
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroelectrochemical properties of a Ru(II) complex with a thiazolo[5,4-d]thiazole triarylamine ligand†
Carol
Hua
a,
Felix J.
Rizzuto
a,
Xuan
Zhang
ab,
Floriana
Tuna
c,
David
Collison
c and
Deanna M.
D'Alessandro
*a
aSchool of Chemistry, The University of Sydney, New South Wales 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au; Fax: +61 (2) 9351 3329; Tel: +61 (2) 9351 3777
bSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210093, P. R. China
cSchool of Chemistry, University of Manchester, Manchester M13 9PL, UK
First published on 16th November 2016
Abstract
A new bis-chelating ligand containing a triarylamine electron donor core fused with a thiazolo-thiazole electron acceptor, N,N′-(thiazolo[5,4-d]thiazole-2,5-diylbis(4,1-phenylene))bis(N-(pyridin-2-yl)pyridin-2-amine) (1) has been synthesised. This non-innocent ligand exhibits interesting electronic and spectral properties that can be tuned as a function of its redox state. In particular, modulation of the electronic state can be used to turn the fluorescence on and off. A dinuclear Ru(II) terpyridine complex, [Ru2(tpy)2Cl2(1)](PF6)2 (2) was subsequently synthesised and the properties of each of the accessible redox states explored using in situ spectroelectrochemical techniques.
Introduction
In the quest for more renewable sources of energy, the development of devices capable of harnessing the energy provided by solar radiation is a subject of continuing extensive research efforts.1–4 Ru(II) terpyridine (tpy) complexes, [RuII(tpy)]2+, have been widely studied as a result of their favourable photoactive and charge transfer properties which facilitate the conversion of light into chemical energy.5–8 Such complexes have also been employed as effective catalysts for a wide range of organic transformations.9,10Towards the development of multifunctional systems, the combination of a [RuII(tpy)]2+ species with a non-innocent redox-active ligand may provide a viable platform for new catalysts and light harvesting devices.11,12 In this context, ligands containing donor and acceptor functionalities are particularly attractive owing to their potential to display multiple electronic states.13–19 The use of triarylamines as redox-active moieties and electron donors has been extensively explored,20–25 and their potential to undergo a one electron oxidation to form a radical cation26,27 has enabled studies on organic mixed-valence compounds.28–30 Meanwhile, thiazolo[5,4-d]thiazoles, consisting of two fused heterocycles containing nitrogen and sulfur atoms are well-known acceptors in organic polymers.31–34 It has previously been shown that the thiazolo-thiazole moiety is planar and capable of undergoing two sequential one electron reduction processes.35,36
Herein, the synthesis of a new donor–acceptor ligand comprising the triarylamine and thiazolo-thiazole moieties is described. To facilitate chelation of a metal centre, 2,2′-bipyridine N,N′-bidentate coordination sites were also introduced into N,N′-(thiazolo[5,4-d]thiazole-2,5-diylbis(4,1-phenylene))bis(N-(pyridin-2-yl)pyridin-2-amine) (1). Such donor–acceptor ligands have previously been shown to support strong electronic delocalisation over relatively large distances.13,37 The new Ru(II) terpyridine complex, [Ru2(tpy)2Cl2(1)](PF6)2 (2), was synthesised to investigate the potential multifunctionality. The electronic, spectral and fluorescence properties of both 1 and 2 have been investigated using in situ UV/Vis/NIR, EPR and fluorescence spectroelectrochemistry to investigate the physical properties as a function of the multiple accessible redox states.
Results and discussion
