†), it was observed that the B150D64B150 (0.35 wt%) gel self-assembled in a fibrous structures. The average height of fibrous structures was about 3.5 nm, which also indicated that the individual fibrous structure was not cylindrical but had nanoribbon morphology. Therefore, the observed nanofibril structure should be formed by the planar stacking of the BCPs which was consistent with the nanoribbon mechanism for the self-assembly of random coil-PBLG block copolymers reported in literature.21,37,48 The PBLG helixes of PBLG-b-PDMS-b-PBLG copolymer chains arranged in an anti-parallel fashion in toluene at a concentration above their critical gelation threshold, wherein the strong dipolar π–π interactions involving phenyl groups between the PBLG helices stabilized the nanoribbon structure and provided the driving force for the self-assembly.13,49 And the PDMS block protruded outside of the ribbon which prevented the aggregation of the nanoribbons due to the high solubility of PDMS chains in toluene.37
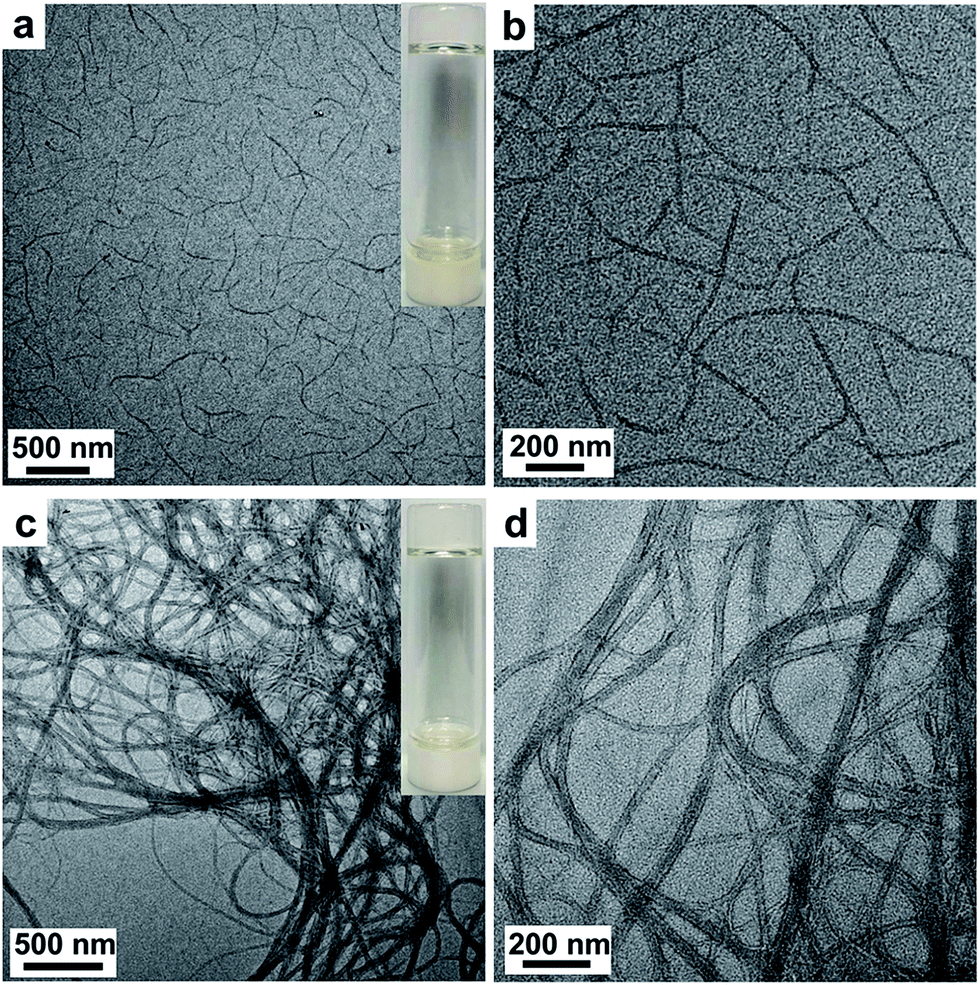 | ||
| Fig. 5 (a, b) TEM image of B61D45B61 organogel (2.20 wt% concentration in toluene) with a digital photograph of the gel in the inset. (c, d) TEM image of B150D64B150 (0.50 wt%) organogel in toluene with a digital photograph of the gel in the inset. | ||
Furthermore, for the B61D45B61, the calculated length of the PBLG helix as shown in Table 1 was 9.2 nm and the fPBLG was 85%, while the observed width of PBLG was about 22 nm. Thus, the nanoribbon of B61D45B61 triblock copolymer should be staked in head-to-head model of the PBLG helical rods in toluene illustrated by the arrangement mode shown in Fig. 7a (left). According to the head-to-head morphology of the PBLG helical rods and the helical length as well as the fPBLG values, the width of the self-assembled nanoribbon was calculated to be 21.6 nm (by the formula 