
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene/polyaniline electrodeposited needle trap device for the determination of volatile organic compounds in human exhaled breath vapor and A549 cell†
Yu Li,
JingHong Li and
Hui Xu *
*
Key Laboratory of Pesticide & Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan, 430079, People's Republic of China. E-mail: huixu@mail.ccnu.edu.cn
First published on 20th February 2017
Abstract
In this work, a graphene/polyaniline (G/PANI) electrodeposited coating was introduced as a novel extraction phase of needle trap microextraction (NTME). The needle trap coating was prepared by in situ electrodepositing G/PANI on the inner surface of a 21-gauge stainless steel needle, and utilized for the dynamic extraction of volatile organic compounds (VOCs) from human exhaled breath vapor (EBV) and headspace gas in cell lines. The coating exhibited enhanced mechanical stability, long lifespan, large specific surface area and good biocompatibility. The parameters for the extraction and analysis of analytes including extraction mode, sampling temperature, sampling speed, desorption parameters (temperature and time) were evaluated and optimized. The extraction performance of the needle trap was also compared with three commercially available SPME fibers. Low detection limits (0.002–0.003 μg L−1) and good linearity (R2 ≥ 0.9905) were obtained for the analysis of eight VOCs. The developed needle trap microextraction-gas chromatography-mass spectrometry (NTME-GC-MS) method has been successfully applied to the analysis of VOCs from human exhaled breath and lung cancer cell lines (A549) with good recovery (70–120%) and acceptable reproducibility (relative standard deviation: intra-day 4.8–15.3%, inter-day, 4.2–14.3%). The proposed method is simple, sensitive and environmentally friendly, it provides a new possibility for the determination of VOCs of exhaled breath both in vivo and in vitro.
Introduction
Sample preparation is the cornerstone of chemical analysis. Preconcentration is a key step that takes 2/3 of the whole analysis process, especially when dealing with a trace complex sample. The development of novel sample preparation methods is becoming increasingly important in analytical chemistry. Conventional methods used in analytical laboratories are usually not compatible with the need for routine and extensive monitoring.1–4 In recent years, there has been a growing demand for rapid, sensitive, single-step and solvent-free microextraction sampling and analysis methods for volatile organic compounds.4,5 Solid-phase microextraction (SPME) and NTME are the examples of solventless sample preparation and introduction methods being developed today.6–9 A needle trap (NT) is an extraction device that contains a single-layer or multilayer sorbent bed packed inside a needle. The NTME method combines sampling, sample preparation, and sample introduction as SPME does. For air/headspace analysis, NTME are applicable for sampling a wide range of volatiles such as benzene, alkanes, and semi-volatile particulate bound compounds.10,11 SPME was first reported by Pawliszyn et al. in the early 1990s. In the method, solid polymer was coated on a small-diameter fused-silica rod (∼1 cm) as the stationary phase. It is a simple, time-efficient, and solvent-free sample preparation technique,12,13 and has been successfully applied to the extraction of various compounds at trace levels such as organic pollutants in environmental, food, biological, pharmaceutical, and clinical samples.14,15 Despite its advantages, SPME has some drawbacks. One of these is that, when applied to air samples, the extraction capacity of SPME fiber is limited.11 In contrast, the internally coated needles or needles packed with commercially available sorbents, can provide greater extracting phase loading, enhanced sample capacity and improved extraction sensitivity.16 Another drawback of SPME is the fragility of the fiber. The needle trap device (NTD) is more robust because the extraction phase is protected inside a stainless steel shield.17–19Sorbent-packed NTD is a traditional NTD mode.20–22 Until now, several sorbents such as polydimethylsiloxane (PDMS), divinylbenzene (DVB), Carboxen 1000, Carbopack X, Tenax and Porapak Q are conventionally used for NTD.11 For example, needle packed with activated-carbon-based sorbents was used for the analysis of very volatile organic compounds23 and needle packed with Tenax TA and Carboxen 1000 particles was used for analysis of priority volatile organic compounds.20 However, it is a time-consuming work to pack the sorbent materials, in addition, the flow rates between packed needles are not reproducible.24 Various new nano-materials with outstanding properties are emerging nowadays. The first application of new nano-material sorbents (carbon nanotube) in NTD was introduced by Heidari et al. in 2012.25,26 Among the new nano-material sorbents, graphene, a single-atom-thick, two dimensional carbon material, is considered as the basic building block of all graphitic forms (including carbon nanotubes, graphite and fullerene (C60)).27 Graphene doped into polyaniline could combine the electrical characteristics of graphene and metals, the mechanical and processing properties of polymers and provide hydrophilic functional NH2 groups.28 What's more, donor–acceptor complex can form between PANI and graphene via electrostatic interaction, π–π and p–π interaction, which render the composite better chemical stability.29,30 However, nanomaterial-packed NTD is easy to encounter with the problem of high column pressure, making it necessary to use very clean samples and low flow rate. In addition, some materials are expensive, and the service life is short. In order to improve the extraction efficiency and permeability, simplify the packing process of needle trap, wall-coated NTD with novel nanomaterial as sorbent is a good choice. It can ensure simple performance and fast analysis, protect the coating from scrapping and allow for the convenient automation of the extraction process. In addition, in tube SPME with graphene/polyaniline (G/PANI) electrodeposited coating has successfully been developed and online coupled with liquid chromatography for the analysis of aldehydes in human exhaled breath condensates.31 But so far, no G/PANI electrodeposited composite has been reported as NTME coating for GC-MS analysis of VOCs.
Breath analysis is a promising approach for the non-invasive assessment of inflammatory and oxidative stress biomarkers involved in the etiopathogenesis of various respiratory conditions.32 However, the sample matrix of human exhaled breath vapor (EBV) is complex, the existence of nitrogen, oxygen, carbon dioxide, inert gases and water may seriously interfere with the analytical signal.33 Therefore, sample preparation or preconcentration step is always adopted before chromatographic analysis. To further explain the origin of cancer-related VOC metabolites, our group has attempted to detect the VOCs produced by the cancer cell lines in vitro. It is an important method to study the mechanism of carcinogenesis and the biological characteristics of cancer cells.34 The aim of this study is to confirm the existence of volatile organic compounds (VOC) specifically released or consumed by the lung cancer cell line A549, which could be used in future as biomarkers for the early detection of lung cancer.
In this report, we present the evaluation of a new needle trap coated with graphene/polyaniline for sampling and analysis of VOCs metabolites in human exhaled breath and lung cancer cell A549 by NTME-GC-MS. The morphology, microstructure and extraction performances of the resultant composites were investigated. The experimental parameters that may influence sampling and extraction were studied. At the same time, the NT methodology was compared with other microextraction techniques in terms of method validation parameters, and the proposed method was applied for the analysis of VOCs in EBV and cellular metabolites.