Synthesis and characterisation
The N,N′-(thiazolo[5,4-d]thiazole-2,5-diylbis(4,1-phenylene))bis(N-(pyridine-2-yl)pyridin-2-amine) ligand (1) was synthesised as a bright yellow solid via a Goldberg reaction of 2,5-bis(4-bromophenyl)thiazolo[5,4-d]thiazole with excess 2,2′-dipyridylamine in 34% yield (Scheme 1a). The ruthenium complex 2 was synthesised by reaction of 1 and Ru(tpy)Cl3 in the presence of N-methylmorpholine (NMM). The complex was isolated as a deep maroon solid in 34% yield after purification by column chromatography (Scheme 1b). The complex was characterised by 1D and 2D NMR experiments, mass spectrometry and elemental analysis. The 1H NMR spectrum in DMSO-d6 revealed the C2 symmetry of 2: the complex displayed chemically equivalent signals for the two terpyridine ligands and the two halves of the central core of 1. The two pyridyl rings on the triarylamine have chemically distinct resonances due to the asymmetric environment about the ruthenium metal centre, resulting from the symmetry-breaking coordination of the chloride ligand. The set of resonances corresponding to one of the triarylamine pyridyl rings was shifted significantly downfield when compared to those of the other triarylamine ring, reflecting the different proximities to the chloride or terpyridine ligands. | ||
| Scheme 1 Synthetic scheme for (a) 1 and (b) 2. | ||
Probing the redox properties
The redox properties of 1 were investigated using solution state cyclic voltammetry in [(n-C4H9)4N]PF6/CH3CN electrolyte. Three processes were observed in the anodic region at 0.035, 0.52 and 0.81 V vs. Fc/Fc+ (Fig. 1a). The first process at 0.035 V may be attributed to oxidation of the triarylamine pyridyl groups to pyridyl N-oxide species, which was supported by the absence of this redox process upon coordination of the ligand to a metal centre (vide infra). The quasi reversible process at 0.52 V was assigned to the one electron oxidation of a single triarylamine core to its radical cation while the quasi reversible process at 0.81 V is consistent with a one electron oxidation of the adjacent triarylamine core.26 In the cathodic region, four processes were observed at −1.24, −1.45, −2.04 and −2.37 V. The first two processes are due to reduction of the TzTz core while the third and fourth processes were attributed to reduction of the pyridyl rings. These processes are consistent with the previously reported 4-Py isomer of 1.13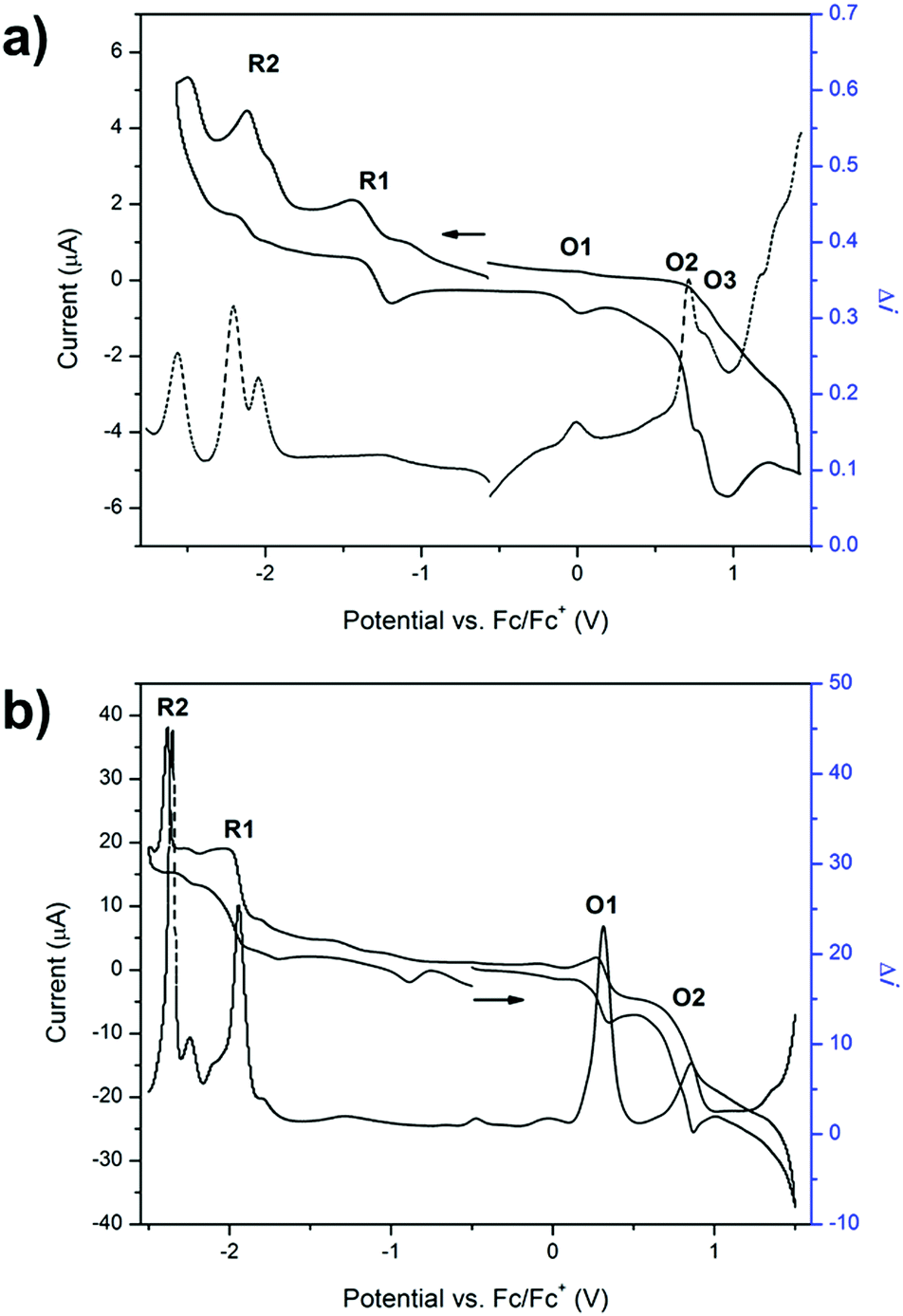 | ||
| Fig. 1 Square wave (at an amplitude of 10 mV and frequency of 9 Hz) vs. cyclic voltammograms at 100 mV s−1 in 0.1 M [(n-C4H9)4N]PF6/CH3CN electrolyte of (a) 1 and (b) 2 where the arrow indicates the direction of the forward scan. | ||
The cyclic voltammogram of 2 displayed two main processes in the anodic region (Fig. 1b). The reversible process at 0.32 V was assigned to the one electron oxidation of Ru(II) to Ru(III) while the quasi reversible process at 0.79 V corresponded well with the process in the ligand for the one electron oxidation of the triarylamine core to its radical cation. The absence of the peak at 0.036 V is consistent with the coordination of the pyridyl groups to the Ru2+ metal centre, which precludes their oxidation to the N-oxide species. A broad peak in the cathodic region of the CV was further explored by performing square wave voltammetry, which showed two main processes in the cathodic region that were assigned to the two sequential one electron reductions of the TzTz core to its radical anion and dianion states (Fig. 1b). The reduction processes due to the free ligand lie outside the potential window afforded by acetonitrile due to stabilisation upon coordination to the ruthenium metal centre.
