
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEncapsulation of an organometallic cationic catalyst by direct exchange into an anionic MOF†
Alexios
Grigoropoulos
a,
George F. S.
Whitehead
a,
Noémie
Perret
a,
Alexandros P.
Katsoulidis
a,
F. Mark
Chadwick
b,
Robert P.
Davies
c,
Anthony
Haynes
d,
Lee
Brammer
d,
Andrew S.
Weller
*b,
Jianliang
Xiao
a and
Matthew J.
Rosseinsky
*a
aDepartment of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK. E-mail: M.J.Rosseinsky@liverpool.ac.uk
bDepartment of Chemistry, Chemistry Research Laboratories, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: andrew.weller@chem.ox.ac.uk
cDepartment of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK
dDepartment of Chemistry, University of Sheffield, Brook Hill, Sheffield S3 7HF, UK
First published on 8th December 2015
Abstract
Metal–Organic Frameworks (MOFs) are porous crystalline materials that have emerged as promising hosts for the heterogenization of homogeneous organometallic catalysts, forming hybrid materials which combine the benefits of both classes of catalysts. Herein, we report the encapsulation of the organometallic cationic Lewis acidic catalyst [CpFe(CO)2(L)]+ ([Fp–L]+, Cp = η5-C5H5, L = weakly bound solvent) inside the pores of the anionic [Et4N]3[In3(BTC)4] MOF (H3BTC = benzenetricarboxylic acid) via a direct one-step cation exchange process. To conclusively validate this methodology, initially [Cp2Co]+ was used as an inert spatial probe to (i) test the stability of the selected host; (ii) monitor the stoichiometry of the cation exchange process and (iii) assess pore dimensions, spatial location of the cationic species and guest-accessible space by single crystal X-ray crystallography. Subsequently, the quasi-isosteric [Fp–L]+ was encapsulated inside the pores via partial cation exchange to form [(Fp–L)0.6(Et4N)2.4][In3(BTC)4]. The latter was rigorously characterized and benchmarked as a heterogeneous catalyst in a simple Diels–Alder reaction, thus verifying the integrity and reactivity of the encapsulated molecular catalyst. These results provide a platform for the development of heterogeneous catalysts with chemically and spatially well-defined catalytic sites by direct exchange of cationic catalysts into anionic MOFs.
Introduction
Homogeneous catalysts mainly comprise well-defined reactive transition metal (TM) complexes with carefully tailored organic ligands, the design of which provides control over the mechanism, rate and selectivity of the respective catalytic process. Moreover, their discrete molecular structures and solubility allow for ready characterization by common spectroscopic techniques and systematic investigation of the overall catalytic mechanism by detailed solution-based kinetic analysis methods. Mechanism can be also studied by computational chemistry methods which are now commonplace for isolated molecular systems. Despite these benefits, separation of the catalyst from the products in homogeneous systems after turnover is challenging, while the chemical robustness of such molecular species is often low under harsher conditions of temperature and pressure. By contrast, heterogeneous catalysts are more robust and can be readily separated from the reaction mixture. They are usually less reactive, necessitating higher temperature and pressure, and can be less selective although progress has been achieved in this field with the introduction of solid-state porous catalysts.1,2 Limitations of experimental and computational methods complicate the identification and monitoring of the actual catalytic species, which in turn means that opportunities for tuning activity and selectivity based on mechanistic understanding are less well-developed. Considerable effort has thus been directed towards immobilizing TM catalysts onto solid-state supports to develop new materials that combine the merits of homogenous and heterogeneous catalysis.3 Examples of supports include amorphous silica,4,5 organic polymers6–8 and porous ordered materials like zeolites or mesoporous silicates9–11 and more recently metal–organic frameworks (MOFs).MOFs are crystalline porous materials consisting of metal-based nodes and multitopic organic linkers which are interconnected by coordination bonds.12–14 They provide a well-defined confined environment with permanent porosity spanning from micro- to mesoporous. Moreover, the size, shape and chemical composition of the pores can be pre-engineered. Therefore, MOFs have emerged as promising hosts for the introduction of catalytic sites inside their uniform pores.15–24 Such hybrid materials could potentially outperform both homogeneous and heterogeneous catalysts since they contain spatially isolated, but accessible, highly reactive and selective catalytic sites and they can be easily recycled.
There are two common approaches for the incorporation of a homogeneous catalyst into a MOF (Scheme 1): (i) covalent bonding to the linker or to the metal-based node having appropriate grafting sites; (ii) direct encapsulation inside the pores via non-covalent interactions. Covalent bonding is the most common approach used and has been extensively investigated, including direct synthesis of the targeted material or post-synthetic modification that allows for milder reaction conditions.18,25–41 However, usually multiple synthetic steps and proper adjustment of the local coordination environment of the TM catalyst are required which may alter its overall catalytic properties. On the contrary, direct encapsulation does not perturb the catalyst's first coordination sphere, in principle allowing for the direct transfer of solution-based chemistry into the MOF and straightforward evaluation of the effect of the cavity.
 | ||
| Scheme 1 Different methods for incorporating a TM catalyst (grey sphere) inside the pores of a MOF (cube). | ||
Although simple in concept, encapsulation is synthetically challenging since the catalyst must be firmly trapped inside the pores in order to avoid leaching into the liquid phase. TM catalysts have been used as templates around which the MOF is built (“bottle around ship” method)42–47 but this approach requires very robust catalysts that must survive the solvothermal conditions used for MOF synthesis. The reverse route (“ship in a bottle” method) in which the TM catalyst is assembled in situ inside the pores of the host under milder conditions, was originally employed in zeolites48–53 and has been recently also demonstrated in MOFs.54,55 However, more than one step is again required (encapsulation of the desired TM species followed by ligand assembly and coordination) and the presence of catalytic species with different coordination motifs due to side reactions is potentially problematic. It should be also noted that both methods, although chemically elegant, require a very specific host–guest combination since the catalyst must be small enough to fit inside the pores but still large enough to avoid leaching through the pore windows. An interesting alternative is the one-step cation exchange in which a positively charged TM catalyst is directly encapsulated as a whole and held inside the pores of an anionic MOF through complementary electrostatic interactions to form a [catalyst]+@[MOF]− material, if a host with large enough pore windows is selected. This approach is conceptually linked to the one employed by Raymond, Bergman, Toste et al. for the encapsulation of positively charged TM catalysts inside discrete soluble anionic coordination cages by cation exchange.56–59 Recently, Sanford et al. reported the viability of such an approach using the anionic ZJU-28 MOF as a host.60 However, systematic characterization and spatial location of the actual catalytic species encapsulated was not provided. Since the Coulomb field that holds the cations in the anionic MOF is likely less localised than the covalent interactions used in grafting approaches, it is the resulting location of the organometallic cations that defines the molecular-level uniformity of the catalyst.
We now report the encapsulation and rigorous characterization of a well-defined organometallic cationic Lewis acidic catalyst, namely [CpFe(CO)2(L)]+ (Cp = η5-C5H5, L = weakly bound solvent), inside the pores of an anionic indium-based MOF (In-MOF) via a one-step cation exchange and demonstrate proof of concept of encapsulation by its use in a benchmark Diels–Alder reaction. To achieve this, a stepwise approach was developed, in which the stability of various anionic MOFs in solvents suitable for homogeneous organometallic catalytic reactions was first evaluated. Cation exchange on successful MOF candidates was then probed using [Cp2Co]+, a robust cation of comparable size to [CpFe(CO)2(L)]+, leading to the selection of [Et4N]3[In3(BTC)4] (H3BTC = benzene-1,3,5-tricarboxylic acid)61 as the most suitable host. The spatial location of the cations and the pore dimensions of the selected host were then determined by single crystal X-ray crystallography via the independent synthesis and isolation of single crystals of [Cp2Co]3[In3(BTC)4], using [Cp2Co]+ as a template. Finally, exchange of [Et4N]+ with the cationic TM catalyst [CpFe(CO)2(L)]+ ([Fp–L]+) inside the pores of the [In3(BTC)4]3− anionic framework resulted in [(Fp–L)0.6(Et4N)2.4][In3(BTC)4], which was benchmarked as a recyclable, heterogeneous catalyst in the Diels–Alder reaction between isoprene and methyl vinyl ketone.
