
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards understanding of laccase-catalysed oxidative oligomerisation of dimeric lignin model compounds†
B. Ramalingam *a,
B. Sanab,
J. Seayada,
F. J. Ghadessyb and
M. B. Sullivanc
*a,
B. Sanab,
J. Seayada,
F. J. Ghadessyb and
M. B. Sullivanc
aOrganic Chemistry, Institute of Chemical and Engineering Sciences, #07-01/02 Neuros, 8 Biomedical Grove, Singapore 138 665. E-mail: balamurugan_ramalingam@ices.a-star.edu.sg; Fax: +65 6464 2102; Tel: +65 6799 8529
bp53 Laboratory, #06-04/05 Neuros/Immunos, 8A Biomedical Grove, Singapore 138 648
cInstitute of High Performance Computing, 1 Fusionopolis Way, #16-16 Connexis, Singapore 138 632
First published on 20th February 2017
Abstract
Laccase-catalysed oligomerisation of dimeric β-O-4 linked lignin model compounds was studied in detail to understand the oligomerisation process by monitoring the reaction progress using high performance liquid chromatography (HPLC) and mass spectroscopy (MS). The initial oxidation intermediates of oligomerisation were isolated for the first time and characterised by spectroscopic methods sucessfully. The experimental observations indicated that C5–C5′ biphenyl linkages, one of the most thermodynamically stable linkages present in the native lignin, are formed exclusively during the early stage of the oligomerisation process. The experimental observations were supported by density functional theory (DFT) calculations of relative free energies of possible products. The C5–C5′ biphenyl tetramer is the thermodynamically more favoured product compared to the C5–O–C4′ product by a free energy difference of 10.0 kcal mol−1 in water. Among the various linking possibilities for further formation of hexamers, the thermodynamically more stable product with a similar C–C linkage is proposed as a plausible structure based on the mass of the hexamer isolated and DFT calculations. The current study demonstrates that laccase catalyzes the oligomerisation more preferentially than oxidative bond cleavage in β-O-4 linkages and that product formation is likely controlled by the thermodynamic stability of the resultant oligomers.
Introduction
Lignin, the second most abundant natural polymer has attracted much attention as one of the potential feedstocks for the sustainable production of fuels,1 chemicals1,2 and materials.3 The lignin polymer mainly consists of phenylpropane units that are bonded through various types of C–O and C–C linkages,3 formed from monolignol such as p-coumaryl, coniferyl and sinapyl alcohols (Scheme 1). The polymerisation of monolignols is believed to be catalysed by enzyme cocktails comprising laccase and peroxidases by the preliminary formation of a phenoxy radical followed by uncontrolled radical polymerisation.4 Synthetically, laccase, a multi-copper containing metalloenzyme has been used in the polymerisation of phenol and amine containing aromatics5 as well as lignin model compounds in the presence of molecular oxygen. The redox potential of the substrates and the nature of substituents on the aromatic ring have been shown to affect the polymerisation. For instance, in the case of phenols,6 naphthols7 and methoxy phenols,8 