UV/Vis/NIR spectroelectrochemistry
The solution state UV/Vis/NIR spectrum of 1 in acetonitrile displayed two bands at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 and 32900 cm−1 which are assigned to π → π* (LUMO–HOMO) transitions.35,38 It was presumed that the molecular orbitals of 1 were similar to those of the analogous 4-pyridyl terminated system previously reported.13 As a potential of −1.1 V was applied to 1 during the spectroelectrochemical experiment in [(n-C4H9)4N]PF6/CH3CN electrolyte, the bands at 25600 and 32900 cm−1 increased due to reduction of the TzTz core to its radical anion (Fig. S5, ESI†).
600 and 32900 cm−1 which are assigned to π → π* (LUMO–HOMO) transitions.35,38 It was presumed that the molecular orbitals of 1 were similar to those of the analogous 4-pyridyl terminated system previously reported.13 As a potential of −1.1 V was applied to 1 during the spectroelectrochemical experiment in [(n-C4H9)4N]PF6/CH3CN electrolyte, the bands at 25600 and 32900 cm−1 increased due to reduction of the TzTz core to its radical anion (Fig. S5, ESI†).
Oxidation of 1 provided access to the radical cation state of the triarylamine moiety. As the potential was increased from 0.8 to 0.9 V, the bands at 25600 and 32900 cm−1 decreased (Fig. S5, ESI†). As the potential was further increased from 1.1 to 1.6 V, these bands further decreased in intensity, while a new band at 23800 cm−1 formed (Fig. S5, ESI†). The decrease of the bands at 25000 and 32900 cm−1 resulted from the loss of electron density throughout the entire ligand due to the high degree of delocalisation in the system which was a consequence of the coplanarity of the TzTz and phenyl functionalities. This arrangement results in a highly delocalised HOMO over the ligand backbone, and a localised HOMO−1 on the triarylamine core.13
The UV/Vis/NIR spectrum of 2 exhibits three major features (Fig. 2a). The band at 19000 cm−1 was assigned to the d → π* MLCT transition between the terpyridine ligand and Ru2+ metal centre, and the bands at 25000 and 32000 cm−1 are attributed to the π → π* transitions of the aromatic moieties in the terpyridine ligands and in the bridging ligand 1.
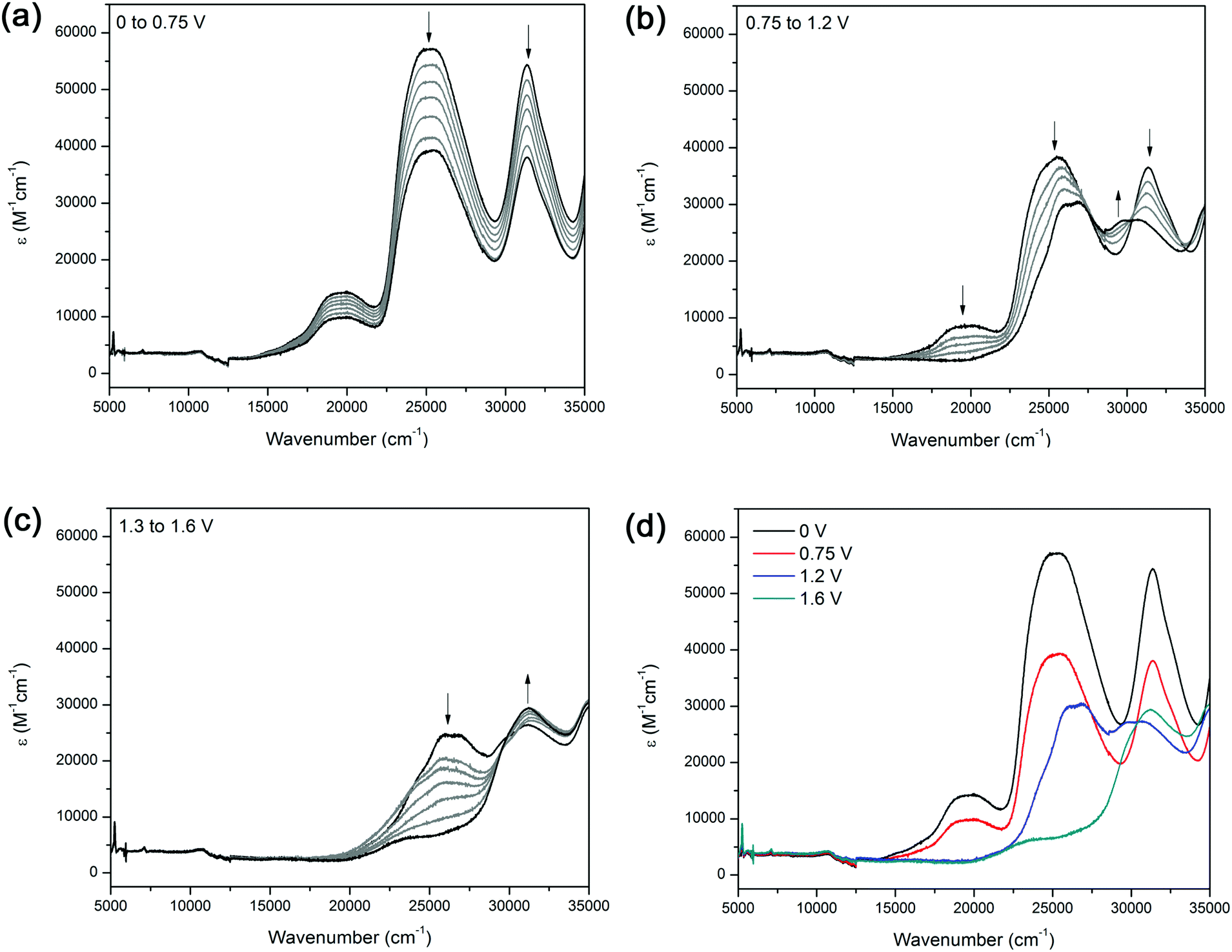 | ||
| Fig. 2 Solution state spectroelectrochemistry on 2 in [(n-C4H9)4N]PF6/CH3CN electrolyte over the potential range of (a) 0 to 0.75 V, (b) 0.75 to 1.2, (c) 1.3 to 1.6 V and (d) summary of the changes in the absorption spectrum as a function of potential applied. | ||