All synthesized materials were thoroughly characterised, thereby providing a rare example in which a cationic TM catalyst is encapsulated intact inside the pores of a stable MOF which survives the catalytic process. Comprehensive identification of the catalytically active species is essential to enable the design of materials in which homogeneous catalysts are encapsulated in MOFs by direct cation exchange. Rigorous protocols, as described here, for assessing the stability of the selected hosts, the precise stoichiometry of the cation exchange process, the spatial location of cationic species and their effect in pore dimensions have not been previously established for these [catalyst]+@[MOF]− hybrid systems.
Results and discussion
Selection of anionic MOFs
A set of criteria were formulated to identify suitable anionic MOFs as hosts for cationic TM catalysts. The anionic MOFs should (i) be microporous with a pore window ideally larger than 7 Å for the cationic TM catalyst to pass through (vide infra); (ii) be stable under the conditions used for catalysis; (iii) possess a well-ordered structure to simplify analysis at the atomic scale; (iv) be readily synthesized in high yield on at least a 0.1 g scale; (v) contain readily exchangeable cations; (vi) not contain any paramagnetic metal centres which would prevent characterisation of the encapsulated TM catalyst by NMR spectroscopy and (vii) not contain any Lewis acidic sites which could complicate the investigation of catalytic activity of the encapsulated catalyst. With these guiding criteria in mind, a search of the Cambridge Structural Database62 narrowed down the selection to the following anionic indium-based MOFs: [(Me2NH2){In3O(BTC)2(H2O)3}2][In3(BTC)4] (CPM-5),63 [Me2NH2]3[In3(BTB)4] [(ZJU-28), H3BTB = benzene-1,3,5-tribenzoic acid]64 and [R4N]3[In3(BTC)4] [R = Et (TEA), nPr (TPA), nBu (TBA)].61Indium is known to form stable microporous anionic frameworks with carboxylate-based linkers since it can accommodate four chelating carboxylate groups, giving rise to mononuclear diamagnetic eight-coordinated anionic [InIII(O2CR)4]− secondary building units (SBUs) of distorted tetrahedral geometry. These serve as tetrahedral nodes for the construction of three-dimensional anionic frameworks with various polytopic linkers. The negative charge is balanced by organic cations which are either generated in situ upon solvent decomposition during the solvothermal synthesis or added in the reaction mixture as templates.65–81
Structures of selected anionic MOFs
All three MOFs comprise monometallic [InIII(O2CR)4]− SBUs interconnected via tritopic linkers (BTB or BTC) forming an anionic [In3(linker)4]3− framework. The negative charge of the framework in [In3(BTC)4]3− is balanced by the [R4N]+ cations, added as templates during solvothermal synthesis and giving rise to a crystal structure of cubic (R = Et) or trigonal (R = nPr, nBu) symmetry, depending on the size of the cationic template.61 In the absence of any such external template, CPM-5 is formed instead in which the charge of the [In3(BTC)4]3− framework is balanced by a smaller positively charged In-containing cage bound to the outer framework and [Me2NH2]+ cations, produced upon decomposition of DMF solvent used in synthesis.63 In ZJU-28, the charge is again balanced by [Me2NH2]+ cations generated in situ, but the introduction of the larger and less rigid BTB linker gives rise to an anionic [In3(BTB)4]3− framework of hexagonal symmetry.64 The chemical and structural stability of these In-MOFs had not been established under conditions appropriate for homogeneous organometallic catalytic reactions, in particular stability in solvents such as CH2Cl2 and acetone. Therefore, a series of experiments was undertaken in order to identify an anionic In-MOF which is chemically and structurally stable in these solvents and whose associated organic cations can be exchanged in a stoichiometrically controlled manner.Stability tests in solvents
The chemical and structural stability of all the selected MOFs was tested in acetone or CH2Cl2 solvent under dry and anaerobic conditions. In a typical experiment, each MOF was soaked for 24 h at room temperature in the respective dry solvent. ICP-OES measurements of indium concentration in the supernatant verified that indium is not leaching ([In] < 0.1 ppm corresponding to <0.05% leaching). However, comparison of the PXRD patterns before and after soaking revealed that both solvents have a detrimental effect on the crystallinity of ZJU-28 (Fig. S1 and S2†). On the contrary, [R4N]3[In3(BTC)4] (Fig. 1) and CPM-5 (Fig. S3†) retain their crystallinity in acetone and CH2Cl2. These two MOFs are stable even in non-dried reagent-grade solvents. Le Bail fitting of the PXRD patterns indicates that the unit cell size does not change under these conditions, further confirming that the structure of the anionic framework remains intact in these solvents (Fig. S4–S10†).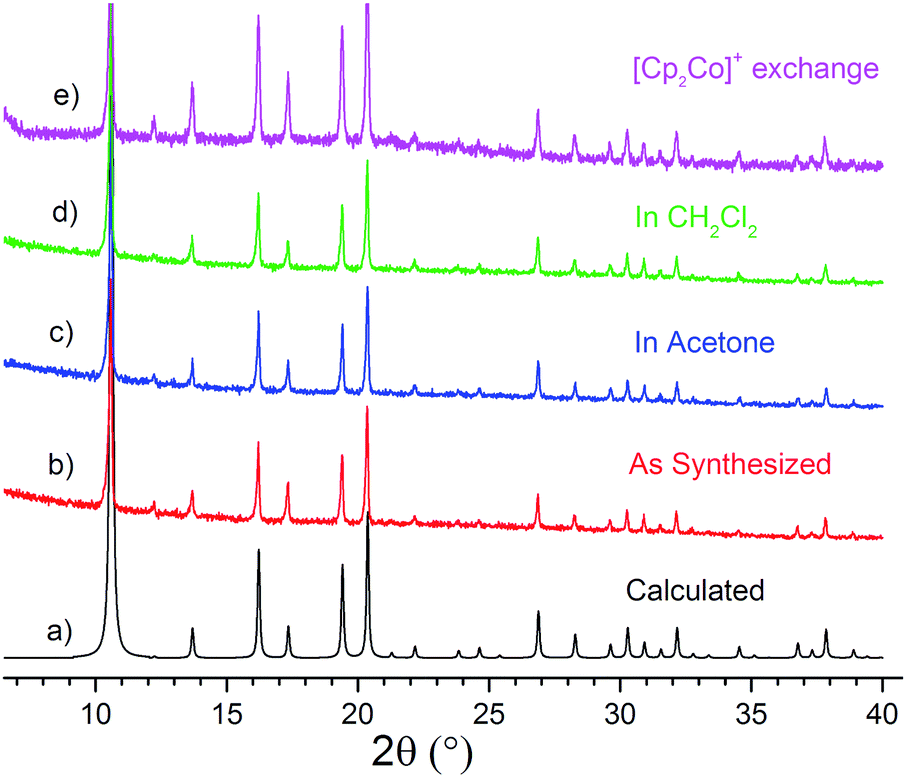 | ||
| Fig. 1 Comparison of Powder X-ray Diffraction (PXRD) patterns of [TEA]3[In3(BTC)4]: (a) calculated from the single crystal structure, (b) as synthesized, (c) after soaking for one day at 298 K in acetone or (d) CH2Cl2 and (e) after [Cp2Co]+ cation exchange in acetone. | ||
Cobaltocenium cation exchange
Once the stability of CPM-5 and [R4N]3[In3(BTC)4] in CH2Cl2 and acetone solvents was established, cation exchange experiments with [Cp2Co][PF6] were performed in order to establish the ideal exchange conditions. [Cp2Co]+ is a robust cation with a characteristic 1H and 13C NMR spectroscopic signature. Moreover, its cylindrical shape and small size makes it an excellent proxy for a series of positively charged TM catalysts bearing cyclopentadienyl (Cp) ligands with a [CpML3]+ molecular formula (e.g. L = CO, PR3, solvent) and a piano stool geometry.82 In a typical experiment, a solution of [Cp2Co][PF6] in acetone (0.02 M) was added to a vial containing each MOF and gentle shaking was applied up to 72 h. Both MOFs remain crystalline after partial [Cp2Co]+ cation exchange and the crystallinity of the anionic framework is retained in the bulk material, as deduced from the qualitative comparison (Fig. 1 and S3†) and the quantitative Le Bail fitting of the respective PRXD patterns (Fig. S7, S11–S13†).Cation exchange was monitored by ICP-OES and NMR spectroscopy. The Co/In ratio of the recovered material was measured by ICP-OES after digestion in diluted HNO3 (1/10 v/v). In the case of CPM-5, cation exchange cannot be controlled and ICP-OES results show that it exceeds 100% (replacement of all the organic [Me2NH2]+ cations by [Cp2Co]+, Table S1†). This indicates that most likely encapsulation of the entire, overall neutral, ion pair [Cp2Co][PF6] also takes place. Although this is an interesting result, it does not serve our purpose of establishing a stoichiometrically controlled cation exchange process, as it leads to multiple organometallic species encapsulated inside the pores of the anionic host.