Experimental
Chemicals and materials
Acetone was provided by Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Nonanal (97%) was obtained from Alfa Aesar (Tianjin, China). Octanal (98%) was purchased from Dixiai Chemical (Shanghai, China). Hexanal (98%) and heptanal (97%) were received from ABCR GmbH & Co. KG (Germany). Decane (99.5%) was purchased from Dr Ehrenstorfer GmbH (Germany). Benzene (99.5%) and undecane (99%) were provided from TCI Development Co. Ltd (Shanghai, China). Pentanal (98.5%) was purchased from Amethyst Chemicals (Beijing, China). Ultra-pure water was used in all experiments (arium®pro Ultrapure Water Systems, Sartorius Stedim Biotech, Gottingen, Germany). HPLC-grade ethanol was purchased from Fisher Chemicals (Fair Lawn, NJ, USA). NaOH was bought from Sailboats Chemical Reagent Co., Ltd (Tianjin, China). Graphite powder (99.95%) was bought from Jinrilai (Qingdao, China) and other chemical reagents were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Gas-tight syringe and 21 stainless steel needles (Hamilton) were from Agilent Technologies Inc. (USA). Five microliter microsyringe was bought from Feige, China. Tedlar bags were obtained from economic and trade co., Ltd (Shanghai, China).Equipment and chromatographic conditions
The GC-MS QP2010 Plus system (Shimadzu, Japan) coupled with a split/splitless injector was employed for all chromatographic analysis experiments. The mass spectrometry was operated in EI mode at 70 eV. All separations were carried out on a CP-Pora bond Q column (30 m × 0.32 mm × 0.5 mm, Varian Chrompack, USA). Helium was used as carrier gas at a constant velocity of 1.62 mL min−1. GC oven temperature was programmed as follows: initially maintained at 80 °C for 2 min, then up to 140 °C at a rate of 8 °C min−1 and held for 2 min, subsequently programmed at 5 °C min−1 to 250 °C and held for 12 min. The transfer line and the ion source temperatures were set at 250 °C and 200 °C, respectively. Data acquisition was performed in selected ion monitoring (SIM) mode. The qualitative ions for each analyte were as follows, acetone: m/z 58, 42; benzene: m/z 77, 52; pentanal: m/z 41, 58; hexanal: m/z 41, 56; heptanal: m/z 41, 70; decane: m/z 43, 71; nonanal: m/z 41, 56; undecane: m/z 43, 71.Surface morphology of the prepared nanoparticles was photographed by scanning electron microscope (SEM, JEOL, JSM-5610LV, Japan). FTIR spectra of the nanoparticles were recorded on a Thermo Nicolet Nexus FTIR model 470 spectrometer. A laboratorial syringe infusion pump (Lsp02-1B) was supplied by Baoding Longer Precision Pump Co., Ltd (Hebei, China).
Preparation of standard solution
Stock solutions of 8 analytes (acetone, benzene, pentanal, hexanal, heptanal, decane, nonanal, undecane) were prepared in ethanol at 1650 mg mL−1. The daily standard working solutions were prepared by diluting the corresponding stock solution with ethanol. All of them were stored at −20 °C in the dark. The mixed gas calibration samples, involving 8 analytes were prepared by vaporization of 2.0 μL working solution in a 25 mL evacuated sealed vial in 45 °C water bath for 20 min.EBV sample collection and preparation
Before collection, all Tedlar bags were cleaned by flushing with nitrogen gas and then filled with nitrogen and heated at 70 °C for 10 h to get rid of any contaminations. Breath samples, involving 8 lung cancer patients (Hubei Cancer Hospital, Hubei, China) and 8 healthy volunteers (Central China Normal University, Hubei, China) who had fasted overnight were collected in 1 L Tedlar bags through a disposable mouthpiece. Then, a 25 mL sample was transferred into a crimped and evacuated glass vial by a gas tight syringe. The institutional committee(s) of our department has approved the experiments. The statement that informed consent was obtained has also been obtained. Ethical approval for the study was obtained from the Ethics Committee of Hubei Cancer Hospital prior to the collection and analysis of breath samples. Our all experiments were performed in compliance with relevant laws or guidelines.A549 cell culturing conditions
The human non-small cell lung cancer (NSCLC) A549 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in a humidified atmosphere containing 5% CO2 at 37 °C. A549 cells were routinely cultured in DMEM/F12 media (Invitrogen, Grand Island, NY, USA) supplemented with 5% and 10% heat-inactivated fetal bovine serum (Hyclone Laboratories, Logan, UT, USA), respectively, and penicillin–streptomycin 100× solution and 2 mM L-glutamine from GIBCO (Invitrogen).Preparation of G/PANI composite coating
The procedure for fabrication of G/PANI composite coating involves three steps, (1) synthesis of graphene, (2) preparation of graphene–polyaniline electrodeposition dispersion, (3) electrodeposition of composite coating. In the first step, graphene oxide was prepared from graphite powder according to the Hummers method.35 60 mg of the prepared graphene oxide was ultrasonically dispersed in 16 mL pure water and reduced with 45 μL hydrazine for 24 h to achieve a black graphene dispersion. In the second step, 100 μL aniline (in 2 mL ethanol) was added into 8 mL of the above dispersion to yield a homogeneous G/PANI electrodeposition dispersion (3 mg L−1).In the third step, 21-gauge stainless steel needle was ultrasonically cleaned with acetone, ethanol and pure water, subsequently, and dried under a stream of N2 in an oven (60 °C). Polymerization process was carried out thereafter using a two-electrode system with the SS needle as cathode and a stainless steel wire as anode. A syringe pump was used to deliver the graphene–polyaniline electrodeposition dispersion through stainless steel needle at the rate of 0.1 mL h−1. The stainless steel needle and stainless steel wire, then, were placed in the G/PANI solution in the depth of about 1 cm and a constant potential of 9 V was applied for dynamic electrodeposition (30 s). Next, the residual solution was released by air, and the fresh coating on the outer surface of the needle was immediately scrubbed off with a clean filter paper and flushed with methanol and water, afterwards, the tube was dried at 60 °C for 2 h under a stream of N2. The above electrodeposition procedures were repeated twice to obtain a proper coating thickness. The prepared needle was washed with methanol and water completely and dried at 60 °C. For comparison, PANI coated needle was prepared following the same procedure, expect with 10 mL water containing 0.05 mol L−1 aniline and 0.5 M H2SO4 as the electrodeposition solution.