The solution state UV/Vis/NIR spectroelectrochemical experiment on 2 in [(n-C4H9)4N]PF6/CH3CN electrolyte displayed several processes upon application of a positive potential. At 1.0 V, a decrease in the MLCT band at 19600 cm−1 was observed, corresponding to a loss of electron density from the metal centre due to oxidation of the Ru(II) centre to Ru(III) (Fig. 2a). The decrease in intensity of the band at 25500 cm−1 was due to electron density being transferred towards the ruthenium centre, resulting in a depopulation of the LUMO. As the potential was further increased from 1.3 to 1.6 V, a band at 31200 cm−1 appeared as the band at 25500 cm−1 continued to decrease (Fig. 2c). The original spectrum of the complex was regained upon decreasing the potential to 0 V, indicating that the processes were spectrally reversible (Fig. 2d).
As the potential was changed from −1.05 to −1.5 V, the bands at 25200 and 31390 cm−1 decreased in intensity, while a narrow band at 21300 cm−1 appeared, presumably due to reduction of the thiazolo[5,4-d]thiazole moiety (Fig. S6, ESI†). The blue shift of the MLCT band at 19000 to 21300 cm−1 may arise from the increase in electron density on the ligand backbone.
EPR spectroelectrochemistry
EPR spectroelectrochemical experiments were conducted on 1 in [(n-C4H9)4N]PF6/CH3CN electrolyte (Fig. 3a). An EPR signal at g = 2.004 was obtained upon the application of a potential of 1.7 V, which corresponds to the formation of the triarylamine radical cation.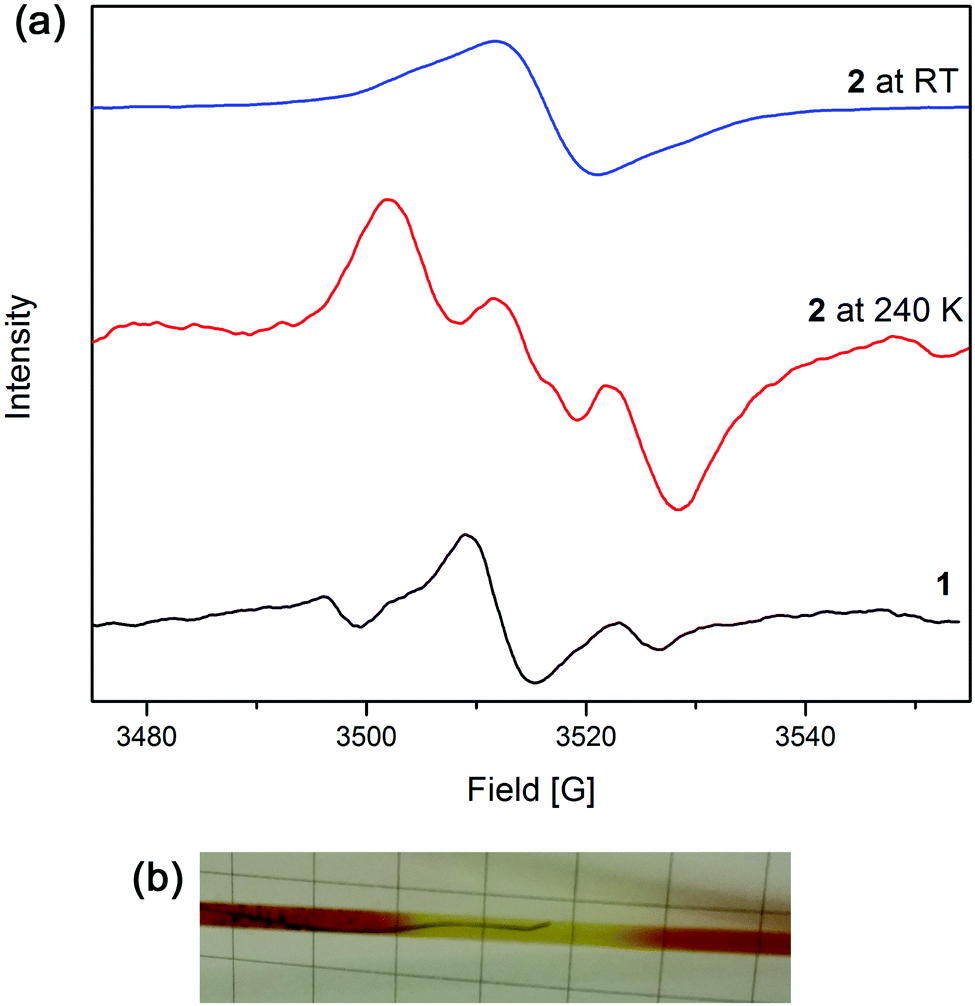 | ||
| Fig. 3 (a) EPR spectra in [(n-C4H9)4N]PF6/CH3CN electrolyte of 1 and 2 at 240 K and room temperature and (b) photo of the EPR cell with electrodes during the experiment showing the colour change from deep red to yellow. | ||
The solution-state EPR spectroelectrochemical experiment on 2 in [(n-C4H9)4N]PF6/CH3CN electrolyte at 240 K yielded a signal at g = 2.002 with a 1:1:1 hyperfine pattern at 1.8 V, where the radical is primarily localised on the triarylamine nitrogen centre and has a hyperfine coupling value of 28 MHz (Fig. 3a). The hyperfine information of the signal was lost upon warming the reaction mixture to room temperature to yield one broad peak. This is due to the faster rate of molecular tumbling at higher temperatures, whereas the lower temperatures resulted in slower molecular movement, allowing the hyperfine coupling to be elucidated. A colour change from deep red to light yellow was observed during the experiment, corresponding to formation of the Ru3+ species and subsequently, oxidation of the triarylamine to its radical cation (Fig. 3b).