9.2 × 2 + 9.2 × 2 × 0.15/0.85), which was consistent with the observed result. For B150D64B150, the calculated length of the PBLG helix was 22.5 nm (as shown in Table 1) and the fPBLG was 91%, and the observed width of PBLG was about 26 nm. Thus, the B150D64B150 triblock copolymer should be arranged in a monolayer morphology which was illustrated by the arrangement mode as shown in Fig. 7a (right).
The morphologies of the GO-g-PBLG/BDB hybrid gels were further investigated by TEM experiments. The TEM images of the GO-g-PBLG (0.10 wt%)/B61D45B61 (2.10 wt%) hybrid organogel (2.20 wt%) were shown in Fig. 6a and b, and the TEM images of GO-g-PBLG (0.10 wt%)/B150D64B150 (0.40 wt%) hybrid organogel were shown in Fig. 6c and d. It was observed that the BDB triblock copolymer still self-assembled into a nanoribbon-like structure as the BDB triblock copolymer native gels, and the graphene sheets were coated by BDB fibers and were well dispersed in the hybrid gels. The width of the nanoribbon in the hybrid gel was similar with that of the corresponding triblock copolymer native gel. It was observed that some of the BDB nanofibrils were on the surface of the GO nanosheets, which indicated that GO nanosheets had a good interaction with the gel nanofibers in the hybrid organogel as a nanocomposite system. The nanostructure and the molecular arrangement of the GO-g-PBLG/BDB hybrid gel were schematically illustrated in Fig. 7b. From the above investigations on the gelation behaviors and the gel morphologies of the GO-g-PBLG/BDB hybrid complex, it could be concluded that the hybrid organogel preserved the basic characteristics of native organogel and the nanoribbon structure still played a key role in the gelation ability of the nanocomposite. The well distribution of the functionalized graphene sheets within the BDB nanofibrils network generated the new gel composites.