Needle trap microextraction procedure
A G/PANI coated 21-gauge stainless steel needle (length: 10 cm, inner diameter: 0.39 mm) was conveniently attached with glass syringes (10 mL, Hamilton) (as shown in Fig. 1). Each NTD was conditioned in the GC injector at 300 °C for 2–3 h with a permanent helium flow to remove impurities. Then, the tip end was sealed with a Teflon septum and the upper part of the needle was closed with a push-button syringe valve (SGE Europe Ltd, Milton Keynes, UK) to prevent contamination during storage. This procedure avoids the use of epoxy resin, which leads to significant levels of impurities in ultra-trace analysis.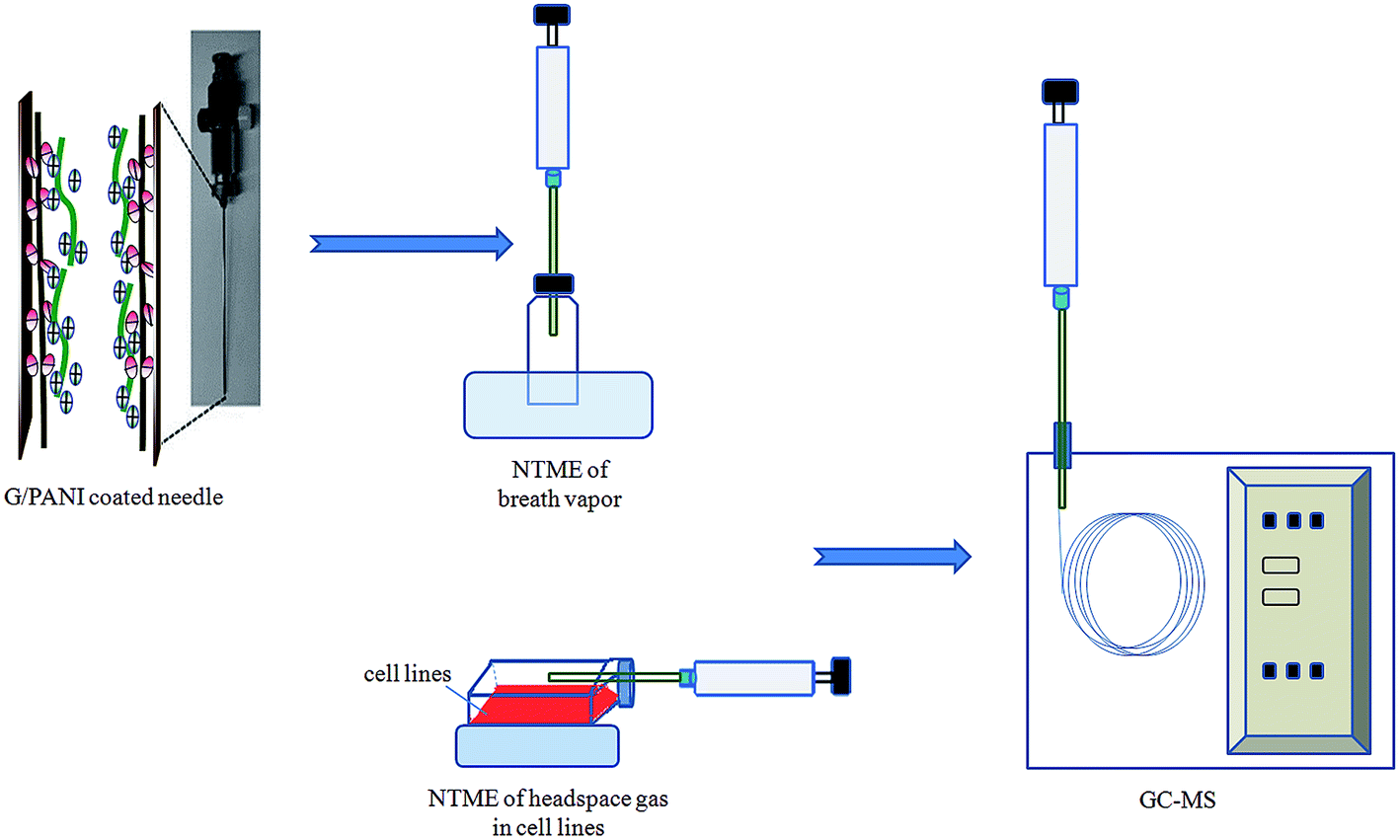 | ||
| Fig. 1 Schematic diagram of NTME-GC-MS. | ||
During exposure, 2 μL working solution was injected into a 25 mL sealed glass vial with a PTFE lined septum cap and evaporated in water bath (45 °C, 20 min). The needle with G/PANI sorbent was inserted in the vial to perform the headspace dynamic extraction by pumping up through the sorbent coating at a withdraw and ejection speed of 5 mL min−1 for 20 min (5 pull–push cycles). Afterward, the syringes was immediately injected into GC for thermal desorption at 265 °C for 5 min and the compounds were subsequently analyzed by GC-MS.
For comparison, the extraction with G/PANI coated fiber, commercial PDMS/CAR, PDMS/DVB and PA fibers were carried out at the static mode with the same sampling and extraction steps. Dynamic extraction procedure with PANI coated needle was the same as that of G/PANI coated needle. After cell growing, the NTD was immediately inserted into the headspace gas phase of the initiated cell culture for analysis, the other conditions were the same with NTME.
Results and discussion
SEM characterization of G/PANI coating
The surface characteristics of the G/PANI coating were investigated by SEM. To convenient observation of the surfaces of G/PANI coating, a stainless steel fiber was used as substrate for G/PANI electrodeposition. Fig. 2a was a low-magnification image, which showed that the PANI coating possessed a homogeneous surface. Compared with the PANI coating, the surface of G/PANI fiber coating (Fig. 2b) was more rough which was beneficial for extraction performance. From the Fig. 2c, we could obtain a high-magnification image that PANI and G had a perfect mixing and possessed a porous structure which could offer a larger surface compared with the PANI coating. The cross-sectional SEM image in Fig. 2d indicated that the thickness of G/PANI coating was approximately 40 μm. These results demonstrated that graphene has been successfully incorporated into PANI polymer, which can provide more accessible adsorption sites for the extraction.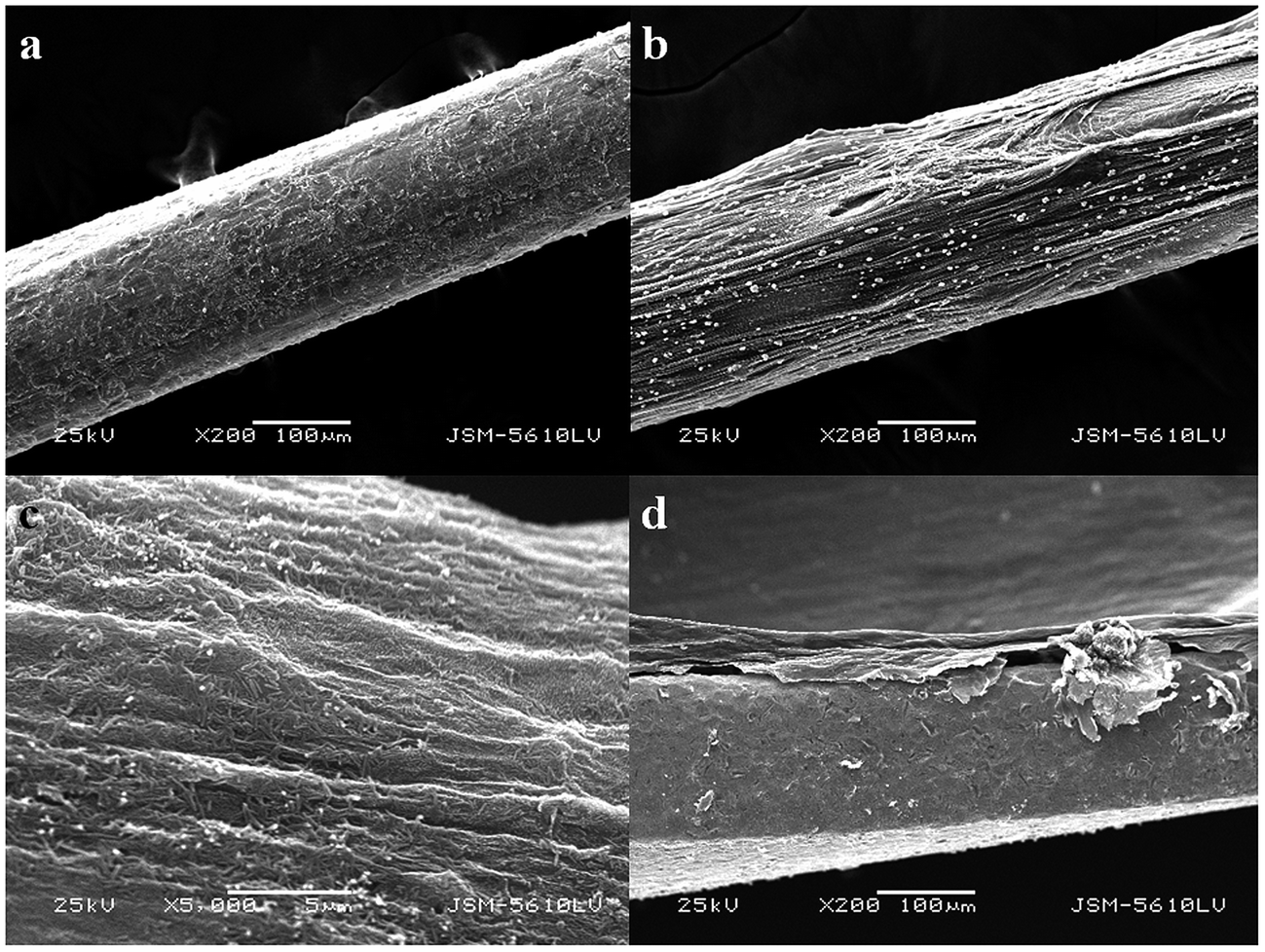 | ||
| Fig. 2 SEM images of images of full view and the surfaces of (a) PANI, 200× and (b) G/PANI, 200×; (c) G/PANI, 5000×; (d) the cross section of coated needle (200×). | ||
Fourier transform infrared (FTIR)
Typical functional groups of the G/PANI coating were investigated by Fourier transform infrared (FTIR) and compared with graphene, the results are shown in the Fig. S1 of ESI.† The presence of strong band sat around 1720, 1645 and 1078 cm−1 in Fig. S1a† (the characteristic of the residual C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O) indicated that the partial reduction of GO.1,2 Many low-intensity peaks ranging from 780 to 580 cm−1 can be assigned to the vibrations of the C–H bonds in the benzene rings (Fig. S1a and S1b†). Compared with graphene, however, several new peaks attributed to PANI were found in the spectrum of G/PANI. Notably, the vibrations of CO at around 1654 cm−1 decreased distinctly, and new peaks around 1567 and 807 cm−1 corresponded the vibrations of CN and C–H appeared, respectively. In addition, a stretching band attributed to C–N in aromatic amine appeared at 1381 cm−1. The characteristic band at around 1135 cm−1 is attributable to the N–Q–N–Q stretch of the quinonoid ring, which clearly indicated the presence and formation of PANI on the graphene surfaces.36 The shift of absorption peak is probably attributed to strong π–π interactions between the conjugated PANI and graphene frameworks. The FTIR result suggested that PANI was successfully grafted into graphene, and G/PANI composite coating was fabricated by the electrodeposition method.