Redox state dependent fluorescence
A light blue fluorescence band was observed for 1 in acetonitrile at 480 nm (20900 cm−1) upon excitation into the π → π* transition at 390 nm (25600 cm−1) with a Stoke's shift of 216 nm (Fig. S7, ESI†). 2 displayed a fluorescence peak at 470 nm (21280 cm−1) upon excitation at 400 nm (25000 cm−1) into the ligand π → π* transition (Fig. S7, ESI†). When the other bands in the absorption spectrum of the metal complex were excited (320 and 512 nm), no fluorescence peaks were observed. These results may suggest a high quantum yield for 1, and a low quantum yield for 2.
The fluorescence was quenched upon oxidation of Ru2+ to Ru3+ in 2 with NOPF6 (0.87 vs. Fc/Fc+) in acetonitrile.39 As shown from the chemical oxidation of 2, the fluorescence is able to be tuned as a function of the redox state. In order to verify that the fluorescence quenching observed ex situ from the chemically oxidised species was due to the formation of the triarylamine radical cation and not due to external environmental factors, in situ fluorescence spectroelectrochemical experiments were performed in [(n-C4H9)4N]PF6/CH3CN electrolyte on both 1 and 2 (Fig. S8 and S9, ESI†).
In 1, increasing the potential to 1.5 V resulted in a decrease of the fluorescence peaks at 504 and 533 nm, corresponding to the formation of one of the triarylamine radical cation centres in the ligand. An additional increase in the potential from 1.7 to 2.0 V, led to a decrease in the fluorescence, corresponding to the oxidation of the second triarylamine core (Fig. S8, ESI†).
Similar trends for the in situ fluorescence spectroelectrochemical experiment were observed for 2 (Fig. S9, ESI†). Upon oxidation, a decrease in the fluorescence band at 475 nm was observed due to formation of the triarylamine radical cation, while application of a reductive potential of −1.0 V resulted in the (re)formation of the band, corresponding to the regeneration of the neutral state. The fluorescence behaviour of the ruthenium complex can therefore be attributed to predominantly ligand based processes.