By contrast, cation exchange for the [R4N]3[In3(BTC)4] MOFs proceeds in a stoichiometrically controlled manner in acetone (eqn (1)) and it depends on the starting [Cp2Co]+/[R4N]+ molar ratio and the size of the organic cation (Fig. 2). Starting with an equimolar [Cp2Co]+/[R4N]+ ratio, exchange reaches 16% for the larger TBA cation after 72 h, increases to 36% for TPA and reaches 60% for the smaller TEA cation. If the initial [Cp2Co]+/TEA ratio is raised to 3/1, then 82% of the TEA cations are replaced after 72 h (Table 1). Moreover, cation exchange is reversible. Mixing [(Cp2Co)1.8(TEA)1.2][In3(BTC)4] (60% exchange) with a solution of [TEA][BF4] in acetone (3-fold excess) results in partial replacement of the [Cp2Co]+ cations by TEA and formation of [(Cp2Co)1.2(TEA)1.8][In3(BTC)4] (40% exchange according to ICP-OES analysis after digestion). By contrast, exchange of TEA cations using the bulkier [Cp*2Co]+ cation (Cp* = η5-C5Me5, Fig. 3) under identical conditions does not take place at all, presumably due to size limitations (vide infra).
| [R4N]3[In3(BTC)4] + x[Cp2Co][PF6] → [(Cp2Co)x(R4N)3−x][In3(BTC)4] + x[R4N][PF6] | (1) |
 | ||
| Fig. 2 Cation exchange (%) of [R4N]+ by [Cp2Co]+ determined by ICP-OES for [(Cp2Co)x(R4N)3−x][In3(BTC)4] (x = 3 corresponds to 100%). Conditions: 0.02 M solution of [Cp2Co][PF6] in acetone, initial ratio [R4N]+/[Cp2Co]+ = 1/1 or 3/1. | ||
 | ||
| Fig. 3 Dimensions of organometallic cations tested for cation exchange (corrected for van der Waals radii).85,86 [Fp–acetone]+ was modelled by truncating the tropone ligand in the single-crystal structure of [Fp–tropone][BF4].103 | ||
Incorporation of the [Cp2Co]+ cation in the [In3(BTC)4]3− anionic framework was also verified by NMR spectroscopy in solution after digestion in DCl/d6-dmso (1/5 v/v). The [Cp2Co]+ cation is particularly stable and survives the digestion conditions, necessary for the holistic analysis of the encapsulated guests by NMR spectroscopy, giving rise to discrete 1H and 13C NMR signals (Fig. S14 and S15†). Therefore, 1H NMR spectroscopy in solution is a useful tool83,84 for quickly establishing the extent of cation exchange via the integration of the relative peak areas of the benzene ring of BTC (δH = 8.52 ppm), [Cp2Co]+ (δH = 5.71 ppm) and TEA (δH = 3.08(q) and 1.05(t) ppm). The results thus obtained are in very good agreement with ICP-OES (Table S2†). In addition, a septet observed at −142.9 ppm in the 31P{1H} NMR spectrum of [Cp2Co][PF6] in DCl/d6-dmso due to the [PF6]− anion (Fig. S14†), is not observed in the respective NMR spectrum of [(Cp2Co)x(TEA)3−x][In3(BTC)4] after digestion, confirming that only the cation and not the ion pair [Cp2Co][PF6] is encapsulated inside the framework, to the detection limit of 31P{1H} NMR spectroscopy (∼5%). TGA measurements for [(Cp2Co)x(TEA)3−x][In3(BTC)4] on heating in air show a higher inorganic residue compared to the parent [TEA]3[In3(BTC)4] MOF and suggest that solvent molecules are still trapped inside the pores of [(Cp2Co)x(TEA)3−x][In3(BTC)4] (Fig. S16†).
If the [Cp2Co]+ and [Cp*2Co]+ cations are described as cylinders, then the cylinder's height corresponds to the distance between the centres of the two Cp rings and the cylinder's radius corresponds to the distance between the centre of a Cp ring and the atoms at its periphery. Dimensions from the published single crystal structures (Fig. 3)85,86 demonstrate that the height and diameter, corrected for van der Waals radii, are markedly larger for [Cp*2Co]+ (7.7 Å × 8.8 Å) than for [Cp2Co]+ (6.6 Å × 6.6 Å). Taking also into account that the pore window in [TEA]3[In3(BTC)4] after correcting for van der Waals radii is 7.0 Å × 7.6 Å (Fig. S17†), it is evident that [Cp*2Co]+ is too large to pass through the windows, in complete agreement with the unsuccessful cation exchange experiments.
In the published structure of [TEA]3[In3(BTC)4], only the [In3(BTC)4]3− anionic framework is fully modelled.61 Each In(III) ion is coordinated by four carboxylate groups from four different BTC linkers. The [In(O2CR)4]− SBUs are situated on crystallographic ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) axes and adopt a tetragonally elongated tetrahedral geometry (S4 symmetry) with two smaller (98.4°) and four larger (115.3°) C–In–C angles. The interconnection of tetrahedral SBUs with the trigonal planar BTC linkers generates a (3,4)-connected C3N4-type framework. The TEA cations and solvent molecules were reported as disordered within the channels of the framework and their exact location was not determined.
axes and adopt a tetragonally elongated tetrahedral geometry (S4 symmetry) with two smaller (98.4°) and four larger (115.3°) C–In–C angles. The interconnection of tetrahedral SBUs with the trigonal planar BTC linkers generates a (3,4)-connected C3N4-type framework. The TEA cations and solvent molecules were reported as disordered within the channels of the framework and their exact location was not determined.