enhanced polymerisation rates were observed when a methoxy substituent is present at the ortho-position relative to the phenolic OH group. The redox potential of substrates was found9 to be important in the fungal laccase-catalysed oxidation of various lignin model compounds. The reaction rates of oxidation were studied by monitoring the oxygen consumption and compared with physiochemical properties of monomeric and dimeric model compounds without details on isolated products. In a very recent study, encapsulated laccases were used10 as stable catalysts for the oxidative polymerisation of aromatics such as syringic acid, gallic acid, vanillic acid and syringol to obtain lignin-like oligomers with molecular weight ranging between 400–2100 Da. The low molecular oligomeric materials obtained from lignin model compounds, are expected to possess some of the characteristics of the native lignin. Thus, the phenol containing lignin-like oligomers could potentially be utilized as an additive to plastics,11 sunscreen lotions,12 as antioxidants13 and UV absorbers14 similar to lignin. The low molecular weight lignin oil was reported15 to be a potential lubricant additive for commercial engine oils with better miscibility and compatibility than kraft lignin.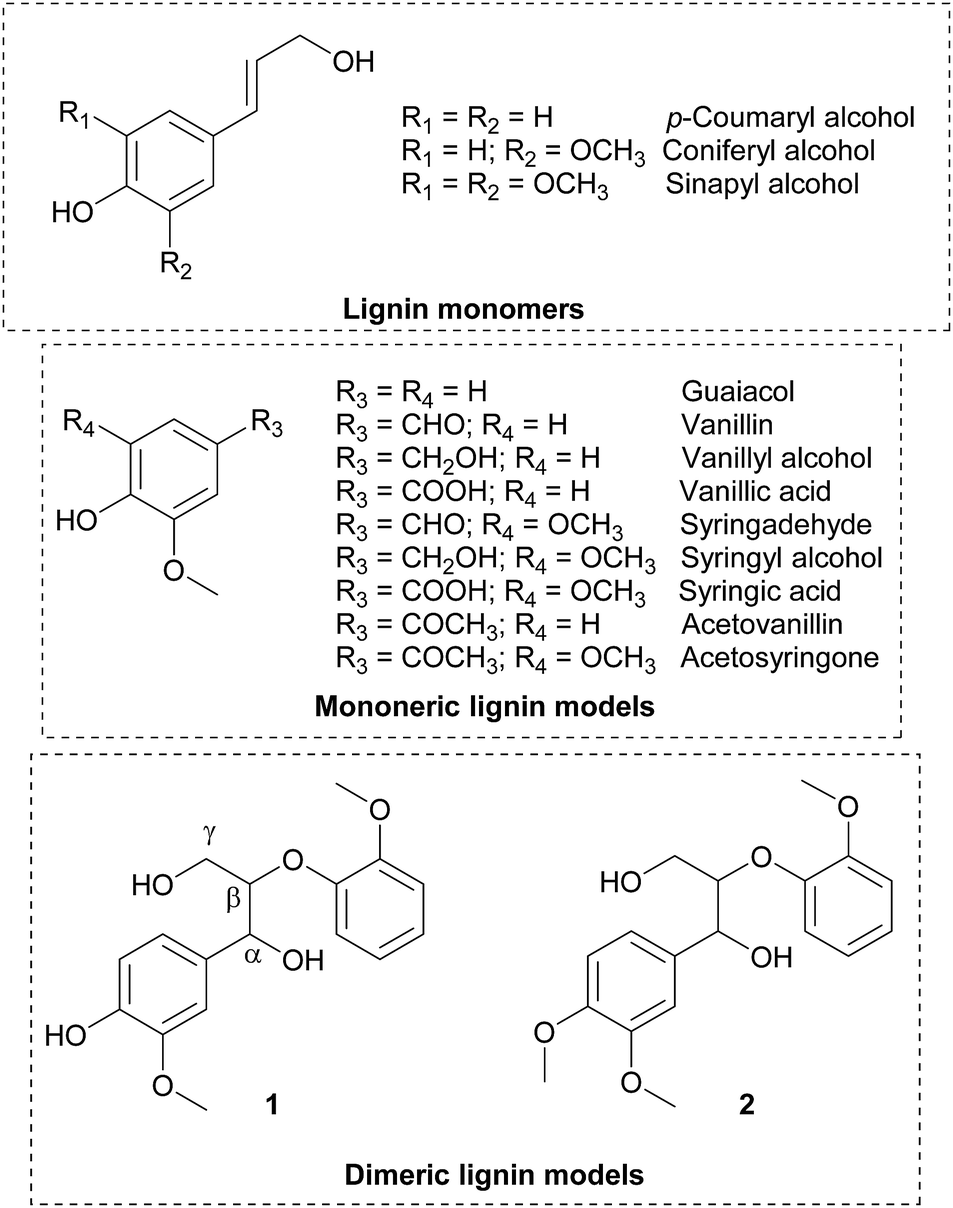 | ||
| Scheme 1 Chemical structure of lignin monomers and models. | ||
As laccase is one of the key enzymes believed to be responsible for the bio-synthesis and degradation of lignins in nature, it is important to understand the mechanism of laccase catalysed oxidations to explore new applications of laccase catalysis. However, in laccase catalysed oxidations, complexity in uncontrolled reactions of phenolic radicals and limitations in isolating the initial oxidation products hinder understanding of the lignin polymerisation and depolymerisation processes. In this regard, Lahtinen et al.16 have studied the product distribution in the laccase catalysed oxidation of monomeric lignin models such as vanillyl alcohol in detail. Computational evaluation of the oxidation products of vanillyl alcohol and its reaction intermediates indicated that the hydrogen bonding provides an additional stabilising effect for formation of the thermodynamically more stable C5–C5′ linkage. Attempts were also made17 to isolate and characterize the products obtained in the fungal laccase catalysed oxidation of guaiacylic and syringylic lignin model compounds. The oxidation of secondary alcohol (Cα) was observed for syringyl alcohol and syringylic β-O-4 dimers, while three types of C5–C5′ coupling products were reported for vanillyl alcohol. The Trametes versicolor laccase (TVL) catalysed selective dimerization of lignin model compounds such as vanillin, acetovanillin, methylvanilllate and eugenol was reported recently.18 The initial oxidation products were characterised by NMR and all of them were found to contain C5–C5′ linkages. However, no details on linkages present in the higher molecular weight oligomers were reported. Laccase-catalysed oxidative phenolic coupling products with C5–C5′ linkages were reported19 for a series of vanillidine derivatives to afford dimeric products in 79–93% yields.