 | ||
| Fig. 6 (a, b) TEM image of GO-g-PBLG (0.10 wt%)/B61D45B61 (2.10 wt%) hybrid organogel (1.40 wt%) with a digital photograph of the gel in the inset. (c, d) TEM image of GO-g-PBLG (0.10 wt%)/B150D64B150 (0.40 wt%) hybrid organogel in toluene with a digital photograph of the gel in the inset. | ||
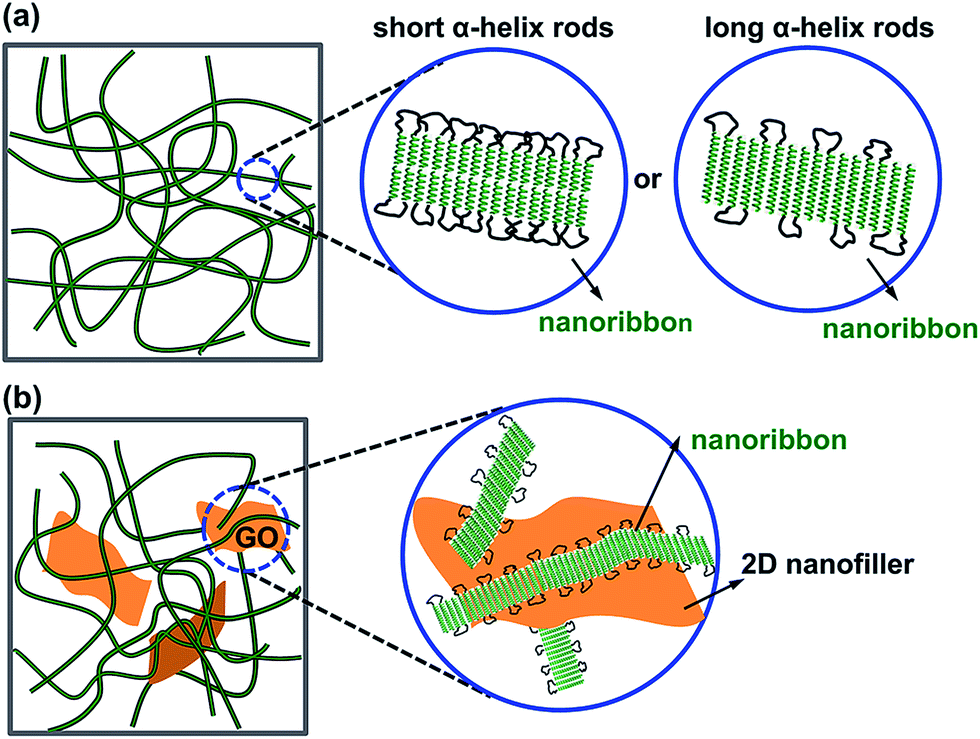 | ||
| Fig. 7 Schematic diagram presentation of the nanoribbon formed in the network structure of (a) the PBLG-b-PDMS-b-PBLG organogel and (b) the GO-g-PBLG/PBLG-b-PDMS-b-PBLG hybrid gel. | ||
The rheological properties of the GO-g-PBLG/BDB hybrid organogels compared with the BDB native gels
Finally, rheological experiments were carried out to investigate the influence of the incorporation of GO platelets on rheological properties of the self-assembled organogels.50 In order to avoid the influence of the concentration on rheological properties of the gels, the same overall concentrated triblock copolymers gels and GO-g-PBLG/BDB hybrid gels were prepared and characterized. The storage modulus (G′, contribution of elastic) and the loss modulus (G′′, contribution of viscous) were measured as a function of stress sweep at a constant frequency of 1 Hz as shown in Fig. 8. In the case of GO-g-PBLG/B61D45B61 hybrid organogel, the G′ values were larger than G′′ values and all the values remained almost constant before the gel was destroyed which indicated that the GO-containing hybrid systems also maintained the free-standing gel state and behaved as solid-like gel materials. Surprisingly, both G′ and G′′ values of hybrid organogels were much larger than those of the corresponding native gel, indicating that the GO increased the elastic properties of the gels and the GO-containing hybrid organogels were more rigid than native gels. For the hybrid gel with 0.05 wt% of