We were able to identify crystals suitable for diffraction from the partially exchanged [(Cp2Co)x(TEA)3−x][In3(BTC)4] MOF. This material adopts the same framework topology as the parent [TEA]3[In3(BTC)4] MOF (Fig. 4A) with very little relaxation of atomic positions. However, there is a large peak in the Fourier difference map on the axes in the centre of the pores, which is not present in the parent structure. With the only difference between the two structures being the cation exchange, it can be inferred that this peak is due to the presence of [Cp2Co]+ and when assigned to Co gives a refined occupancy of 0.28(2). The Co atom is situated in the middle of the cavity which is delimited by six tetrahedral [In(O2CR)4]− SBUs, with the four shortest Co–In distances measured at 6.247(1) Å and the two longest ones, defining the long axis of the cavity, measured at 10.202(1) Å (Fig. 4B). However, the positions of the Cp rings cannot be determined and as a result neither cation present (TEA or [Cp2Co]+) can be reliably modelled after cation exchange (Fig. S18†). Nevertheless, these results unequivocally demonstrate that the framework remains intact after cation exchange and strongly suggest that the [Cp2Co]+ cations are located in or near the centre of the pores. Moreover, cations with similar size and shape to [Cp2Co]+ should undergo cation exchange and be incorporated inside the pores of the [In3(BTC)4]3− anionic framework.
 | ||
| Fig. 4 (A) Overlay of the anionic frameworks of [TEA]3[In3(BTC)4] (blue), [(Cp2Co)x(TEA)3−x][In3(BTC)4] after partial cation exchange (green) and [Cp2Co]3[In3(BTC)4] (red). (B) Single crystal structure of [(Cp2Co)x(TEA)3−x][In3(BTC)4] after partial cation exchange. The Cp rings could not be reliably modelled and only the Co atoms are shown, positioned in the centre of the pores on axes. Co–In distances are depicted with green dashed lines. (C) Guest-accessible space in [Cp2Co]3[In3(BTC)4], calculated with one of the four disordered components of [Cp2Co]+ present. The Cp rings are facing into the channels. (D) Single crystal structure of [Cp2Co]3[In3(BTC)4] showing the encapsulated [Cp2Co]+ cation positioned offset from the center of the pores. Nearest C–H⋯O contacts between the Cp rings and the anionic framework are represented by red dotted lines. | ||
Cobaltocenium as a template
The identification of the spatial position of cations by X-ray crystallography is not always trivial, especially in MOFs in which the crystallographic symmetry of the local channels exceeds that of the encapsulated cations.87 As shown above, the associated cations in the crystal structures of [TEA]3[In3(BTC)4] and [(Cp2Co)x(TEA)3−x][In3(BTC)4] are disordered and cannot be accurately modelled using single crystal X-ray diffraction methods, thus preventing the calculation of the exact guest-accessible space within the framework. In order to remove the disorder between the two cationic species, we exploited the chemical robustness of [Cp2Co]+ by directly synthesising single crystals of the same [In3(BTC)4]3− framework using [Cp2Co]+ as a template which survives the solvothermal synthesis conditions. The reaction in DMF at 120 °C for 24 h between InCl3·4H2O, H3BTC and [Cp2Co][PF6] at the stoichiometrically required 3/4/3 ratio leads to yellow crystals of [Cp2Co]3[In3(BTC)4]·3(DMF)·6(H2O) which retain their colour even after repeated washing with DMF and MeOH. Single crystal X-ray diffraction reveals that [Cp2Co]3[In3(BTC)4] crystallizes in the same space group (I3d) and shows the same framework topology with parent [TEA]3[In3(BTC)4] and partially exchanged [(Cp2Co)x(TEA)3−x][In3(BTC)4] (Fig. 4A). More importantly, the location of the cations within the anionic framework can now be determined.
The negative charge of the [In3(BTC)4]3− framework is balanced by [Cp2Co]+ cations which reside slightly offset from the axes in the centre of the pores and the entire [Cp2Co]+ unit can be modelled satisfactorily despite being disordered across the four equivalent sites (Fig. S19†). Mapping of the remaining voids reveals a three-dimensional guest-accessible network of channels running through the framework (Fig. 4C). The [Cp2Co]+ cations sit close to the mid-point between two [In(O2CR)4]− SBUs (Co–In distances of 5.720(7) and 5.845(7) Å) interacting weakly with the framework via hydrogen bonding between the H atoms of the Cp rings and the O atoms of the framework, with the respective C–H⋯O shortest distances ranging from 2.399(4) to 2.802(4) Å. These weak supramolecular interactions help to position the cylindrical [Cp2Co]+ cations slightly away from the centre of the channel, but still almost in parallel with the long axis of the cavity (Fig. 4D). This well-defined orientation exposes the plane of one Cp ring to the guest-accessible space, with its normal oriented perpendicular to the main channel axis within the framework.
The guest-accessible space in [Cp2Co]3[In3(BTC)4] is 27%, as calculated with the OLEX2 programme,88 using a 1.8 Å spherical probe and accounting for the positional disorder of the cations (cf. 66% guest-accessible space when considering only the anionic framework, Fig. S17†). In the remaining pore space there are many peaks in the Fourier difference map, which are likely due to disordered solvent molecules. However, these are not uniquely identified and attempts to quantify the electron count using SQUEEZE89 are hampered by the disordered nature of the [Cp2Co]+ cations. Nevertheless, we have determined from the combination of TGA, ICP-OES, NMR spectroscopy and CHN elemental analysis that the remaining pore space is occupied by disordered DMF and H2O solvent molecules, leading to an overall formula of [Cp2Co]3[In3(BTC)4]·3(DMF)·6(H2O).
TGA shows the expected weight loss for H2O (5.1%) and DMF (10.5%) molecules trapped inside the pores (Fig. S20†). The 1H and 13C NMR spectra (Fig. S21†) after digestion in DCl/d6-dmso (1/5 v/v) show only the expected peaks for [Cp2Co]+ (δH = 5.75 ppm, 30H; δC = 85.6 ppm), BTC (δH = 8.54 ppm, 12H; δC = 132.5, 134.5 and 166.6 ppm) and DMF solvent (δH = 7.87, 2.83 and 2.66 ppm; δC = 163.9, 37.1 and 32.0 ppm). Moreover, no signals are observed in the corresponding 31P{1H} NMR spectrum, verifying that encapsulation of the ion pair [Cp2Co][PF6] does not take place.
Le Bail fitting of the PXRD pattern of the bulk material proves that a single crystalline phase is produced (Fig. S22†). Comparison of the PXRD patterns of [TEA]3[In3(BTC)4], [(Cp2Co)x(TEA)3−x][In3(BTC)4] and [Cp2Co]3[In3(BTC)4] suggests that the framework structure does not change as the number of [Cp2Co]+ cations encapsulated inside the pores is increased (Fig. S23†). Activation of [TEA]3[In3(BTC)4] and [Cp2Co]3[In3(BTC)4] at room temperature after solvent exchange with acetone and measurement of N2 adsorption–desorption isotherms at 77 K show that the BET surface area and the pore volume also do not vary substantially upon exchanging cations (Fig. S24–S28†). The BET surface area decreases from 592 m2 g−1 for [TEA]3[In3(BTC)4] to 505 m2 g−1 for [Cp2Co]3[In3(BTC)4], whereas pore volume is almost the same, measured at 0.235 cm3 g−1 for [TEA]3[In3(BTC)4] and 0.241 cm3 g−1 for [Cp2Co]3[In3(BTC)4]. These results imply that the shape and the size of the pores do not change dramatically when TEA cations are replaced by [Cp2Co]+, consistent with the similarity of the anionic frameworks in the respective crystal structures (Fig. 4A). Overall, [Cp2Co]+ is an excellent surrogate for determining the spatial positioning of cylindrical cations inside the framework. Finally, it should be noted that when the bulkier [Cp*2Co]+ cation is used instead as a template, only an amorphous material is obtained, consistent with the unsuccessful cation exchange experiments involving crystalline [TEA]3[In3(BTC)4].