The polymerisation of lignin model compounds including β-O-4 linked dimer 1 was reported by Rittstieg et al. using T. hirsuta laccase as a catalyst in the absence20 and presence21 of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS). The molecular weight distribution (MWD) 4000 Da was reported in the absence of ABTS, while increase in MWD was observed in presence of ABTS. About 21% of initially used ABTS was found to present in the resulting polymeric product. Although laccase-catalysed oligomerisation of dimeric lignin model compounds has been reported in the literature, details of the oligomerisation process and linkages present in the oxidation products have not been reported so far. The objective of the current study was to gain insight into the laccase catalysed oxidation process and the mode of linkages present in the initial oxidation products using β-O-4 linked dimeric lignin model compounds that are considered close mimetics of native lignin. Unlike the monomeric lignin model compounds, the dimeric model compounds possess β-O-4 linkages, the most abundant (45–60%) linkage present in native lignin. The structural information gained on the oxidation products of such dimeric model compounds would enable (i) improved knowledge on the role of laccase in enzyme-based lignin polymerisation and depolymerisation processes, (ii) use of the oligomeric materials for specific applications with improved understanding on the linkages present and (iii) to broaden the scope of laccase-catalysed reactions for the greener synthesis of organic molecules in a more sustainable manner.
Results and discussion
In this study, the oxidation of β-O-4 linked dimeric lignin model compounds 1-(4-hydroxy-3-methoxyphenyl)-2(2-methoxy phenoxy)-propane-1,3-diol (1) and 1-(3,4-dimethoxyphenyl)-2(2-methoxyphenoxy)-propane-1,3-diol (2) were investigated adopting a similar reaction protocol reported by Rittstieg et al.20 Commercially available laccases from Trametes versicolor laccase (TVL), Rhus vernicifera laccase (RVL) and Pleurotus ostreatus laccase (POL) were used as the oxidation catalysts in the current study. Compounds 1 and 2 were independently taken in sodium acetate buffer (pH ∼5.2) under oxygen atmosphere and stirred at room temperature for 3 hours with 20 units per mL of TVL. A pale brown precipitate formed from the flask containing compound 1 was collected and found to be soluble in common organic solvents such as acetone, ethyl acetate and chloroform. Preliminary analysis by nuclear magnetic resonance (NMR) spectra indicated the complete disappearance of compound 1 and possible formation of oligomeric species. Analysis of the precipitate obtained from compound 1 by gel permeation chromatography (GPC) indicated the formation of higher molecular weight compounds with average molecular weight (Mw) of 2120 Da and polydispersity (Mw/Mn) of 1.1 (Fig. 1). A molecular weight distribution (MWD) up to 4000 Da was reported20 for the polymerisation of compound 1 using T. hirsuta laccase. The observed difference (2120 vs. 4000 Da) in MWD might be due to the variance in source and concentration of the laccase and methods used in molecular weight determination. POL also promoted the oligomerisation of 1 as that of TVL and led to the formation of oligomers with similar MWD (Mn = 2095 Da; Mw = 2165 Da; Mw/Mn = 1.0, Fig. S1, ESI†). Treatment of compound 1 with RVL did not promote any oxidation products and the starting material remained unreacted (Fig. 3) as observed by NMR and HPLC. Treatment of compound 2 with TVL, RVL and POL laccases did not initiate any oligomerisation or oxidative cleavage,22 indicating the specificity of laccase towards the phenolic substrates in the formation of phenoxy radicals that is crucial for subsequent oligomerisation. The reduction in concentration of laccase from 20 units per mL to 2 units per mL did not affect the molecular weight distribution of the oligomer formed (Mn = 2120 Da; Mw = 2170 Da; Mw/Mn = 1.0, Fig. S2, ESI†). However, the initial rate of disappearance of 1 was found to be approximately 5 fold slower (t1/2 = 34.9 min, Fig. S3, ESI†).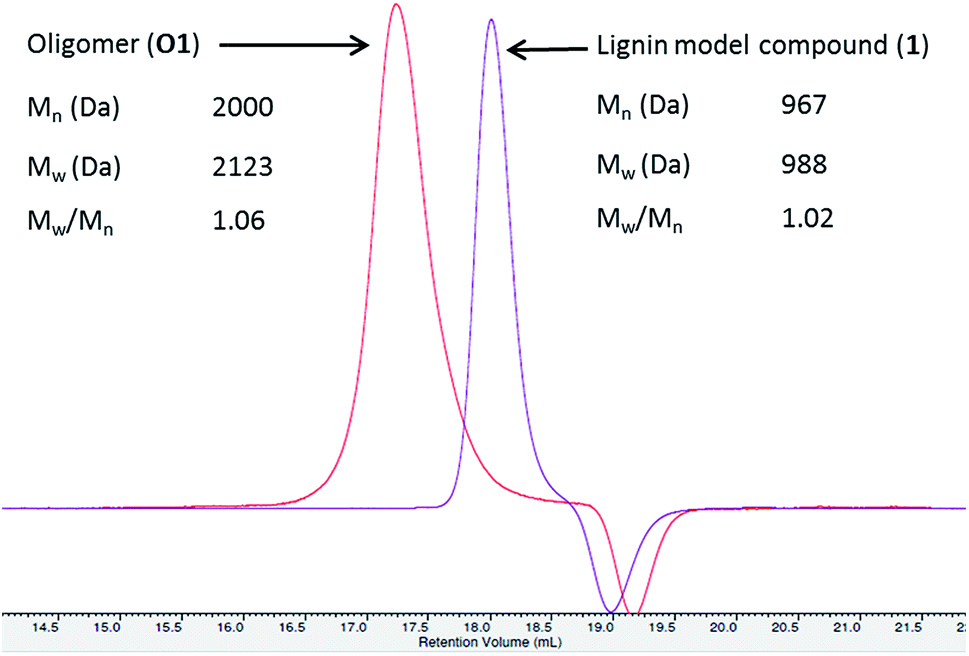 | ||
| Fig. 1 GPC of oligomers (O1). GPC lignin model compound is given for comparison. | ||