functionalized GO, both G′ and G′′ values of hybrid organogels increased to be two times of the corresponding values of the BDB native gels. In addition, the gel was destroyed when the G′′ value was larger than the G′ value and the fracture stress of the gel could be determined at the intersection of the curves. The fracture stress of the GO-g-PBLG (0.05 wt%)/B61D45B61 (2.25 wt%) occurred at 660 Pa and that of GO-g-PBLG (0.10 wt%)/B61D45B61 (2.20 wt%) hybrid gel occurred at 810 Pa, both of which were much larger than the fracture stress (393 Pa) of native B61D45B61 (2.30 wt%) gel.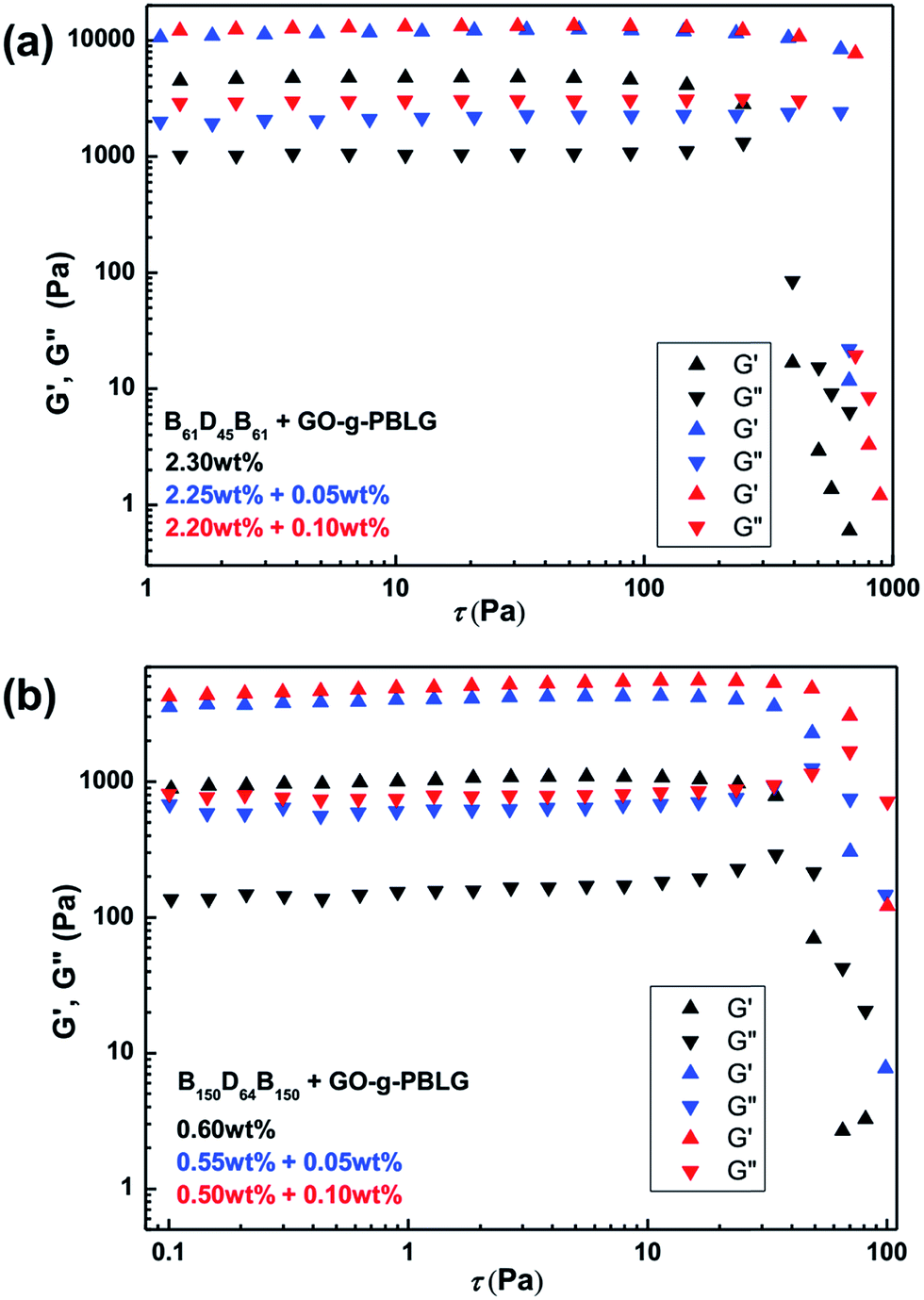 | ||
| Fig. 8 (a) Storage (G′) and loss (G′′) moduli of B61D45B61 gel and GO-g-PBLG/B61D45B61 hybrid gels with indicated compositions at the same overall concentration as a function of stress sweep. (b) Storage (G′) and loss (G′′) moduli of B150D64B150 gel and GO-g-PBLG/B150D64B150 hybrid gels with indicated compositions at the same overall concentration as a function of stress sweep. | ||
The rheological results of GO-g-PBLG/B150D64B150 hybrid gels were shown in Fig. 8b, in which it was also observed that both the moduli (G′ and G′′) and the fracture stresses of the hybrid gels were much larger than the corresponding values of the native gel within the same overall concentration. Moreover, as shown in Fig. 9, the hybrid gels of B16D45B16, B90D45B90 and B101D64B101 with 0.01 wt% GO-g-PBLG showed an increase of storage and loss moduli and fracture stress than the corresponding native BCP gels. Therefore, the rheological results indicated that the incorporation of the GO sheet in organogels significantly induced the increases of the storage modulus, the loss modulus and the fracture stress, which demonstrated the enhanced mechanical strength of the hybrid self-assembled gel composites.