[CpFe(CO)2(L)]+ cation exchange
The [Cp2Co]+ exchange and template synthesis experiments suggest that cations with a similar cylindrical shape and small size could be encapsulated by cation exchange in the [R4N]3[In3(BTC)4] family of MOFs. Therefore, we turned our attention to small positively charged [CpML3]+ organometallic catalysts with piano-stool geometry, where one of the Cp rings is replaced by three monodentate ligands. [CpFe(CO)2(THF)][BF4] ([Fp–THF][BF4]) was selected as a suitable candidate as the CO ligands serve as a spectroscopic handle allowing for inspection of the catalyst's integrity by IR spectroscopy after cation exchange. THF is readily displaced by various donor ligands, including coordinating solvent. [Fp–L]+ species (L = weakly bound solvent) are thermally unstable, decomposing via CO and solvent dissociation (vide infra). Therefore, encapsulation of [Fp–L]+ inside the [In3(BTC)4]3− anionic framework provides a potential means of suppressing this intrinsic instability and prolong the catalyst's lifetime.90As the solvent molecule is only weakly bound, [Fp–L]+ is a latent 16-electron Lewis acidic complex, potentially capable of activating substrates through single-point coordination.91,92 It is a well-documented homogeneous catalyst for a range of transformations such as the hydrosilylation of aldehydes, ketones and esters93,94 the cyclopropanation and aziridination of alkenes,95–97 the epoxidation of aromatic aldehydes,98,99 and in particular the classical Diels–Alder (DA) [4 + 2] cycloaddition of dienes and dieneophiles.100,101 In this regard, it is only a moderate homogeneous catalyst, often requiring loadings between 1 and 5 mol%. Nevertheless, it is ideally suited for our proof of principle objective to demonstrate catalyst's integrity and activity after encapsulation via cation exchange, rather than full optimisation of catalytic efficiency.
Cation exchange was carried out under strictly dry and anaerobic conditions. The highest exchange ratio was obtained using dry acetone as the solvent, which readily replaces THF in [Fp–THF]+,102 leaving [Fp–acetone]+ as the most probable cation encapsulated in the MOF. Even though a structure containing [Fp–acetone]+ has not yet been reported, examination of many reported crystal structures that contain [Fp–L]+ cations revealed that the diameter of the Cp ring, corrected for van der Waals radii, ranges between 6.64 Å and 6.71 Å. Moreover, the distance between the O atoms of the CO ligands and the mean plane containing the Cp ring lies between 6.4 and 6.7 Å, corrected for van der Waals radii.93,94,103,104 Recognising that the weakly-bound acetone ligand will be quite flexible in its binding orientation, the [Fp–acetone]+ cation can be viewed as approximately cylindrical in shape with dimensions between those of [Cp2Co]+ and [Cp*2Co]+ but closer to the former (Fig. 3).
As shown above, the [TEA]3[In3(BTC)4] MOF remains porous after cation exchange, even if all the TEA cations are exchanged with [Cp2Co]+. Calculation of the guest-accessible space for [Cp2Co]3[In3(BTC)4] reveals that the [Cp2Co]+ cations are located in the centre of the pores with the Cp rings facing the channels (Fig. 4). It is reasonable to suppose that the cylindrical [Fp–L]+ cation will be also positioned near the centre of the pores, adopting a similar orientation, due to the supramolecular interaction between the Cp rings of [Fp–L]+ and the SBUs of the framework. This will expose the opposite side, bearing the CO ligands and the labile L site to the guest-accessible space, allowing for substrates to access the catalytic sites. Therefore, [TEA]3[In3(BTC)4] was selected as the most suitable member of the [R4N]3[In3(BTC)4] family to enable cation exchange for the encapsulation of the catalytically active [Fp–L]+ cation.
Following our previously established protocol in which 53% of the TEA cations were exchanged with [Cp2Co]+ in 24 h, a 0.02 M solution of [Fp–THF][BF4] in dry acetone was combined with [TEA]3[In3(BTC)4] (initial molar ratio [Fp–L]+/TEA = 1/1) and gentle shaking was applied for 24 h during which the colour of the material changed from white to bright red (i.e. the colour of [Fp–L]+). ICP-OES verified the presence of iron in the isolated material after digestion in diluted HNO3, indicating 24% cation exchange. The IR spectrum revealed three bands in the ν(CO) region (2123, 2065 and 2016 cm−1) with the central peak showing higher intensity (Fig. 5c).
 | ||
| Fig. 5 Solid-state ATR IR spectra of (a) [Fp–THF][BF4], (b) [TEA]3[In3(BTC)4] and [(Fp–L)x(TEA)3−x][In3(BTC)4] prepared via cation exchange in acetone under different conditions: (c) shaking for 1 day at 25 °C, (d) soaking for 1 day at 25 °C and (e) soaking for 2 days at 5 °C. | ||
It has been shown that [Fp–L]+ species are unstable at room temperature and slowly decompose to form the tricarbonyl complex [CpFe(CO)3]+ ([Fp–CO]+).105 This redistribution of π-electron density to a total of three CO ligands shifts the ν(CO) peaks to higher frequencies (literature values of 2124 and 2074 cm−1).106 Therefore, the presence of three bands in the IR spectrum of the isolated material after cation exchange is consistent with partial decomposition of [Fp–L]+ to [Fp–CO]+ and encapsulation of both cationic species inside the pores of [In3(BTC)4]3−. Although this should give rise to four bands (i.e. A′, A′′ for [Fp–L]+ and A1, E for [Fp–CO]+), coincidence of the high frequency band of [Fp–L]+ and the low frequency band of [Fp–CO]+ leads to the observation of only three bands in this region of the spectrum, the middle of which has a higher observed intensity. This conclusion is supported by the fact that the intensity of the higher frequency band increases, while that of the lower frequency band decreases if the exchange process is extended to 72 h (Fig. S29†).
In contrast, when the exchange reaction was carried out by simply soaking and not shaking the MOF in a 0.02 M solution of [Fp–THF][BF4] in acetone for 24 h, formation of the undesired [Fp–CO]+ complex was not observed by IR spectroscopy (Fig. 5d). ICP-OES analysis of this material showed 23% exchange of TEA by [Fp–L]+ (Table 2). The exchange process can be carried out by soaking at 5 °C, in order to further suppress the formation of [Fp–CO]+. Under these conditions, cation exchange drops to 15% after 24 h, increasing to 20% if the reaction is extended to 48 h (Table 2), forming [(Fp–L)0.6(TEA)2.4][In3(BTC)4]. The reaction is reproducible and consistently 17–23% of the TEA cations were exchanged by [Fp–L]+ in the course of five different experiments. The IR spectrum of the isolated material shows only two bands at 2067 and 2018 cm−1 due to encapsulated [Fp–L]+ (Fig. 5e). Interestingly, the ν(CO) bands do not shift significantly compared to [Fp–THF][BF]4 implying that THF or a ligand of comparable donor strength is coordinated to iron. Nevertheless, THF is most likely replaced in the presence of a large excess of acetone as the solvent (also an O-donor ligand).102 Moreover, THF is not detected by IR spectroscopy. Instead, two bands are observed at 1711 and 1221 cm−1 (Fig. 5e), whereas all the other bands coincide with those of the parent MOF (Fig. 5b). The band at 1711 cm−1 could be assigned to acetone, which is also exchanged in the pores, or protonated carboxylate groups of the BTC linker, formed during cation exchange.107,108 However, the observation of a second peak at 1221 cm−1 suggests that both peaks originate from acetone (Fig. S30†). The presence of acetone as the only solvent inside the pores of [(Fp–L)0.6(TEA)2.4][In3(BTC)4] is corroborated by 1H NMR spectroscopy (vide infra), suggesting that most likely acetone occupies the vacant coordination site of the [Fp]+ cation.