To gain understanding on the progress of the oxidation reaction, the reaction mixture was analysed by high performance liquid chromatography equipped with mass spectroscopy (LC-MS) after quenching the reaction with sodium azide at regular intervals. The percentage of unreacted compound 1 was quantified using 9-acetylphenanthrene as an internal standard. The initial reaction progress for the oxidation of 1 with laccases TVL, POL and RVL is presented in Fig. 3. The amount of 1 decreased dramatically upon treatment with TVL and POL, while RVL did not show any noticeable activity. The higher activity of TVL and POL could be attributed respectively to their higher redox potentials (790 mV and 650 mV) than that of RVL (430 mV). The HPLC chromatograms obtained for the reaction progress of 1 with TVL is presented in Fig. 2. Analysis of two new major peaks appeared (Fig. 2) at 25.2 min and 26.4 min by LC-MS indicated the mass of the tetramer (3, 661.3 Da (M+ + Na), Fig. S5, ESI†) and the hexamer (4, 979.3 Da (M+ + Na), Fig. S7, ESI†) respectively. As the reaction proceeded, the concentration of 1 decreased progressively and the formation of 3 and 4 (Fig. 2) were found to be enriched. The detection of 4 (Fig. 2) by LC-MS suggested that the oligomerisation likely took place by a step-wise association of 1 to 3. Otherwise, the self-coupling of 3 would lead to the formation of octamer directly without the formation of the hexamer 4 (Fig. 2). In contrast to the T. hirsuta laccase catalysed polymerisation of 1,20 we did not observe the formation of any low molecular weight monomeric products as evidenced by LC-MS. The results indicated that laccase promoted the oligomerisation of 1 directly rather than depolymerisation of the dimeric or higher molecular weight products into monomeric products followed by re-polymerisation. The minor peak appeared at 25.6 min (Fig. 2) exhibited the mass value of 3 (661.2 Da, M+ + Na, Fig. S6, ESI†), indicating possible formation of diastereomeric isomers of tetramer 3. The bond rotation of newly formed aryl–aryl bond (cf. below, 3C, Fig. 4) is likely restricted due to highly substituted nature of lignin model compound (1) that resulted in the development of R- and S- diastereoisomers. More possibilities could be anticipated while the next molecule of 1 couple with 3C. Consequently, analysis of peak at 27.19 min (Fig. 2) by LC-MS indicated the mass value of hexamer 4 (979.1 Da, M+ + Na, Fig. S8, ESI†). The appearance of minor peaks at 25.6 min and 27.19 min, might also be due to the formation of tetramers and hexamers with different linkages, although DFT calculation did not favour such possibilities (cf. below).
 | ||
| Fig. 2 LC-MS traces of laccase-catalysed oxidation of 1 at reaction times 0 min and 30 min. 9-Acetylphenanthrene was used as an internal standard for quantification. Mass obtained for the major and minor peaks are indicated. LC-MS traces at regular time intervals are depicted in ESI (Fig. S4†). | ||
 | ||
| Fig. 3 Comparison of initial reactivity of three different laccases. | ||
 | ||
| Fig. 4 (a) HSQC correlations of aromatic region of compounds 1 and 3C. (b) Important HMBC correlations present in compound 3C. | ||
The formation of a phenoxy radical by one electron oxidation with concomitant reduction of oxygen to water by laccase is well known.23 The phenolic radical (1R1) could undergo resonance and consequently various radical couplings are possible (Scheme 2). Simple oxidation of primary (Cγ) and secondary alcohol (Cα) could also be possible22 under the present oxidative conditions (Scheme 2). However, we did not observe any detectable amount of Cα and/or Cγ oxidised products under these conditions. In order to isolate the product that is being formed at the initial stage of the oligomerisation, the reaction was carried out with a lower concentration of laccase (2 units per mL).24 After 30 min, the reaction was quenched with an aqueous solution of NaN3. LC-MS analysis indicated the presence of the tetramer (3) predominantly (∼50%) together with compound 1 and other higher molecular weight oligomers. Tetramer (3) was separated from the other oligomers and compound 1 by semi-preparative thin layer chromatography (TLC) using 10% dichloromethane in hexane. The observed mass value of 661.39 (M + Na) by ESI mass spectroscopy indicated the isolation of tetramer 3. For the isolation of hexamer 4, the reaction was carried out with 20 units per mL of laccase and the reaction was quenched after 1 h. Isolation of hexamer was carried out by semi preparative HPLC and confirmed by mass (m/z, 979.60, M + Na) analysis.