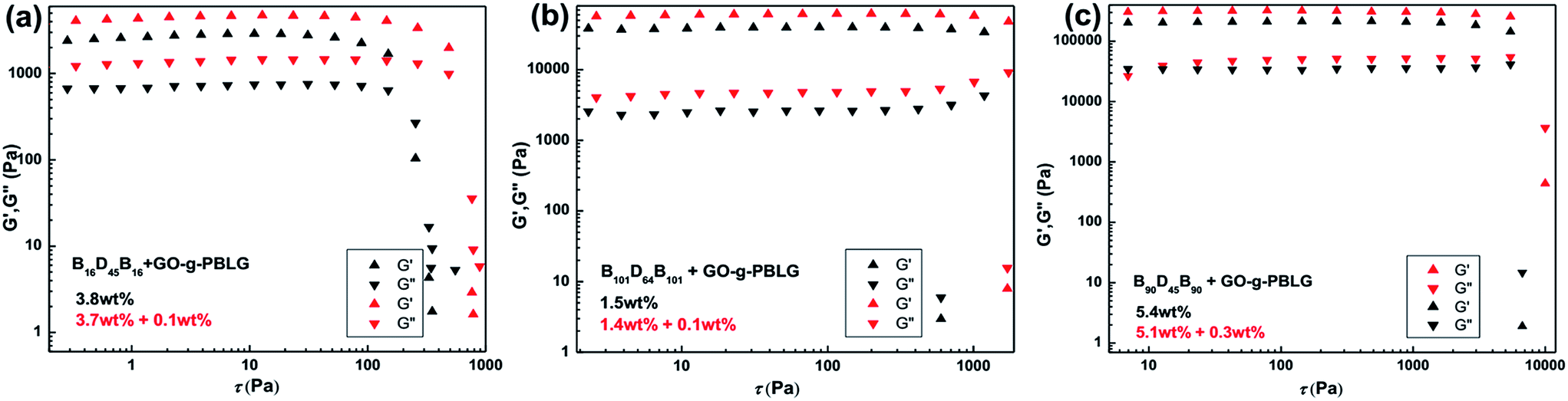 | ||
| Fig. 9 Storage (G′) and loss (G′′) moduli of (a) B16D45B16 gel and GO-g-PBLG/B61D45B61 hybrid gels, (b) B101D64B101 gel and GO-g-PBLG/B101D64B101 hybrid gels, and (c) B90D45B90 gel and GO-g-PBLG/B90D45B90 hybrid gels with indicated compositions at the same overall concentration as a function of stress sweep. | ||
According to the gelation behavior and morphologies of the hybrid gels studied above, the improvement of the mechanical strength of the hybrid organogels should be contributed to the homogeneous hybrid complex with hierarchical 3D network structures consisted of BDB self-assembled nanoribbon-like network and the closely incorporated 2D planar sheets of the functionalized graphene. The GO sheets efficiently interacted with gel nanoribbon and behaved as 2D nanofillers in the hybrid gel, which contributed to the enhancement of the mechanical properties of the gels.