| [TEA]3[In3(BTC)4] + x[Fp–L][BF4] → [(Fp–L)x(TEA)3−x][In3(BTC)4] + x[TEA][BF4] | ||
|---|---|---|
| 24 h (soaking, 25 °C) | 24 h (soaking, 5 °C) | 48 h (soaking, 5 °C) |
| a Conditions: 0.02 M solution of [Fp–THF][BF4] in dry acetone, initial ratio [(Fp–L)+]/[TEA] = 1/1. b Digestion in diluted HNO3 (1/10 v/v). | ||
| 23% (x = 0.69) | 15% (x = 0.45) | 20% (x = 0.60) |
In order to establish whether a solvent molecule is indeed weakly coordinated to iron, [(Fp–L)0.6(TEA)2.4][In3(BTC)4] was treated with CO gas for 15 min in a solid/gas reaction,109,110 during which the colour of the material changed from deep red to orange-yellow. Three bands (2123, 2066 and 2017 cm−1) were observed in the ν(CO) region of the respective IR spectrum, due to partial formation of [Fp–CO]+,105,106 confirming the presence of a single labile site in the coordination sphere of the encapsulated catalyst (Fig. S31†).
Attempts to remove the solvent molecules from [(Fp–L)0.6(TEA)2.4][In3(BTC)4] to measure gas sorption were not successful since the encapsulated catalyst decomposes under prolonged dynamic vacuum even at room temperature via CO and solvent dissociation, as evidenced by IR spectroscopy (Fig. S32†) with a concomitant colour change of the material from deep red to black. As shown, exchange of TEA with [Cp2CO]+ does not essentially affect N2 uptake of the respective MOFs (Fig. S24†). Likewise, exchange of TEA with [Fp–L]+, which is quasi-isosteric with [Cp2Co]+ (Fig. 3), is not expected to significantly alter pore size distribution. The shape of the particles does not change after cation exchange, as determined by SEM imaging (Fig. S33†). Qualitative comparison (Fig. 6) and Le Bail fitting of the PXRD patterns (Fig. S34†) before and after cation exchange demonstrate that the framework is intact and the dimensions of the unit cell do not change.
 | ||
| Fig. 6 Comparison of PXRD patterns between (a) [TEA]3[In3(BTC)4] and [(Fp–L)0.6(TEA)2.4][In3(BTC)4] (b) as synthesized, (c) after one and (d) after three catalytic cycles. | ||
The cation exchange can be monitored by 1H and 13C NMR spectroscopy in solution after digestion in DCl/d6-dmso (1/5 v/v). 1H NMR spectroscopy reveals that encapsulated [Fp–L]+ decomposes under digestion conditions, giving rise to two sets of peaks: a singlet at 5.12 ppm and two peaks of minor intensity at 6.29 and 6.37 ppm (Fig. 7). These observations are consistent with the 1H NMR spectrum of [Fp–THF][BF4] in DCl/d6-dmso (Fig. S37†). The lower field peaks emanate from an unknown decomposition product, thus complicating a reliable calculation of the cation exchange percentage. However, integration of the peak areas corresponding to the benzene ring of BTC (δH = 8.44 ppm, 12H) and TEA (δH = 3.05(q) ppm, 19.4H, CH2; δH = 1.00(t) ppm, 29.1H, CH3) is still possible, demonstrating 19% cation exchange (x = 0.57) and verifying that a stoichiometric process is taking place, in excellent agreement with ICP-OES results as well as IR evidence. Acetone is the only solvent detected (δH = 1.91 ppm, 5.4H, CH3) and importantly no peaks originating from THF are observed (cf. δH = 3.43 and 1.59 ppm for [Fp–THF][BF4] in DCl/d6-dmso, Fig. S37†). Finally, no 19F signals were observed in the 19F{1H} NMR spectrum of [(Fp–L)0.6(TEA)2.4][In3(BTC)4] after digestion, verifying that encapsulation of the entire ion pair [Fp–L][BF4] does not take place (cf. δF = −148 ppm for [Fp–THF][BF4] in DCl/d6-dmso).
 | ||
| Fig. 7 1H NMR spectrum of [(Fp–L)0.6(TEA)2.4][In3(BTC)4] after digestion in DCl/d6-dmso (1/5 v/v). NMR solvent peaks are marked with an asterisk. | ||
The 13C NMR spectrum of [(Fp–L)0.6(TEA)2.4][In3(BTC)4] after digestion (Fig. S38†) shows only the expected peaks for BTC (δC = 132.8, 134.8 and 166.9 ppm), TEA (δC = 52.8 and 8.4 ppm), Cp (δC = 86.9 ppm) and acetone solvent (δC = 31.4 ppm, CH3). Finally, TGA demonstrates the presence of Fe through a higher inorganic residue than expected for [TEA]3[In3(BTC)4] and a slight weight loss between 150–250 °C corresponding to catalyst decomposition (Fig. S39†). The identification of signals assigned to acetone in both NMR and IR spectra strongly suggests that acetone is the solvent molecule occupying the labile site in the iron's coordination sphere. However, a definite assignment has not been possible and the weakly bound ligand is referred to as L. To summarize, all spectroscopic evidence concurs that [Fp–L]+ remains intact after cation exchange, replacing 20% of the TEA cations and forming [(Fp–L)0.6(TEA)2.4][In3(BTC)4] where the labile ligand (L) is most likely acetone.
Diels–Alder reaction
For the final step in our approach, having established encapsulation of the catalyst inside the MOF, we explored whether [(Fp–L)0.6(TEA)2.4][In3(BTC)4] acts as a heterogenised catalyst in the DA reaction between isoprene (Iso) and methyl vinyl ketone (MVK) to form the 1,4-(para) and 1,5-(meta) isomers of methylacetylcyclohexene (Scheme 2). This is a well-established, but rather sluggish [4 + 2] cycloaddition which only takes place either under reflux or in the presence of a catalyst at room temperature.100,101 Both cationic and neutral TM complexes are known to mediate this transformation, operating however at relatively high loadings ranging between 1 and 5 mol%.111–113 Notably, Fujita et al. have demonstrated that carrying out a DA reaction inside a soluble organopalladium coordination cage may induce highly unusual regioselectivity.114 Jeong and co-workers have shown that S-KUMOF-1 MOF, comprising Ti(IV)-chelating chiral biphenol linker groups, catalyses a hetero-DA between Danishefsky's diene and benzaldehyde,115 whereas Zhou et al. have recently reported that PCN-223 MOF, containing an [Fe(III) (TCPP)Cl] metallolinker (TCPP = tetrakis-4-carboxy-phenyl porphyrin), catalyses a hetero-DA reaction between 2,3-dimethyl-1,4-butadiene and benzaldehyde in the presence of AgBF4 as the co-catalyst.116 In this contribution our choice of catalyst and substrates was dictated by the necessity to demonstrate proof of principle for the encapsulation of a discrete cationic catalyst rather than to optimise a particular catalytic transformation and arises from the quasi-isosteric nature of [Fp–L]+ and [Cp2Co]+.Control experiments showed that the parent MOF does not catalyse this reaction, as expected since all the In(III) ions of the framework are coordinatively saturated (entry 1, Table 3). By contrast, if [(Fp–L)0.6(TEA)2.4][In3(BTC)4] is used as the catalyst (10 mol% [Fe]/[MVK] loading), 26% yield is observed in CH2Cl2 after 96 h under gentle shaking (entry 2). Further optimization experiments show that the yield considerably increases if a higher [Iso]/[MVK] ratio is introduced (entries 3–5). The yield of the heterogeneous reaction reaches 45% after 96 h if four equivalents of isoprene are added. The yield of the homogeneous counterpart under the same Fe loading and substrate concentration reaches 67% after 48 h and does not increase any further (Fig. 8A and S42†). The regioselectivity of the heterogeneous reaction is slightly different than the homogeneous counterpart and a 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 ratio between the 1,4- and 1,5-isomers is obtained, compared with 97:3 for the homogeneous system.