 | ||
| Scheme 2 Laccase-catalysed oxidative dimerization of 1. Radical formation and their different coupling possibilities are shown. The more favoured structure 3C is shown. The protonation and re-aromatization sequence are not presented for clarity. Relative free energies were calculated in gas phase and water by DFT and are given respectively within parentheses. | ||
The linkage present in 3 was established by 1H, 13C and 2D NMR spectroscopy. The 1H and 13C NMR spectra were measured in deuterated acetone-D6 (Fig. S9–S12, ESI†) and the following observations and interpretations were made. (i) The presence of phenolic OH (δ, 7.39 ppm), Cα–OH (δ, 4.51 ppm) and Cγ–OH (δ, 3.69 ppm) groups observed in deuterated acetone, which ruled out the possibility of dimer formation through C–O or O–O linkages; (ii) the observation of only one set of signals for Cα–H (δ, 4.93 ppm), Cβ–H (δ, 4.33 ppm), Cγ–H (δ, 3.76 and 3.76 ppm) and methoxy (δ, 3.79 and 3.86 ppm) protons indicated that the tetramer formed from compound 1 is C2-symmetric; (iii) Attached Proton Test (APT) experiment in 13C spectrum, the number of quaternary carbons in the aromatic region was increased while the number of aromatic CH carbons decreased compared to the spectrum of compound 1 (Fig. S13, ESI†). All the above information indicated the possibility of a C2-symmetric molecule with a linkage through the aromatic carbons. The above fact was supported by the observation of only six aromatic C–H correlations in the heteronuclear single quantum coherence (HSQC) spectra compared to the seven C–H correlations found for compound 1 (Fig. 4a). The structure of tetramer 3C was finally confirmed by the heteronuclear multiple bond correlation (HMBC) spectrum (Fig. 4b). The phenolic –OH group showed both J2 and J3 correlations with three quaternary carbon atoms with δ (ppm) values 148.5, 143.7 and 126.0, which indicated the formation of a new bond at the C5 position of ‘ring A’ of compound 1. The proton assigned to one of the aromatic protons of ‘ring A’ at δ, 6.95 ppm showed strong HMBC (J3) correlations with four different carbons as indicated in Fig. 4b. The J3 correlation of the proton (δ, 6.95 ppm) at ‘ring A’ to the carbon (126.0 ppm) of ‘ring A*’ indicated that both rings are connected through a C5–C5′ linkage. Based on the above rationale, the structure of the tetramer was confirmed as 3C (Fig. 4). To our knowledge, the complete NMR interpretation for the tetramer 3C is reported for the first time.25
The exclusive formation of 3C under the experimental conditions was supported with results obtained by DFT studies. The relative free energies of hypothetically favoured products that are being formed through linkages C5–C5′ (3C) and C5–O–C4 (3B) were calculated. The formation of 3C was found to be more favoured by 5.4 kcal mol−1 than that of 3B. Solvation with water was found to destabilize 3B further by 4.6 kcal mol−1, indicating the possible role of hydrogen bonding in the formation of C5–C5′ linkage. DFT calculation for the formation 3A in gas phase (74.2 kcal mol−1) and in water (71.4 kcal mol−1) indicated that the O–O linkage would be highly unfavourable. The observations could be related to 5–5′ biphenyl linkages present in lignin. In the biosynthesis of lignin, the more stable biphenyl linkages (115–118 kcal mol−1)26 might be formed initially by the oxidative polymerisation of p-coumaryl, coniferyl and sinapyl alcohols.