Conclusions
The combination of the electron donating triarylamine core with the electron accepting thiazolo-thiazole moiety within ligand 1 and Ru(II) complex 2 has resulted in a versatile donor–acceptor system. The accessibility of the different redox states has been demonstrated through spectroelectrochemical experiments, with the properties of each redox state characterised. The tunability of the complex's fluorescence properties as a function of redox state may lend to its application in photoactive and catalytic processes.Experimental
General considerations and synthetic procedures
All chemicals and solvents were used as obtained without further purification. 2,2′-Dipyridylamine,40 Ru(tpy)Cl3,41,42 and 2,5-bis(4-bromophenyl)thiazolo[5,4-d]thiazole43,44 were synthesised according to literature procedures. For experiments requiring dry solvents, acetonitrile was dried over CaH2 and methanol was dried over magnesium/magnesium methoxide before being distilled under nitrogen. Toluene was obtained from a PuraSolv solvent purification system and stored over activated 4 Å molecular sieves.Solution state 1H and 13C{1H} NMR spectra were recorded on either a Bruker AVANCE300 or AVANCE500 spectrometer operating at 300, 500 MHz for 1H and 75, 125 MHz for 13C, respectively. 1H and 13C NMR chemical shifts were referenced internally to residual solvent resonances. Spectra were recorded at 298 K and chemical shifts (δ), with uncertainties of ±0.01 Hz for 1H and ±0.05 Hz for 13C are quoted in ppm. Coupling constants (J) are quoted in Hz and have uncertainties of ±0.05 Hz for 1H–1H. Deuterated solvents were obtained from Cambridge Stable Isotopes and used as received. Mass spectrometry was carried out at the mass spectrometry analysis facility at the University of Sydney on a Finnigan LCQ mass spectrometer. Microanalyses were carried out at the Chemical Analysis Facility – Elemental Analysis Service in the Department of Chemistry and Biomolecular Science at Macquarie University, Australia.
N,N′-(Thiazolo[5,4-d]thiazole-2,5-diylbis(4,1-phenylene))bis(N-(pyridine-2-yl)pyridin-2-amine) (1)
2,2′-Dipyridylamine (430 mg, 2.50 mmol), 2,5-bis(4-bromophenyl)thiazolo[5,4-d]thiazole (450 mg, 1.00 mmol), anhydrous potassium carbonate (690 mg, 5.00 mmol), anhydrous copper sulfate (150 mg, 0.900 mmol), 18-crown-6 (36.0 mg, 0.150 mmol) and diphenyl ether (10.0 mL) were heated at 200 °C under nitrogen for 48 h. The reaction mixture was cooled to room temperature before water (20.0 mL) was added. The aqueous layer was extracted with dichloromethane (3 × 50 mL), and the organic layer dried over Na2SO4. The solvent was removed under reduced pressure and the residue purified by silica gel column chromatography (using an elution gradient of 20:1 CH2Cl2:Et3N to 20:1:1 CH2Cl2:Et3N:MeOH) to yield the pure product as a yellow powder (180 mg, 34%), m.p. >260 °C. 1H NMR (300 MHz, d6-DMSO): 8.31 (dd, 3JH–H = 4.8 Hz, 4JH–H = 1.2 Hz, 4H, H10), 8.02–7.97 (m, 4H, H9), 7.79–7.73 (m, 4H, H8), 7.20 (d, 3JH–H = 8.7 Hz, 4H, H4), 7.12 (dd, 3JH–H = 7.3, 4JH–H = 4.9 Hz, 4H, H7), 7.06 (d, 3JH–H = 8.3 Hz, 4H, H5) ppm. 13C{1H} NMR (75 MHz, d6-DMSO): δ 168.0 (C2), 157.2 (C7), 150.1 (C1), 148.5 (C3), 147.5 (C10), 138.3 (C9), 129.0 (C4), 127.3 (C6), 126.2 (C9), 119.3 (C8), 117.7 (C5) ppm. ESI-MS (ESI+, MeOH): 633.00 (calculated [M + H]+ = 633.16, 100%) amu. HR-MS (MeOH): found 655.14559 (calculated [M + Na]+ = 655.14576) amu. Elemental analysis; found: C, 68.46, H, 3.31, N, 17.75, S, 10.04%. Calculated for C36H24N8S2: C, 68.33, H, 3.82, N, 17.71, S, 10.13%.