O and C–O) indicated that the partial reduction of GO.1,2 Many low-intensity peaks ranging from 780 to 580 cm−1 can be assigned to the vibrations of the C–H bonds in the benzene rings (Fig. S1a and S1b†). Compared with graphene, however, several new peaks attributed to PANI were found in the spectrum of G/PANI. Notably, the vibrations of CO at around 1654 cm−1 decreased distinctly, and new peaks around 1567 and 807 cm−1 corresponded the vibrations of CN and C–H appeared, respectively. In addition, a stretching band attributed to C–N in aromatic amine appeared at 1381 cm−1. The characteristic band at around 1135 cm−1 is attributable to the N–Q–N–Q stretch of the quinonoid ring, which clearly indicated the presence and formation of PANI on the graphene surfaces.36 The shift of absorption peak is probably attributed to strong π–π interactions between the conjugated PANI and graphene frameworks. The FTIR result suggested that PANI was successfully grafted into graphene, and G/PANI composite coating was fabricated by the electrodeposition method.
Stability and robustness of G/PANI coating
In order to use in NTME-GC-MS, the mechanical stability and thermal stability are vital for the coating. It has been reported that the thermal stability of PANI coating was poor,37 and it is not suitable for GC-MS analysis. In this work, graphene was selected to incorporate with PANI to improve the thermal stability of the polymeric coating. The robustness and thermal stability of the coating was investigated by comparison of the peak responses of the target analytes after different use times (1, 50, 150, 200 and 300 times). As shown in Fig. 3, almost no any significant decline of peak responses was found for eight VOCs. The above results demonstrated that G/PANI coating possessed excellent mechanical and thermal stability and superior robustness, it is suitable to use in NTME-GC-MS analysis.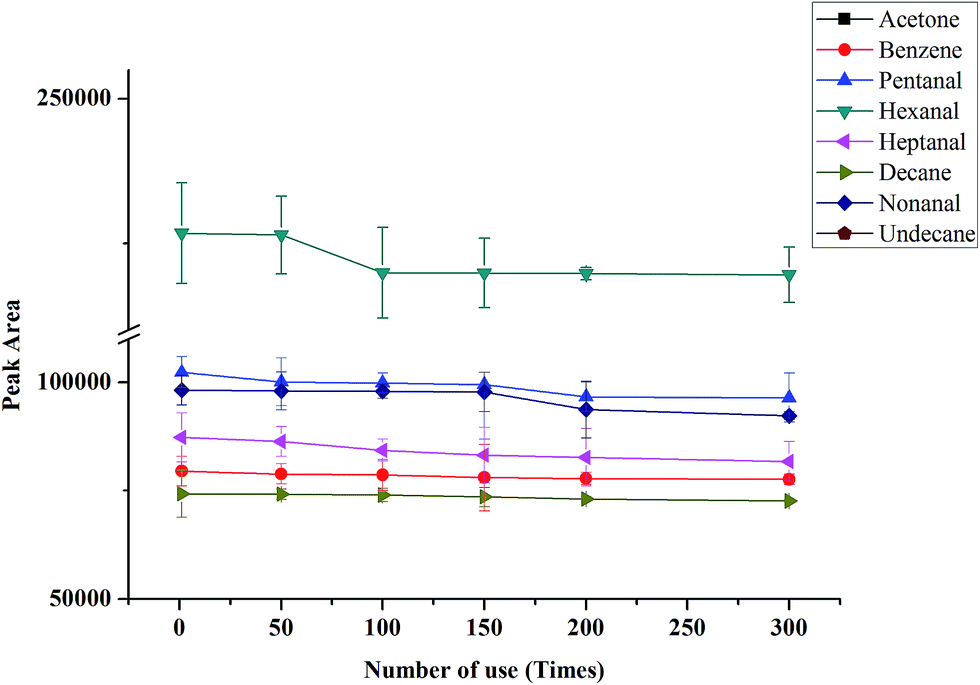 | ||
| Fig. 3 Durability and lifetime of the coating. VOCs concentration: 2 μg L−1. | ||
Enrichment performance
Since this is the first report on the use of G/PANI coating in NTD, the performance aspects of the proposed sampler needed to be evaluated by comparing it with some well-established sampling techniques. For this reason, some different commercial SPME fibers including PDMS/CAR, PDMS/DVB and PA were compared to evaluate the extraction efficiency (per unit area) of the novel coating toward eight VOCs. From Fig. 4, it is showed that G/PANI coating presented higher extraction efficiency compared with PDMS/DVB and PDMS/CAR coatings, especially for polar aldehydes, which might be explained for the enhanced π–π stacking interaction and larger surface area of G/PANI coating.30 What's more, it's worth mentioning that the comparable extraction efficiency was obtained with PA coating. At the same time, the extraction efficiency of the G/PANI coated needle was also compared with G/PANI coated fiber and PANI coated needle. The results showed that G/PANI needle presented higher peak area compared with G/PANI fiber (almost 2.1–20 folds) and PANI coated needle (about 1.2–1.9 folds). The better extraction performance can be explained for the increased surface area of coating (for the former) and the enhanced π–π stacking conjugated interaction and hydrogen bonding interaction between conjugated aldehyde derivatives and the composite G/PANI coating (for the latter). The above results indicated that G/PANI needle presented higher extraction capacity.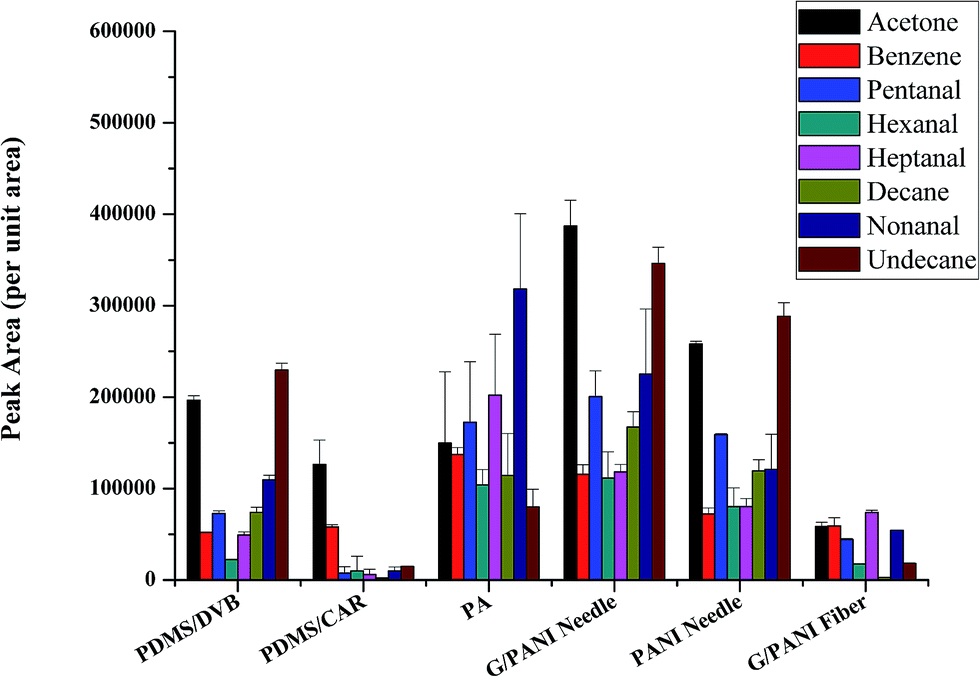 | ||
| Fig. 4 Comparison with different commercial SPME fibers (PDMS/CAR, PDMS/DVB and PA), PANI needle and G/PANI fiber. VOCs concentration: 2 μg L−1. | ||
Selection of experimental conditions
In the experiment, to evaluate the performance of the NTD-G/PANI, several important parameters, including sampling methods, the sample flow rate through the needle, extraction temperature and time, desorption time and temperature were optimized systematically with the standard solution of eight VOCs (2 μg L−1).Comparison of sampling methods