NMR spectra of the hexamer 4 in acetone-D6 and chloroform-D showed a complex unresolved spectral pattern, possibly due to the existence of hexamers with different linkages that were not separable under the HPLC conditions. Based on the limited experimental details (mass, m/z = 979.60, M + Na) available for the hexamers, we envision that calculating the free energies of most possible structures would help to identify the linkages present in the hexamer. In fact, in the case of the tetramer (3C), the prediction by DFT is in good agreement with the experimentally observed product. After the initial formation of 3, radicals originating from both 1 and 3 could possibly be present in the reaction mixture. The phenoxy radical of tetramer (3R1) undergoes delocalisation as indicated in the Scheme 3. The potential transition states that are originated from the radicals centred at C1, C3 and C5 on the aromatic ‘ring A’ of 3 are not taken into consideration, as the dimerization through those carbons ultimately leads to formation of the less favoured de-aromatized product. In the LC-MS analysis, we did not observe significant formation (Fig. 2) of octamers that would possibly be formed by homo or hetero radical coupling of 3R2 and/or 3R3. Thus, based on the above rational assumptions and understanding of radical chemistry, the five most possible modes of linkages for the hexamers were considered for the DFT calculation (4A–4E, Scheme 3). Relative free energy changes (Fig. 5) for hexamers 4A–4E were compared to the most stable product. The thermodynamic stability of the hexamers follows the order 4B (0 kcal mol−1) > 4A (4.1 kcal mol−1) > 4D (5.8 kcal mol−1) > 4C (13.7 kcal mol−1) > 4E (29.3 kcal mol−1). We propose that the hexamer might be formed through a C–C linkage as illustrated in hexamer 4B. However, the difference in energy between 4A vs. 4B (4.1 kcal mol−1) and 4A vs. 4D (1.7 kcal mol−1) is not remarkably different. DFT calculation of 4A–4E was performed using water as a solvent. The observed data indicated that solvation significantly enhance the overall stability of product formation (Fig. 5). In the oxidation of vanillyl alcohol, the formation of a thermodynamically less stable C5–C5′ (by 5.6 kcal mol−1) dimer was observed16 over the most stable vanillin due to hydrogen bonding present in the former.
 | ||
| Scheme 3 Proposed pathways for the formation of hexamers (4A–4E). The unflavoured structures originated from radical resonances at C3, C1 and C5 carbons of ‘ring A’ are not presented; relative free energies were calculated in gas phase and water by DFT and are given respectively within parentheses. | ||
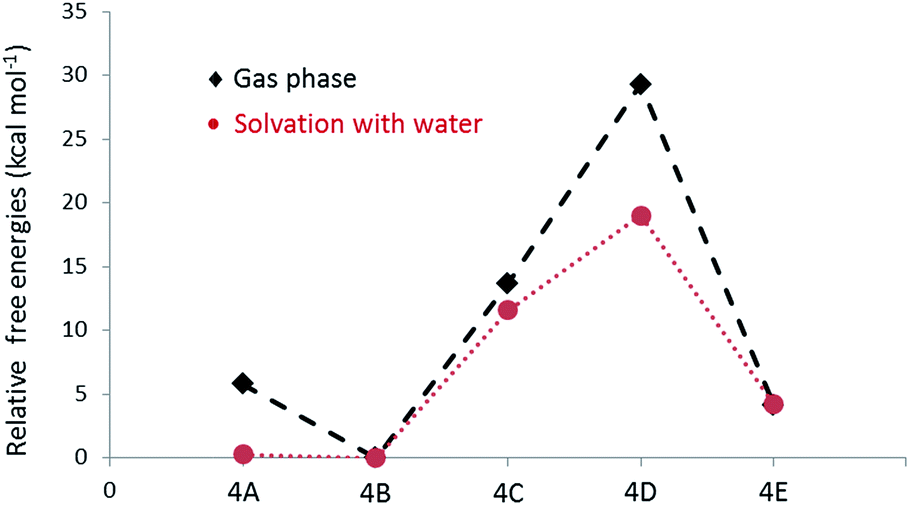 | ||
| Fig. 5 Relative free energy changes for the formation of hexamers with different linkages in gas phase and under solvation. | ||
Experimental
Chemicals and materials
Lignin model compounds 1-(4-hydroxy-3-methoxyphenyl)-2(2-methoxyphenoxy)-propane-1,3-diol (1) and 1-(3,4-dimethoxyphenyl)-2(2-methoxyphenoxy)-propane-1,3-diol (2) were purchased from TCI and Azatech chemicals respectively and used as such for all the experiments. Trametes versicolor laccase (TVL), Rhus vernicifera laccase (RVL) and Pleurotus ostreatus laccase (POL) were purchased from Aldrich and used as such for enzymatic reactions.Conclusions
The initial progress of the laccase-catalysed oxidative oligomerisation reaction for β-O-4 linked dimeric model lignin compounds was carefully studied. The redox potential and concentration of laccase were found to be important in the formation of products. The initial oligomerisation products were isolated successfully and the mode of linkages was established. The current study indicated that the oligomerisation of β-O-4 linked lignin model compound led to the formation of a tetramer through C–C linkages exclusively similar to C5–C5′ linkages present in the native lignin. The initial stage of oligomerisation process was found to be governed by the thermodynamic stability of products although different linkages are possible for the radicals to couple. The current understanding on the oligomerisation of dimeric lignin model compounds would pave the way to make important decisions in the use of laccase in the oxidative cleavage of lignin. Laccase has been