[Ru2(tpy)2Cl2(1)](PF6)2 (2)
Ru(tpy)Cl3 (139 mg, 0.316 mmol) and 1 (100 mg, 0.158 mmol) were suspended in dry methanol (80.0 mL) and N-methylmorpholine (5.0 mL) was added. The green suspension was heated at reflux under nitrogen for 24 hours to yield a deep green solution, then cooled to room temperature. The solvent was reduced and a saturated solution of KPF6 in water was added to the reaction mixture to yield a dark purple-brown precipitate. The solid was isolated by filtration and washed with water before being dried. The crude product was purified by a silica gel column (19:1:0.1 CH3CN:H2O:KNO3 (sat. solution in water)), before the product fractions were combined and a saturated solution of NH4PF6 in water added to yield the precipitation of a dark red solid. The solid was dried under vacuum to yield the product (77.0 mg, 34%). 1H NMR (500 MHz, d6-DMSO): 9.75 (d, 3JH–H = 4.5 Hz, 2H, H8a), 8.76 (d, 3JH–H = 8.5 Hz, 4H, H18), 8.67 (d, 3JH–H = 8.0 Hz, 4H, H15), 8.42 (td, 3JH–H = 8.0 Hz, 4JH–H = 1.5 Hz, 2H, H10a), 8.28 (d, 3JH–H = 8.0 Hz, 2H, H11a), 8.17 (t, 3JH–H = 8.0 Hz, 2H, H19), 7.98 (d, 3JH–H = 6.0 Hz, 4H, H12), 7.96 (t, 3JH–H = 7.5 Hz, 4H, H13), 7.91–7.88 (m, 6H, H4 and H9a), 7.75 (td, 3JH–H = 7.5 Hz, 4JH–H = 1.5 Hz, 2H, H9), 7.63 (d, 3JH–H = 8.0 Hz, 2H, H8), 7.15 (t, 3JH–H = 6.5 Hz, 4H, H14), 6.98 (d, 3JH–H = 8.0 Hz, 4H, H5), 6.96 (d, 3JH–H = 6.0 Hz, 2H, H11), 6.92 (t, 3JH–H = 8.0 Hz, 2H, H10) ppm. 13C{1H} NMR (125 MHz, d6-DMSO): δ 167.6 (C2), 158.7 (C16), 158.6 (C17), 156.7 (C11), 154.0 (C7a), 152.6 (C13), 150.83 (C7), 149.9 (C1), 146.3 (C3), 139.4 (C10a), 138.5 (C9), 136.9 (C14), 133.72 (C19), 127.7 (C4), 127.3 (C6), 126.5 (C12), 124.1 (C11a), 123.8 (C9a), 123.7 (C8a, C8, C10), 123.5 (C15), 122.7 (C18), 116.3 (C5) ppm. ESI-MS (ESI+, MeOH): 685.93 (calculated [M-2PF6]2+ = 686.045, 100%) amu. Elemental analysis; found: C, 48.77, H, 3.45, N, 11.54, S, 5.04. Calculated for C69H54Cl2F12N14P2Ru2S2: C, 48.57, H, 3.19, N, 11.49, S, 5.10%.
Physical measurements
000 cm−1 using a CARY5000 Spectrophotometer interfaced with Varian WinUV software. The baseline was taken with the solvent that the sample was to be dissolved in prior to measurement of the sample of interest. A 3 mL (1 cm pathlength) quartz cuvette was used.
000 cm−1 was performed using a CARY5000 spectrophotometer interfaced with Varian WinUV software. In the solution state, the absorption spectra of the electrogenerated species were obtained in situ by the use of an Optically Semi-Transparent Thin-Layer Electrosynthetic (OSTLE) cell, path length 0.65 mm, mounted in the path of the spectrophotometer. Solutions for the spectroelectrochemical experiment contained 0.1 M [(n-C4H9)4N]PF6/CH3CN supporting electrolyte and ca. 1 mM of the compound. Appropriate potentials were applied by using an eDAQ e-corder 410 potentiostat and the current was carefully monitored throughout the electrolysis. By this method, the electrogenerated species were obtained in situ, and their absorption spectra were recorded at regular intervals throughout the electrolysis. The attainment of a steady-state spectrum and the decay of the current to a constant minimum at a potential appropriately beyond E1/2 (for the redox process in question) was indicative of the complete conversion of the starting material.
Acknowledgements
We gratefully acknowledge support from the Australian Research Council and the EPRSRC UK National Electron Paramagnetic Resonance Service at the University of Manchester.Notes and references
- M. C. So, G. P. Wiederrecht, J. E. Mondloch, J. T. Hupp and O. K. Farha, Chem. Commun., 2015, 51, 3501–3510 RSC.
- A. Bahadori and C. Nwaoha, Renewable Sustainable Energy Rev., 2013, 18, 1–5 CrossRef.
- T. G. Deepak, G. S. Anjusree, S. Thomas, T. A. Arun, S. V. Nair and A. Sreekumaran Nair, RSC Adv., 2014, 4, 17615–17638 RSC.
- T. Ameri, P. Khoram, J. Min and C. J. Brabec, Adv. Mater., 2013, 25, 4245–4266 CrossRef CAS PubMed.
- J. Choi, H. M. Nguyen, S. Yoon, N. Kim, J.-W. Oh and F. S. Kim, Mol. Cryst. Liq. Cryst., 2014, 600, 22–27 CrossRef CAS.
- P. Hrobárik, V. Hrobáriková, I. Sigmundová, P. Zahradník, M. Fakis, I. Polyzos and P. Persephonis, J. Org. Chem., 2011, 76, 8726–8736 CrossRef PubMed.
- A. O. Adeloye and P. A. Ajibade, Molecules, 2014, 19, 12421–12460 CrossRef PubMed.
- L. Hammarstroem and O. Johansson, Coord. Chem. Rev., 2010, 254, 2546–2559 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. Murakami Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954–1965 CrossRef CAS PubMed.
- A. Winter, G. R. Newkome and U. S. Schubert, ChemCatChem, 2011, 3, 1384–1406 CrossRef CAS.
- W. Kaim and J. Fiedler, Chem. Soc. Rev., 2009, 38, 3373–3382 RSC.
- M. Venturi, Lect. Notes Chem., 2012, 78, 209–225 CAS.