:9 ratio between the 1,4- and 1,5-isomers is obtained, compared with 97:3 for the homogeneous system.
 | ||
| Scheme 2 Diels–Alder reaction between Iso and MVK. | ||
| Catalyst | mol% [Fe]/[MVK] | [Iso]/[MVK] | % Yield (1,4:1,5)b |
|
|---|---|---|---|---|
| a [MVK] = 0.1 M, solvent = CH2Cl2, 25 °C, t = 96 h. b Yield and regioselectivity (in parenthesis) were determined by GC. The major product, 1-methyl-4-acetyl-cyclohexene (1,4-MeAcCyHe) was further identified by 1H and 13C NMR spectroscopy (Fig. S40 and S41). c Same amount of parent MOF used. d t = 48 h. | ||||
| 1 | [TEA]3[In3(BTC)4]c | 0 | 4 | 0 |
| 2 | [(Fp–L)0.6(TEA)2.4][In3(BTC)4] | 10 | 1 | 26 (91:9) |
| 3 | [(Fp–L)0.6(TEA)2.4][In3(BTC)4] | 10 | 2 | 40 (92:8) |
| 4 | [(Fp–L)0.6(TEA)2.4][In3(BTC)4] | 10 | 4 | 45 (91:9) |
| 5 | [(Fp–L)0.6(TEA)2.4][In3(BTC)4] | 5 | 4 | 21 (93:7) |
| 7 | [Fp–THF][BF4]d | 10 | 4 | 67 (97:3) |
| 8 | [Fp–THF][BF4]d | 1 | 4 | 24 (98:2) |
As separating catalyst and products is challenging in the homogeneous system the reaction flask was recharged with more substrates without separation, which lead to a further 33% product yield. This suggests that product inhibition plays a role in the observed temporal profile. By contrast the encapsulated catalyst is very simply physically separated from the reaction mixture by cannula-filtration, washed thoroughly with dry CH2Cl2 and exposed to a fresh batch of substrates. These recycling experiments (Fig. 8B) showed that the encapsulated catalyst could be recycled twice (42% yield for the 2nd cycle, 27% for the 3rd cycle). Thus, although both catalyst systems give very similar TON (10 for the homogeneous and 12 for the heterogeneous reaction), the encapsulated version shows the significant benefit of the ability to physically separate the active catalyst from the reaction mixture, while retaining activity.
 | ||
| Fig. 8 (A) Yield vs. time plot of heterogeneous (black) and homogeneous (red) DA reaction under identical conditions (10 mol% [Fe]/[MVK] in CH2Cl2, [Iso] = 0.4 M, [MVK] = 0.1 M). (B) Recycling of [(Fp–L)0.6(TEA)2.4][In3(BTC)4] under the same conditions. Each cycle lasts 96 h. | ||
Qualitative comparison (Fig. 6) and Le Bail fitting (Fig. S34–S36†) of the PXRD patterns for [(Fp–L)0.6(TEA)2.4][In3(BTC)4] before and after turnover confirm that the [In3(BTC)4]3− anionic framework remains crystalline and the dimensions of the unit cell do not change after three catalytic cycles. SEM images show that the shape and size of the particles also does not change (Fig. S33†). However, the IR spectrum of the isolated catalyst shows a decrease of the intensity of the ν(CO) and the ν(Me2CO) bands during turnover (Fig. 9A and S43†). This suggests that the encapsulated [Fp–L]+ cation is slowly decomposing during turnover, as already noted in homogeneous catalysis studies where the loss of activity is more pronounced. Moreover, the ν(CO) bands are shifted to higher frequencies after turnover, indicating a change in the coordination sphere of the catalyst, possibly by substrate, product or CH2Cl2 solvent replacing the labile acetone ligand. These two observations regarding the ν(CO) bands (decreasing of intensity due to partial decomposition and shifting to higher frequencies due to a different coordination environment) are consistent with the catalyst's intrinsic instability and justify the lower catalytic activity during the 3rd cycle. The fate of the catalyst, however, is not resolved.
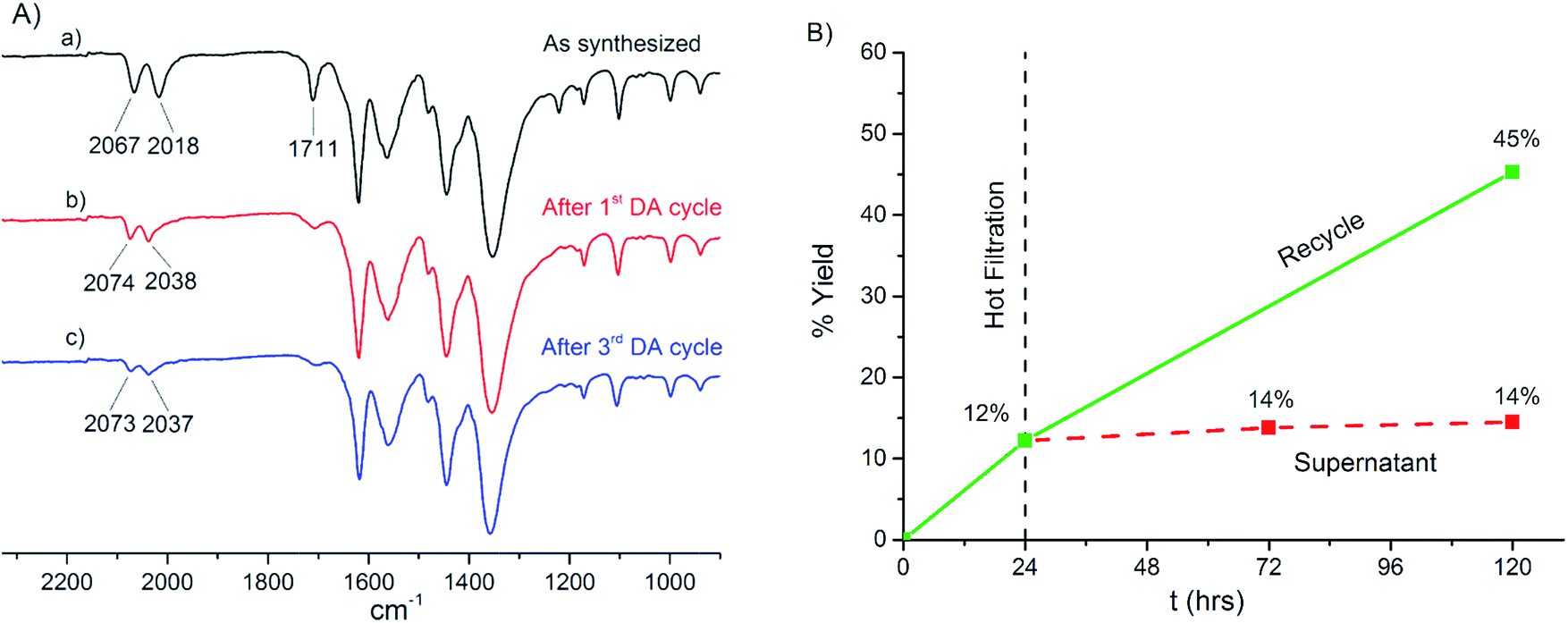 | ||
| Fig. 9 (A) IR spectrum of [(Fp–L)0.6(TEA)2.4][In3(BTC)4] (a) as synthesized, (b) after one and (c) after three catalytic cycles. Each cycle lasts 96 h. Spectra have been normalized to the highest intensity peak for the sake of comparison. (B) Leaching test for [(Fp–L)0.6(TEA)2.4][In3(BTC)4]. The DA reaction essentially stops in the supernatant after filtering off the catalyst (red dashed line). The solid catalyst is still active if a fresh batch of reactants is added (green solid line). | ||
ICP-OES confirms that neither iron or indium leach into the liquid phase ([Fe] < 0.5 ppm corresponding to less than 0.05 mol% Fe loading in the supernatant, Table S3†). Moreover, the Fe/In ratio of the catalyst does not change after 2 cycles (Table S4†). The heterogeneous nature of the reaction was further established by filtering off the supernatant after 24 h of reaction and allowing it to react for further 96 h. Catalytic turnover in the supernatant essentially stops in the absence of the heterogeneous catalyst (Fig. 9B). By contrast, the isolated [(Fp–L)0.6(TEA)2.4][In3(BTC)4] catalyst is still active and conversion again reaches 45% if a fresh batch of reactants is added to the solid previously separated from the supernatant.