considered30 as one of the potential enzymes for the depolymerisation of lignin. Our current studies indicate that oligomerisation by oxidative coupling is more preferred over the oxidative cleavages even in the case of readily cleavable β-O-4 linkages (54–69 kcal mol−1).26 Although the laccase has a potential to cleave lignin or lignin model compounds in the presence of mediators22 to phenol-containing monomers or dimers, these phenolic compounds may undergo re-polymerisation more readily under similar reaction conditions. As a result, the thermodynamically more stable oligomers or polymers will be formed at the end of the overall process. Meyer et al.26 reviewed laccase-catalysed bond cleavage in lignin and made similar conclusions earlier. Thus, laccase alone would not be able to cleave lignin under oxidative conditions. Altering the redox potential of laccase by mutation in order to facilitate the oxidation of non-phenolic hydroxyl groups, may help lignin depolymerisation in the presence of suitable redox mediators. The current observations might help to exploit the readily accessible stable laccase enzymes for aromatic C–C coupling, which is currently under exploration in our laboratory.Acknowledgements
The work was funded by Agency for Science, Technology and Research (A*STAR), Singapore under the ‘Biomass to Chemicals’ programme. Mr Zhao Wenguang and Dr Saravanan Gowrisankar are acknowledged for GPC analysis and HPLC calibration respectively. Dr Michael B. Sullivan acknowledges A*STAR Computational Resource Centre (ACRC), Singapore for the use of high performance computing facilities for the DFT computations performed in this work.Notes and references
- (a) C. Xu, R. Arneil, D. Arancon, J. Labidid and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC; (b) R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed; (c) R. Katahira, A. Mittal, K. McKinney, P. N. Ciesielski, B. S. Donohoe, S. K. Black, D. K. Johnson, M. J. Biddy and G. T. Beckham, ACS Sustainable Chem. Eng., 2014, 2, 1364–1376 CrossRef CAS.
- (a) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed; (b) C. Amen-Chen, H. Pakdel and C. Roy, Bioresour. Technol., 2001, 79, 277–299 CrossRef CAS PubMed.
- C. W. Dence and S. Y. Lin, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer-Verlang, Berlin, Heidelberg, 1992, pp. 3–19 Search PubMed.
- (a) J. M. Humphreys and C. Chapple, Curr. Opin. Plant Biol., 2002, 5, 224–229 CrossRef CAS PubMedJ. F. D. Dean and K. E. L. Eriksson, Holzforschung, 1994, 48, 21–33 CrossRef CAS; (b) D. M. O'Malley, R. Whetten, W. Bao, C. L. Chen and R. Sederoff, Plant J., 1993, 4, 751–757 Search PubMed.
- (a) J. R. Jeon, P. Baldrian, K. Murugesan and Y. S. Chang, Micobial Biotech., 2012, 5, 318–332 CrossRef PubMed; (b) N. Mita, S. Tawaki, H. Uyama and S. Kobayashi, Macromol. Biosci., 2003, 3, 253–257 CrossRef CAS; (c) A. M. Mayer and R. C. Staples, Phytochemistry, 2002, 60, 551–565 CrossRef CAS PubMed; (d) L. Gianfreda, F. Xu and J. M. Bollag, Biochem. J., 1999, 3, 1–25 CAS.
- (a) X. Sun, R. Ba, Y. Zhang, Q. Wang, X. Fan, J. Yuan, L. Cui and P. Wang, Appl. Biochem. Biotechnol., 2013, 171, 1673–1680 CrossRef CAS PubMed; (b) R. Ikeda, J. Sugihara, H. Uyama and S. Kobayashi, Macromolecules, 1996, 29, 8702–8705 CrossRef CAS.
- N. Aktas, G. Kibarer and A. Tanyolac, J. Chem. Technol. Biotechnol., 2000, 75, 840–846 CrossRef CAS.
- T. Tanaka, M. Takahashi, H. Hagino, S. I. Nudejima, H. Usui, T. Fujii and M. Taniguchi, Chem. Engg. Sci., 2010, 65, 569–573 CrossRef CAS.
- M. Lahtinen, K. Kruus, H. Boer, M. Kemell, M. Andberg, L. Viikari and J. Sipila, J. Mol. Catal. B: Enzym., 2009, 57, 204–210 CrossRef CAS.
- L. Pistone, G. Ottolina, S. De, A. A. Romero, L. O. Martins and R. Luque, ChemSusChem, 2016, 9, 756–762 CrossRef CAS PubMed.
- (a) D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC; (b) V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef CAS.
- Y. Qian, X. Q. Qiu and S. P. Zhu, Green Chem., 2015, 17, 320–324 RSC.
- (a) K. Crouvisier-Urion, P. R. Bodart, P. Winckler, J. Raya, R. D. Gougeon, P. Cayot, S. Domenek, F. Debeaufort and T. Karbowiak, ACS Sustainable Chem. Eng., 2016, 4, 6371–6381 CrossRef CAS; (b) D. Kai, Y. K. Chua, L. Jiang, C. Owh, S. Y. Chan and X. J. Loh, RSC Adv., 2016, 6, 86420–86427 RSC.