- F. J. Rizzuto, T. B. Faust, B. Chan, C. Hua, D. M. D'Alessandro and C. J. Kepert, Chem. – Eur. J., 2014, 20, 17597–17605 CrossRef CAS PubMed.
- D. M. D'Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- C. F. Leong, B. Chan, T. B. Faust and D. M. D'Alessandro, Chem. Sci., 2014, 5, 4724–4728 RSC.
- C. L. Ramírez, C. N. Pegoraro, O. Filevich, A. Bruttomeso, R. Etchenique and A. R. Parise, Inorg. Chem., 2012, 51, 1261–1268 CrossRef PubMed.
- C.-J. Yao, R.-H. Zheng, Q. Shi, Y.-W. Zhong and J. Yao, Chem. Commun., 2012, 48, 5680–5682 RSC.
- H.-J. Nie, W.-W. Yang, J.-Y. Shao and Y.-W. Zhong, Dalton Trans., 2016, 45, 10136–10140 RSC.
- C.-J. Yao, Y.-W. Zhong and J. Yao, Inorg. Chem., 2013, 52, 4040–4045 CrossRef CAS PubMed.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D'Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- C. Hua, P. Turner and D. M. D'Alessandro, Dalton Trans., 2015, 44, 15297–15303 RSC.
- C. Hua and D. M. D'Alessandro, Supramol. Chem., 2015, 27, 792–797 CrossRef CAS.
- C. Hua, A. Rawal, T. B. Faust, P. D. Southon, R. Babarao, J. M. Hook and D. M. D'Alessandro, J. Mater. Chem. A, 2014, 2, 12466–12474 CAS.
- C. Hua and D. M. D'Alessandro, CrystEngComm, 2014, 16, 6331–6334 RSC.
- C. Hua, P. Turner and D. M. D'Alessandro, Dalton Trans., 2013, 42, 6310–6313 RSC.
- S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141–152 CrossRef CAS.
- C. Lambert and G. Nöll, J. Am. Chem. Soc., 1999, 121, 8434–8442 CrossRef CAS.
- A. Heckmann and C. Lambert, Angew. Chem., Int. Ed., 2012, 51, 326–392 CrossRef CAS PubMed.
- C. Lambert and G. Nöll, Synth. Met., 2003, 139, 57–62 CrossRef CAS.
- C. Lambert, G. Noll and J. Schelter, Nat. Mater., 2002, 1, 69–73 CrossRef CAS PubMed.
- I. Osaka, R. Zhang, J. Liu, D.-M. Smilgies, T. Kowalewski and R. D. McCullough, Chem. Mater., 2010, 22, 4191–4196 CrossRef CAS.
- T. W. Lee, N. S. Kang, J. W. Yu, M. H. Hoang, K. H. Kim, J.-I. Jin and D. H. Choi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5921–5929 CrossRef CAS.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761–1775 CrossRef CAS.
- S. Ando, J.-I. Nishida, H. Tada, Y. Inoue, S. Tokito and Y. Yamashita, J. Am. Chem. Soc., 2005, 127, 5336–5337 CrossRef CAS PubMed.
- M. R. Pinto, Y. Takahata and T. D. Z. Atvars, J. Photochem. Photobiol., A, 2001, 143, 119–127 CrossRef CAS.
- S. Van Mierloo, S. Chambon, A. E. Boyukbayram, P. Adriaensens, L. Lutsen, T. J. Cleij and D. Vanderzande, Magn. Reson. Chem., 2010, 48, 362–369 CAS.
- F. J. Rizzuto, C. Hua, B. Chan, T. B. Faust, A. Rawal, C. F. Leong, J. M. Hook, C. J. Kepert and D. M. D'Alessandro, Phys. Chem. Chem. Phys., 2015, 17, 11252–11259 RSC.
- A. Brillante, B. Samori, C. Stremmenos and P. Zanirato, Mol. Cryst. Liq. Cryst., 1983, 100, 263 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- P. J. Zapf, R. L. LaDuca, R. S. Rarig, K. M. Johnson and J. Zubieta, Inorg. Chem., 1998, 37, 3411–3414 CrossRef CAS.
- D. Hvasanov, A. F. Mason, D. C. Goldstein, M. Bhadbhade and P. Thordarson, Org. Biomol. Chem., 2013, 11, 4602–4612 CAS.
- B. P. Sullivan, J. M. Calvert and T. J. Meyer, Inorg. Chem., 1980, 19, 1404–1407 CrossRef CAS.
- S. Hisamatsu, H. Masu, I. Azumaya, M. Takahashi, K. Kishikawa and S. Kohmoto, Cryst. Growth Des., 2011, 11, 5387 CAS.
- J. R. Johnson and R. Ketcham, J. Am. Chem. Soc., 1960, 82, 2719 CrossRef CAS.
- Y. Liu, J.-R. Li, W. M. Verdegaal, T.-F. Liu and H.-C. Zhou, Chem. – Eur. J., 2013, 19, 5637–5643 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H–1H COSY NMR, electrochemistry, fluorescence, UV/Vis/NIR and fluorescence spectroelectrochemistry. See DOI: 10.1039/c6nj02802k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2017 |