Conventional static headspace (HS) extraction was evaluated and compared with dynamic sampling strategies using NTDs. The main advantage of NTME is that VOCs are retained by an adsorption mechanism.38 By increasing the extraction temperature, the amount of analytes in the headspace is increased so improving the sensitivity of the method. However, some sensitivity is also lost due to the fact that the increased temperature results in a partial desorption of the VOCs from the sorbent particles of the NTD. The combined adsorption/desorption mechanism therefore requires an accurate determination of the thermostating temperature to find the optimum sampling conditions when working with NTDs.Dynamic HS extraction, more commonly known as purge-and-trap sampling, can be used to increase the sensitivity of headspace sampling. What's more, it can greatly shorten the equilibrium time. By comparing static HS with dynamic sampling strategies using NTDs, the latter possesses higher extraction efficiency, shorter equilibrium time and lower extraction temperature, which might be explained for the enhanced mass transfer. Therefore, dynamic HS mode was selected for the following sampling method in the further studies.
Sampling flow
As is known to us, sampling flow is a vital parameter for NTME. A high sampling rate is desirable for rapid analysis, while it may cause the incomplete adsorption of the target analytes, which is inaccurate for quantitative analysis, especially for trace analysis. The extraction flow rate was varied between 1 mL min−1 and 8 mL min−1 at constant extraction time (10 min) in Fig. 5A. As Fig. 5A shows, the extraction efficiencies increased with the speeded sampling rate at first, and the maximum extraction efficiencies were achieved at a flow rate of 5 mL min−1 for eight VOCs. For decane and undecane, the curve leveled off or decreased slightly. This is probably because the adsorbed analytes generated anti-adsorption at a low sampling flow and residence time. With decreasing extraction flow rate, an increase in the extraction yield occurred indicating a higher degree of non-equilibrium sorption due to rate limiting diffusion into the extraction phase at higher extraction flow rates. As a consequence, a sampling rate of 5 mL min−1 was selected for the following investigation in the further studies.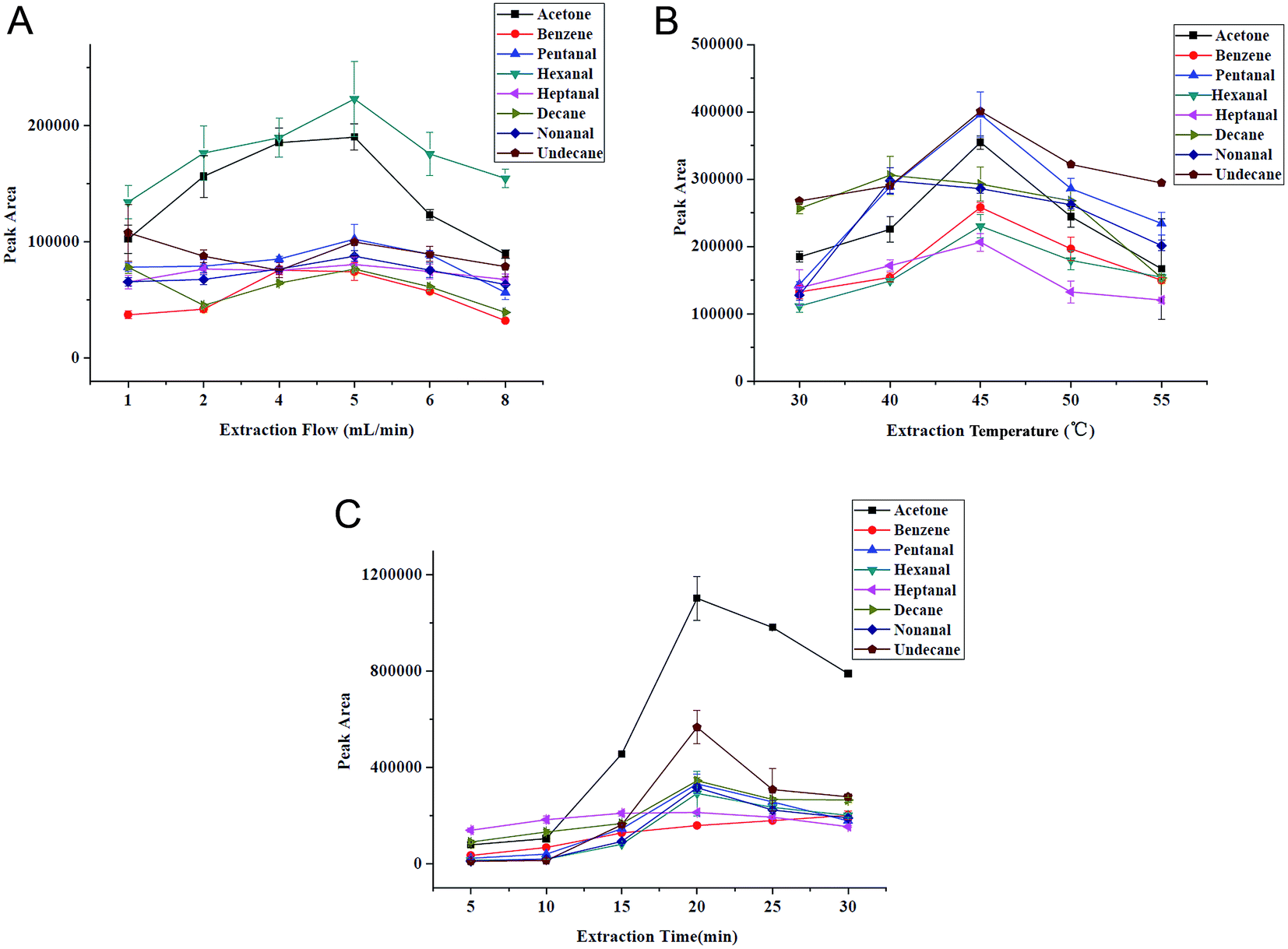 | ||
| Fig. 5 Selection of extraction conditions. (A) Sampling flow, (B) extraction temperature and (C) extraction time. VOCs concentration: 2 μg L−1. | ||
In order to examine whether the target analytes are absorbed completely in dynamic HS mode, the residual gas in the sample vial was absorbed and analyzed by NTD-SPME-GC/MS method a second time. No VOCs was found in residual gas, indicating that the absorption was almost complete.
Sampling temperature and time
Generally, high temperature can enhance distribution velocity, which is benefit for the diffusion of gaseous analytes to needle, thereby improving extraction ability of SPME. But adsorption is an exothermic process, high temperature will decrease partition coefficient of analytes towards the extraction phase. Fig. 5B demonstrates the extraction temperature profiles for VOCs and we can see that the extracted amount increases with the elevated temperature (from 30 to 45 °C) besides decane, but decreases obviously after 45 °C. Therefore, 45 °C was chosen to be the optimum.NTME is an equilibrium-based process. Extraction time, a key factor in NTME is closely related to extraction efficiency. The selection of extraction time has direct impact on the sensitivity and repeatability of the proposed analytical method. The influence of the extraction time on the extraction efficiency was studied at 5, 10, 15, 20, 25 and 30 min with constant sampling rate (5 mL min−1). The extraction time profile is shown in Fig. 5C. We can find that the extraction efficiency increases with time within 20 min and slightly decreases in 25 min. Thus, the extraction time of 20 min was selected for the following studies. The relatively short equilibration time is attributed to the porous structure and thin thickness of G/PANI coating which accelerates mass transfer.