- A. Hambardzumyan, L. Foulon, B. Chabbert and V. Aguie-Beghin, Biomacromolecules, 2012, 13, 4081–4088 CrossRef CAS PubMed.
- M. K. Chan, Q. Ye, Z. M. Png, H. N. Zeng, X. Wang and J. Xu, Waste Biomass Valorization, 2016 DOI:10.1007/s12649-016-9737-4.
- M. Lahtinen, P. Heinonen, M. Oivanen, P. Karhunen, K. Kruus and J. Sipilä, Org. Biomol. Chem., 2013, 11, 5454–5464 CAS.
- M. Lahtinen, K. Kruus, P. Heinonen and J. Sipila, J. Agric. Food Chem., 2009, 57, 8357–8365 CrossRef CAS PubMed.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, J. Mol. Catal. B: Enzym., 2016, 125, 34–41 CrossRef CAS.
- M. A. Constantin, J. Conrad and U. Beifuss, Green Chem., 2012, 14, 2375–2379 RSC.
- K. Rittstieg, A. Suurnäkki, T. Suortti, K. Kruus, G. M. Guebitz and J. Buchert, Enzyme Microb. Technol., 2002, 31, 403–410 CrossRef CAS.
- K. Rittstieg, A. Suurnäkki, T. Suortti, K. Kruus, G. M. Guebitz and J. Buchert, Biotechnol. Prog., 2003, 19, 1505–1509 CrossRef CAS PubMed.
- Oxidative cleavage of β-O-4 containing dimeric model compounds could potentially be possible by the oxidation of Cα- and Cγ-hydroxyl groups, however in the presence of mediator systems. See. (a) R. Bourbonnais and M. G. Paice, FEBS Lett., 1990, 267, 99–102 CrossRef CAS PubMed; (b) S. Kawai, M. Nakagawa and H. Ohashi, FEBS Lett., 1999, 446, 355–358 CrossRef CAS PubMed; (c) S. Kawai, M. Nakagawa and H. Ohashi, Enzyme Microb. Technol., 2002, 30, 482–489 CrossRef CAS; (d) H. Hirai, H. Shibata, S. Kawai and T. Nishida, FEMS Microbiol. Lett., 2006, 265, 56–59 CrossRef CAS PubMed; (e) A. I. Canas and S. Camarero, Biotechnol. Adv., 2010, 28, 694–705 CrossRef CAS PubMed; (f) C. Crestini, L. Jurasek and D. S. Argyropoulos, Chem. –Eur. J., 2003, 9, 5371–5378 CrossRef CAS PubMed.
- G. Carlo, M. Catherine, V. Raffaella, J. Claude and G. Patrizia, ChemBioChem, 2013, 14, 2500–2505 CrossRef PubMed.
- We observed higher product distribution of 3 after 30 min of reaction time when 2 units per mL of TVL was used. While to get higher product distribution of 4, 20 units per mL of TVL was used for 1 h of reaction time.
- To our knowledge, complete NMR data for 3C has not been reported. Incomplete NMR data was provided for the acetylated 3C in the literature without details on the assignments. (a) L. L. Landucci, S. A. Ralph and K. E. Hammel, Holzforschung, 1998, 52, 160–170 CrossRef CAS; (b) K. Syrjänen and G. Brunow, J. Chem. Soc., Perkin Trans. 1, 1998, 3425–3429 RSC . It is practically harder to establish the linkages using the acetylated 3C, as phenolic –OH group gives the strong basis for the identification of the C5–C5′ linkage by HMBC NMR spectrum.
- L. Munk, A. K. Sitarz, D. C. Kalyani, D. Mikkelsen and A. S. Meyer, Biotechnol. Adv., 2015, 33, 13–24 CrossRef CAS PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; J. A. Montgomery Jr; J. E. Peralta; F. Ogliaro; M. Bearpark; J. J. Heyd; E. Brothers; K. N. Kudin; V. N. Staroverov; R. Kobayashi; J. Normand; K. Raghavachari; A. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; N. Rega; M. J. Millam; M. Klene; J. E. Knox; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; R. L. Martin; K. Morokuma; V. G. Zakrzewski; G. A. Voth; P. Salvador; J. J. Dannenberg; S. Dapprich; A. D. Daniels; Ö. Farkas; J. B. Foresman; J. V. Ortiz; J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) S. Majumdar, T. Lukk, J. O. Solbiati, S. Bauer, S. N. Nair, J. E. Cronan and J. A. Gerlt, Biochemistry, 2014, 53, 4047–4058 CrossRef CAS PubMed; (b) H.-D. Youn, Y. C. Hah and S.-O. Kang, FEMS Microbiol. Lett., 1995, 132, 183–188 CrossRef CAS; (c) C. Eggert, U. Temp and K. E. L. Eriksson, FEBS Lett., 1997, 407, 89–92 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete details of GPC, mass and NMR spectra. See DOI: 10.1039/c6ra26975c |
| This journal is © The Royal Society of Chemistry 2017 |