Conclusion
In summary, the thermoreversible self-assembled organogels of peptide-based triblock copolymers (PBLG-b-PDMS-b-PBLG) with different PBLG lengths and nanoribbon-like structures, and the gel composites of the triblock copolymers with polypeptide-functionalized graphene thereof were synthesized and investigated. The hybrid organogels possessed lower gelation concentration and thermoreversibility, and considerably showed increased moduli and fracture stresses than the corresponding native gels with the same overall concentration. It indicated that the gelation ability and mechanical strength of polypeptide block copolymer organogels could be tuned with the inclusion of the polypeptide functionalized graphene sheets, and may provide potential applications in biomaterials and other advanced materials.Acknowledgements
Financial supports from the National Natural Science Foundation of China (Grant 51403132) and Sichuan Ministry of Science, Technology Project (Grant 2015GZX0226), and Open Project Foundation of the Beijing National Laboratory for Molecular Sciences (2016) are gratefully acknowledged.Notes and references
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS PubMed.
- S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim and Y. I. Song, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS PubMed.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- H. Tanisaka, S. Kizaka-Kondoh, A. Makino, S. Tanaka, M. Hiraoka and S. Kimura, Bioconjugate Chem., 2007, 19, 109–117 CrossRef PubMed.
- M. Wang, C. Zhou, J. Chen, Y. Xiao and J. Du, Bioconjugate Chem., 2015, 26, 725–734 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- A. M. Jonker, D. W. Löwik and J. C. van Hest, Chem. Mater., 2012, 24, 759–773 CrossRef CAS.
- J. Huang, C. L. Hastings, G. P. Duffy, H. M. Kelly, J. Raeburn, D. J. Adams and A. Heise, Biomacromolecules, 2012, 14, 200–206 CrossRef PubMed.
- D. S. Poche, M. J. Moore and J. L. Bowles, Synth. Commun., 1999, 29, 843–854 CrossRef CAS.
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC.
- M. Lee, B.-K. Cho and W.-C. Zin, Chem. Rev., 2001, 101, 3869–3892 CrossRef CAS PubMed.
- H.-A. Klok, J. F. Langenwalter and S. Lecommandoux, Macromolecules, 2000, 33, 7819–7826 CrossRef CAS.
- A. Top, S. Zhong, C. Yan, C. J. Roberts, D. J. Pochan and K. L. Kiick, Soft Matter, 2011, 7, 9758–9766 RSC.
- H. Li, L.-Y. Shi, W. Cui, W.-W. Lei, Y.-L. Zhang, Y.-F. Diao, R. Ran and W. Ni, RSC Adv., 2015, 5, 86407–86413 RSC.
- P. Papadopoulos, G. Floudas, H.-A. Klok, I. Schnell and T. Pakula, Biomacromolecules, 2004, 5, 81–91 CrossRef CAS PubMed.
- M. Y. Seungju, V. P. Conticello, G. Zhang, C. Kayser, M. J. Fournier, T. L. Mason and D. A. Tirrell, Nature, 1997, 389, 167–170 CrossRef PubMed.
- C. Robinson and J. Ward, Nature, 1957, 180, 1183 CrossRef CAS PubMed.
- K. Tohyama and W. G. Miller, Nature, 1981, 289, 813–814 CrossRef CAS PubMed.
- K. T. Kim, C. Park, G. W. Vandermeulen, D. A. Rider, C. Kim, M. A. Winnik and I. Manners, Angew. Chem., 2005, 117, 8178–8182 CrossRef.
- H. Gao, Z. Hu, Q. Guan, Y. Liu, F. Zhu and Q. Wu, Polymer, 2013, 54, 4923–4929 CrossRef CAS.
- S. Bhattacharya and S. K. Samanta, Chem. Rev., 2016, 116, 11967–12028 CrossRef CAS PubMed.