Desorption time and temperature
Desorption time and temperature are important performance parameters of the NTME technique, since they have significant effects on the peak response of the GC instrument. Clearly higher desorption temperatures and longer desorption times can result in the complete desorption of analytes with less carryover and memory effect. Desorption times and temperatures were assessed for NTDs in five and six levels of 3–15 min and 220–270 °C (see in ESI Fig. S2†), respectively. Three consecutive analyses were done for each desorption time and temperature. Higher temperatures can cause better desorption with less carryover on the sorbent surface but this is limited by the degree of separation of the compounds of interest. For these reasons, optimum desorption time and temperature were selected at 5 min and 250 °C.Comparison with other methods
The new wall-coated NTD method has been developed and is compared to previously reported literatures11,39 with sorbent-packed NTDs, the results are listed in Table 1. Theoretically, wall-coated NTDs are apt for air/headspace analysis. It possesses good permeability, high analysis speed, no eddy diffusion and small mass transfer resistance. For sorbent-packed NTD, the extraction capacity of sorbents maybe is higher because of the larger sorbent loading amount. So, the LODs in ref. 11 are lower than our method. However, it is a tough and time-consuming work to pack the sorbent materials into narrow-diameter needles. In comparison with packing methods,11,39 the proposed wall-coated NTD is easy to prepare, its precision is better compared with the other two methods, and wider linear range is obtained than that in ref. 11. In addition, the excellent thermal stability of the G/PANI coating enables it can stand up high temperature up to 300 °C. While the optimal desorption temperature for the polymer in ref. 11 was only 200 °C.| Extraction device | Sorbents | Linear range (ng L−1) | LOD (ng L−1) | RSD (%) | Real sample | Ref. |
|---|---|---|---|---|---|---|
| Coated needle | Graphene–polyaniline | 5–2000 | 2–3 | 4.2–15.3 | EBV | This work |
| Packed needle | Carboxen 1000 and Tenax TA | 200–7000 | 20–80 | 15–23 | Blood samples | 39 |
| Packed needle | PDMS, DVB Carbopack X and Carboxen 1000 | 0.37–1373 | 0.01–0.03 | 4.74–18.51 | — | 11 |
Analytical performance of NTD-SPME-GC-MS
The NTME method was developed for the quantification of eight VOCs in EBV samples under the optimized conditions. Parameters for analytical performance were investigated and are listed in Table 2. Good linearity were obtained in the range of 5–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ng L−1 for acetone and benzene and 5–2000 ng L−1 for other VOCs with R2 ≥ 0.9905. The limits of detection (LODs) with signal-to-noise ratio of 3 (S/N = 3) were between 2 and 3 ng L−1. The limits of quantification (LOQ) of eight VOCs were between 7 and 10 ng L−1, which is sufficient to determine trace amounts of VOCs in EBV samples. The reproducibility between five duplicate runs including intra-day and inter-day was determined at three spiked concentration levels and relative standard deviation (RSD) for each peak was calculated. RSD values ranging from 4.2% to 15.3% were found (Table 2), which indicates good reproducibility of the method. Recoveries were also tested in EBV matrix at three concentration levels (5, 150 and 1000 ng L−1) to evaluate the applicability of the proposed method. As Table 2 shows acceptable relative recoveries in the direct injection of spiked EBC sample were achieved for the selected aldehydes ranging from 70 to 120%, indicating that our method was also free from matrix effect. GC-MS-SIM chromatograms of VOCs in EBV samples are shown in Fig. 6 ((A) healthy volunteer, (B) lung cancer patient, (C) lung cancer cell line, (D) spiked EBV of healthy volunteer (0.05 μg L−1), (E) spiked EBV (5 μg L−1)). And a comparison of GC-MS-SIM chromatograms of VOCs extracted by SPME PA fiber and G/PANI needle are shown in the Fig. S3 of ESI.† These results show that the proposed analytical method is effective and reliable.
000 ng L−1 for acetone and benzene and 5–2000 ng L−1 for other VOCs with R2 ≥ 0.9905. The limits of detection (LODs) with signal-to-noise ratio of 3 (S/N = 3) were between 2 and 3 ng L−1. The limits of quantification (LOQ) of eight VOCs were between 7 and 10 ng L−1, which is sufficient to determine trace amounts of VOCs in EBV samples. The reproducibility between five duplicate runs including intra-day and inter-day was determined at three spiked concentration levels and relative standard deviation (RSD) for each peak was calculated. RSD values ranging from 4.2% to 15.3% were found (Table 2), which indicates good reproducibility of the method. Recoveries were also tested in EBV matrix at three concentration levels (5, 150 and 1000 ng L−1) to evaluate the applicability of the proposed method. As Table 2 shows acceptable relative recoveries in the direct injection of spiked EBC sample were achieved for the selected aldehydes ranging from 70 to 120%, indicating that our method was also free from matrix effect. GC-MS-SIM chromatograms of VOCs in EBV samples are shown in Fig. 6 ((A) healthy volunteer, (B) lung cancer patient, (C) lung cancer cell line, (D) spiked EBV of healthy volunteer (0.05 μg L−1), (E) spiked EBV (5 μg L−1)). And a comparison of GC-MS-SIM chromatograms of VOCs extracted by SPME PA fiber and G/PANI needle are shown in the Fig. S3 of ESI.† These results show that the proposed analytical method is effective and reliable.
| Analyte | Regression equation (n = 7) | Linear range (ng L−1) | R2 | LOD (ng L−1) | LOQ (ng L−1) | Precision (RSD, %, n = 5) | Recovery (%) | |
|---|---|---|---|---|---|---|---|---|
| Intra-day | Inter-day | |||||||
| a Spiked aldehydes concentrations: 5 ng L−1.b Spiked aldehydes concentrations: 150 ng L−1.c Spiked aldehydes concentrations: 1000 ng L−1. | ||||||||
| Acetone | y = 92309x + 247592 | 5–20000 |
0.9905 | 3 | 10 | 6.3a | 4.2a | 104a |
| 8.9b | 6.9b | 99b | ||||||
| 12.5c | 9.5c | 104c | ||||||
| Benzene | y = 26916x + 69989 | 5–20000 |
0.9993 | 3 | 10 | 6.6 | 4.8 | 70 |
| 9.6 | 10.2 | 91 | ||||||
| 15.3 | 12.4 | 98 | ||||||
| Pentanal | y = 34882x + 198.41 | 5–2000 | 0.9957 | 2 | 7 | 7.5 | 6.3 | 102 |
| 12.4 | 11.4 | 98 | ||||||
| 14.7 | 14.3 | 98 | ||||||
| Hexanal | y = 25407x + 4793.7 | 5–2000 | 0.9988 | 2 | 7 | 10.8 | 9.3 | 107 |
| 9.2 | 9.5 | 100 | ||||||
| 8.8 | 10.2 | 89 | ||||||
| Heptanal | y = 35652x − 642 | 5–2000 | 0.9934 | 2 | 7 | 10.5 | 11.5 | 81 |
| 4.8 | 8.4 | 119 | ||||||
| 10.7 | 7.9 | 90 | ||||||
| Decane | y = 66835x − 2375.1 | 5–2000 | 0.9934 | 2 | 7 | 10.4 | 11.2 | 97 |
| 6.5 | 7.6 | 120 | ||||||
| 12.7 | 11.5 | 116 | ||||||
| Nonanal | y = 56049x + 2240.7 | 5–2000 | 0.9977 | 3 | 10 | 10.4 | 11.2 | 92 |
| 9.5 | 9.6 | 83 | ||||||
| 11.3 | 12.4 | 102 | ||||||
| Undecane | y = (1.1 × 105)x − 2824 | 5–2000 | 0.9963 | 3 | 10 | 7.5 | 8.3 | 92 |
| 12.4 | 11.5 | 78 | ||||||
| 11.9 | 12.6 | 117 | ||||||
 | ||
| Fig. 6 GC-MS-SIM chromatograms of VOCs in EBV samples ((A) healthy volunteer, (B) lung cancer patient, (C) lung cancer cell line, (D) spiked EBV of healthy volunteer (0.5 μg L−1), (E) spiked EBV (5 μg L−1)). | ||
Application in EBV and A549 cell samples
To evaluate the reliability of the method, the EBV samples of 8 lung cancer patients and 8 healthy volunteers and A549 cells were analyzed under the optimum conditions. The results in Table 3 show that the concentrations of nearly all analytes (except for decane) in the EBV samples of patients were higher than those of healthy people. The statistically analysis using the student t-test was performed at 95% confidence level. The t values of acetone, pentanal, hexanal, heptanal and nonanal were exceed the theoretical value, especially for pentanal and heptanal, demonstrating there existed statically difference between the two groups. In addition, the concentrations in lung cancer subjects are basically in accordance with VOCs in A549 cells. VOCs found in lung cancer cell line are favorable to the cognition of the mechanism of VOCs generating in lung cancer patients. The study of lung cancer markers from the cell level can effectively avoid the interference of exogenous matrix (such as environment, food and smoking). The results further validate that the proposed method is reliable and acceptable for the analysis of VOCs in human EBV and cell lines. Meanwhile, long-term and extensive research with more statistic data is required in further study.| Concentration (μg L−1) | Acetone | Benzene | Pentanal | Hexanal | Heptanal | Decane | Nonanal | Undecane |
|---|---|---|---|---|---|---|---|---|
| a Theoretical t (7, 95%) = 2.3646. | ||||||||
| Average conc. in controls | 9.65 | 5.57 | 0.53 | 1.29 | 0.48 | 0.64 | 1.12 | 0.23 |
| Conc. range in controls | 7.81–12.56 | 2.75–8.22 | 0.22–1.10 | 0.53–2.36 | 0.11–1.31 | 0.21–1.18 | 0.78–1.43 | 0.04–0.54 |
| Average conc. in patients | 10.38 | 6.00 | 1.81 | 1.47 | 1.47 | 0.59 | 1.20 | 0.34 |
| Conc. range in patients | 7.24–16.67 | 4.20–7.48 | 1.30–1.98 | 0.44–2.85 | 0.94–2.05 | 0.29–1.25 | 0.8–1.67 | 0.09–0.75 |
| Average conc. in A549 cell | 10.70 | 6.12 | 2.01 | 1.32 | 1.55 | 0.63 | 1.34 | 0.39 |
| Conc. range in A549 cell | 6.87–16.24 | 3.94–9.13 | 0.98–3.10 | 0.65–1.95 | 1.23–2.7 | 0.32–1.11 | 0.97–1.56 | 0.15–0.70 |
| ta | 2.4627 | 1.6003 | 9.1801 | 2.7026 | 19.5233 | 1.1899 | 2.7007 | 2.1084 |
Conclusions
In this work, we reported the first example of electrochemically deposited G/PANI coating in 21-gauge stainless steel needle for the analysis of the volatile metabolites in the headspace of cancer cell cultures and gaseous samples, such as human breath. This composite coating showed good mechanical and chemical stability, excellent extraction efficiency, long lifespan, superior selectivity and good biocompatibility. The NTD permits the dynamic active analysis of complex exhaled breath vapor sample. NTME is an inexpensive microextraction technique which can be used for the sampling and determination of a wide range of compounds in a single step. Wide linear range, low limits of detection and high recovery can be obtained for the method. The proposed method provides an alternative tool to the determination of VOCs from human exhaled breath and headspace gas of cell lines.Conflict of interest
The authors have declared no conflict of interest.Acknowledgements
This research was financially supported by National Natural Science Foundation of China (No. 21675058) and self-determined research funds of CCNU from the colleges' basic research and operation of MOE (No. CCNU16A02040).References
- J. Pawliszyn, TrAC, Trends Anal. Chem., 1995, 14, 113–122 CAS.