- J. Liu, G. Chen and M. Jiang, Macromolecules, 2011, 44, 7682–7691 CrossRef CAS.
- Q.-Y. Cheng, D. Zhou, Y. Gao, Q. Chen, Z. Zhang and B.-H. Han, Langmuir, 2012, 28, 3005–3010 CrossRef CAS PubMed.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951–954 CrossRef CAS PubMed.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass and A. N. Marchenkov, Science, 2006, 312, 1191–1196 CrossRef CAS PubMed.
- J. C. Meyer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth and S. Roth, Nature, 2007, 446, 60–63 CrossRef CAS PubMed.
- Y. Zhang, T.-T. Tang, C. Girit, Z. Hao, M. C. Martin, A. Zettl, M. F. Crommie, Y. R. Shen and F. Wang, Nature, 2009, 459, 820–823 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- R. Liu, S. Liang, X.-Z. Tang, D. Yan, X. Li and Z.-Z. Yu, J. Mater. Chem., 2012, 22, 14160–14167 RSC.
- S. Sun and P. Wu, J. Mater. Chem., 2011, 21, 4095–4097 RSC.
- P. Xing, X. Chu, G. Du, M. Ma, S. Li and A. Hao, Colloid Polym. Sci., 2014, 292, 3223–3231 CAS.
- A. R. Hirst, D. K. Smith, M. C. Feiters, H. P. Geurts and A. C. Wright, JACS, 2003, 125, 9010–9011 CrossRef CAS PubMed.
- V. K. Kotharangannagari, A. Sánchez-Ferrer, J. Ruokolainen and R. Mezzenga, Macromolecules, 2012, 45, 1982–1990 CrossRef CAS.
- A. Sen and T. Keiderling, Biopolymers, 1984, 23, 1533–1545 CrossRef CAS PubMed.
- A. Dong, P. Huang and W. S. Caughey, Biochemistry, 1990, 29, 3303–3308 CrossRef CAS PubMed.
- W. K. Surewicz and H. H. Mantsch, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1988, 952, 115–130 CrossRef CAS.
- J. K. Kauppinen, D. J. Moffatt, H. H. Mantsch and D. G. Cameron, Anal. Chem., 1981, 53, 1454–1457 CrossRef CAS.
- E. Ibarboure, J. Rodríguez-Hernández and E. Papon, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4668–4679 CrossRef CAS.
- A. Sanchez-Ferrer and R. Mezzenga, Macromolecules, 2009, 43, 1093–1100 CrossRef.
- P. Rohrer and H. G. Elias, Die Makromolekulare Chemie, 1972, 151, 281–283 CrossRef CAS.
- C. Kim, K. T. Kim, Y. Chang, H. H. Song, T.-Y. Cho and H.-J. Jeon, JACS, 2001, 123, 5586–5587 CrossRef CAS PubMed.
- H. Schlaad, B. Smarsly and M. Losik, Macromolecules, 2004, 37, 2210–2214 CrossRef CAS.
- G. H. Michler, in Electron Microscopy of Polymers, Springer-Verlag, Berlin, 2008 Search PubMed.
- S.-W. Kuo, H.-F. Lee, W.-J. Huang, K.-U. Jeong and F.-C. Chang, Macromolecules, 2009, 42, 1619–1626 CrossRef CAS.
- S. Ludwigs, G. Krausch, G. Reiter, M. Losik, M. Antonietti and H. Schlaad, Macromolecules, 2005, 38, 7532–7535 CrossRef CAS.
- S. Das, F. Irin, L. Ma, S. K. Bhattacharia, R. C. Hedden and M. J. Green, ACS Appl. Mater. Interfaces, 2013, 5, 8633–8640 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: DSC curves of PBLG-b-PDMS-b-PBLG triblock copolymers. See DOI: 10.1039/c6ra24677j |
| This journal is © The Royal Society of Chemistry 2017 |