- A. Gjelstad and S. Pedersen-Bjergaard, Anal. Bioanal. Chem., 2014, 406, 375–376 CrossRef CAS PubMed.
- D. Vuckovic, Current trends and challenges in sample preparation for global metabolomics using liquid chromatography-mass spectrometry, Anal. Bioanal. Chem., 2012, 403, 1523–1548 CrossRef CAS PubMed.
- K. Demeestere, J. Dewulf, B. D. Witte and H. V. Langenhove, J. Chromatogr. A, 2007, 1153, 130–144 CrossRef CAS PubMed.
- A. Namera and T. Saito, Bioanalysis, 2013, 5, 915–932 CrossRef CAS PubMed.
- C. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- H. H. Son, S. Bae and D. S. Lee, Anal. Chim. Acta, 2012, 751, 86–93 CrossRef CAS PubMed.
- A. P. Wang, F. Fang and J. Pawliszyn, J. Chromatogr. A, 2005, 1072, 127–135 CrossRef CAS PubMed.
- J. M. Warren and J. Pawliszyn, J. Chromatogr. A, 2011, 1218, 8982–8988 CrossRef CAS PubMed.
- I.-Y. Eom, A.-M. Tugulea and J. Pawliszyn, J. Chromatogr. A, 2008, 1196–1197, 3–9 CrossRef CAS PubMed.
- P. Trefz, S. Kischkel, D. Hein, E. S. James and J. K. Schubert, J. Chromatogr. A, 2012, 1219, 29–38 CrossRef CAS PubMed.
- H. L. Lord and J. Pawliszyn, Anal. Chem., 1997, 69, 3899–3906 CrossRef CAS PubMed.
- H. Yu, L. Xu and P. Wang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 826, 69–74 CrossRef CAS PubMed.
- G. Ouyang and J. Pawliszyn, Anal. Bioanal. Chem., 2006, 386, 1059–1073 CrossRef CAS PubMed.
- H. Kataoka, H. L. Lord and J. Pawliszyn, J. Chromatogr. A, 2000, 880, 35–62 CrossRef CAS PubMed.
- I. Y. Eom, V. H. Niri and J. Pawliszyn, J. Chromatogr. A, 2008, 1196, 10–14 CrossRef PubMed.
- H. Bagheri, H. Piri-Moghadam and A. Es’haghi, J. Chromatogr. A, 2011, 1218, 3952–3957 CrossRef CAS PubMed.
- W. P. Zhang, J. Zhang, T. Bao, W. Zhou, J. W. Meng and Z. L. Chen, Anal. Chem., 2013, 85, 6846–6854 CrossRef CAS PubMed.
- T. T. Wang, Y. H. Chen, J. F. Ma, M. J. Hu, Y. Li, J. H. Fang and H. Q. Gao, Anal. Bioanal. Chem., 2014, 406, 4955–4963 CrossRef CAS PubMed.
- M. Alonso, L. Cerdan, A. Godayol, E. Anticó and J. M. Sanchez, J. Chromatogr. A, 2011, 1218, 8131–8139 CrossRef CAS PubMed.
- M. Heidari, A. Bahrami, A. R. Ghiasvand, M. R. Emam, F. G. Shahna and A. R. Soltanian, Talanta, 2015, 131, 142–148 CrossRef CAS PubMed.
- L. Vallecillos, F. Borrull, J. M. Sanchez and E. Pocurull, Talanta, 2015, 132, 548–556 CrossRef CAS PubMed.
- I. Ueta, E. L. Samsudin, A. Mizuguchi, H. Takeuchi, T. Shinki, S. Kawakubo and Y. Saito, J. Pharm. Biomed. Anal., 2014, 88, 423–428 CrossRef CAS PubMed.
- M. Mieth, J. K. Schubert, T. Groger, B. Sabel, S. Kischkel, P. Fuchs, D. Hein, R. Zimmermann and W. Miekisch, Anal. Chem., 2010, 82, 2541–2551 CrossRef CAS PubMed.
- M. Heidari, A. Bahrami, A. R. Ghiasvand, F. G. Shahna and A. R. Soltanian, Talanta, 2012, 101, 314–321 CrossRef CAS PubMed.
- M. Heidari, A. Bahrami, A. R. Ghiasvand, F. G. Shahna and A. R. Soltanian, Anal. Chim. Acta, 2013, 785, 67–74 CrossRef CAS PubMed.
- L. N. Xu, X. Y. Qi, X. J. Li, Y. Bai and H. W. Liu, Talanta, 2016, 146, 714–726 CrossRef CAS PubMed.
- Z. F. Li, H. Y. Zhang, Q. Liu, Y. D. Liu, L. Stanciu and J. Xie, Carbon, 2014, 71, 257–267 CrossRef CAS.
- J. Li, S. Liu, J. H. Yu, W. J. Lian, M. Cui, W. Xu and J. D. Huang, Sens. Actuators, B, 2013, 188, 99–105 CrossRef CAS.
- A. Mehdinia, H. Khani and S. Mozaffari, Microchim. Acta, 2014, 181, 89–95 CrossRef CAS.
- Y. Li and H. Xu, J. Chromatogr. A, 2015, 1395, 23–31 CrossRef CAS PubMed.
- F. D. Francesco, R. Fuoco, M. G. Trivella and A. Ceccarini, Microchem. J., 2005, 79, 405–410 CrossRef.
- S. M. Gordon, J. P. Szldon, B. K. Krotoszynski, R. D. Gibbons and H. J. O'Neill, Clin. Chem., 1985, 31, 1278–1282 CAS.
- W. Filipiak, A. Sponring, A. Filipiak, C. Ager, J. Schubert, W. Miekisch, A. Amann and J. Troppmair, Cancer Epidemiol., Biomarkers Prev., 2010, 19, 182–195 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- N. A. Kumar, H. J. Choi, Y. R. Shin, D. W. Chang, L. M. Dai and J. B. Baek, ACS Nano, 2012, 6, 1715–1723 CrossRef CAS PubMed.
- J. Yan, T. Wei, Z. J. Fan, W. Z. Qian, M. L. Zhang, X. D. Shen and F. Wei, J. Power Sources, 2010, 195, 3041–3045 CrossRef CAS.
- J. Ai, Anal. Chem., 1997, 69, 1230–1236 CrossRef CAS.
- M. Alonso, M. Castellanos, E. Besalú and J. M. Sanchez, J. Chromatogr. A, 2012, 1252, 23–30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25453e |
| This journal is © The Royal Society of Chemistry 2017 |