Conclusions
We provide here a comprehensive protocol for the encapsulation of positively charged TM catalysts inside the pores of an anionic MOF by direct one-step cation exchange. To achieve this, a stepwise, gated, approach was developed to assess host-MOF stability, cation exchange efficiency, pore dimensions of the selected host, spatial location of the guest cations, encapsulation of an organometallic cationic TM catalyst inside the pores and finally benchmarking as a heterogeneous recyclable Diels–Alder catalyst. Central to this approach is thorough characterisation by IR and NMR spectroscopy, TGA, powder and single-crystal X-ray diffraction, SEM and ICP-OES at each step of the process. Such well characterized, catalyst@MOF hybrid materials are rare.[TEA]3[In3(BTC)4] was selected as a suitable host, after screening its chemical and structural stability in organic solvents suitable for organometallic catalytic reactions. Cation exchange efficiency was monitored using the robust [Cp2Co]+ cation as an inert probe. [Cp2Co]+ replaces TEA inside the pores of the [In3(BTC)4]3− anionic framework in a stoichiometrically controlled manner without perturbing the crystal structure of the framework. However, the encapsulated cations are disordered after partial cation exchange and their exact position cannot be determined by X-ray crystallography.
By contrast, the spatial location of the guest cations is possible in [Cp2Co]3[In3(BTC)4]. This new MOF, synthesized using [Cp2Co]+ as a cationic template, shows identical connectivity and comparable porosity with [TEA]3[In3(BTC)4]. The cylindrically shaped encapsulated [Cp2Co]+ cations are held in place through strong electrostatic attractive forces and weaker H-bonding interactions. They are situated slightly offset from the centre of the pores with their main axis orientated in parallel to the long axis of the cavities.
Finally, the cationic Lewis acidic [CpFe(CO)2(L)]+ TM catalyst was successfully exchanged into the pores of the [In3(BTC)4]3− anionic framework. Rigorous characterisation of the material obtained after cation exchange shows that [Fp–L]+ is encapsulated intact, forming [(Fp–L)0.6(TEA)2.4][In3(BTC)4]. The [Fp–L]+ cations are quasi-isosteric with [Cp2Co]+, therefore they are expected to adopt a similar position and orientation within the pores of the [In3(BTC)4]3− anionic framework. They are most likely also situated near the centre of the pores with the catalytically active side facing into the guest-accessible space, therefore accessible to substrates. [(Fp–L)0.6(TEA)2.4][In3(BTC)4] was benchmarked as a recyclable heterogeneous catalyst in a Diels–Alder reaction between isoprene and MVK. The encapsulated [Fp–L]+ cation does not leach into the liquid phase. Similar catalytic activity is observed for the heterogeneous and the homogeneous systems, demonstrating the feasibility of the method. Importantly, the hybrid catalyst can be easily recycled by simple physical separation from the reaction mixture and it still demonstrates catalytic activity after 12 days.
Overall these data point to a comprehensively characterised example of a cationic molecular TM catalyst encapsulated within the pores of a stable anionic MOF. Such an approach is essential to not only understand and exploit the system at hand, but also to enable the rational, reliable and scalable design of future materials in which well-defined cationic TM catalysts are encapsulated in MOFs by direct cation exchange to form [catalyst]+@[MOF]− hybrid systems. The criteria outlined herein should be widely applicable as general design elements for new hybrid catalyst materials, and establish a platform for further catalyst development by this charge-assisted approach, where supramolecular interactions also play a part in locating the organometallic catalyst in a well-defined orientation. Future work will be directed to encapsulation of more sophisticated and active catalysts or catalytic precursors to perform a variety of challenging transformations.
Experimental section
Detailed report of materials, methods and instrumentation used is provided in the ESI.† Herein we describe only the most important experimental procedures.[(Cp2Co)x(TEA)3−x][In3(BTC)4]
Reaction was carried out in air using reagent-grade solvents. [TEA]3[In3(BTC)4]·2(DMF)·5(H2O) (100 mg, 0.056 mmol) was combined in a vial with a 0.02 M acetone solution of [Cp2Co][PF6] (9 mL, 0.18 mmol). The vial was gently shaken for a specific number of days and the colour of the MOF progressively changed from white to yellow. The crystals were isolated by filtration, washed with acetone until the filtrate was colourless (3 × 10 mL) and dried under vacuum. Cobalt and indium content was determined by ICP-OES after digesting 10 mg of the material in 10 mL of diluted HNO3 (1/10 v/v). Cation exchange was calculated by the respective Co/In ratio and verified by 1H NMR after digestion in DCl/d6-dmso (1/5 v/v).[Cp2Co]3[In3(BTC)4]·3(DMF)·6(H2O)
Reaction was carried out in air using reagent-grade solvents. Stock solutions of InCl3·4H2O (0.13 M), H3BTC (0.20 M) and [Cp2Co][PF6] (0.08 M) in DMF were initially prepared. In a 40 mL vial with a teflon-lined screw cap, InCl3·4H2O (2.35 mL, 0.305 mmol), H3BTC (2.00 mL, 0.400 mmol) and [Cp2Co][PF6] (3.79 mL, 0.304 mmol) were added and then diluted with DMF (1.86 mL) to a total volume of 10 mL. The vial was placed in an oven and heated at 120 °C (1.0 °C min−1), maintained at this temperature for 24 h and subsequently slowly cooled down to room temperature (0.2 °C min−1). Yellow crystals precipitated upon cooling that were collected by filtration, washed thoroughly with DMF and MeOH and dried under vacuum. Detailed characterization is provided in the ESI.†[(Fp–L)0.6(TEA)2.4][In3(BTC)4]
Reaction was carried out using standard Schlenk line techniques or an Ar atmosphere dry box and dry solvents. [TEA]3[In3(BTC)4]·2(DMF)·5(H2O) (100 mg, 0.056 mmol) was immersed in 5 mL of dry acetone inside a Schlenk tube and gentle shaking was applied for 48 h during which the solvent was replenished once after 24 h. The solvent was decanted and the crystals were dried under high vacuum. A 0.02 M solution of [Fp–THF][BF4] (64 mg, 0.190 mmol) in 9 mL of dry and degassed acetone was prepared inside a Schlenk flask at 5 °C. The solution was transferred via a filter cannula inside the Schlenk tube containing the parent MOF, the tube was sealed under N2 and kept in the refrigerator for 48 h. The supernatant was filtered off, the red coloured crystals were washed twice with dry acetone and then soaked in acetone for further 48 h at 5 °C. Subsequently, the supernatant was filtered off and the crystals were dried under vacuum until the particles could move freely. The isolated product was stored in a vial in the refrigerator inside the glovebox where it is indefinitely stable. Detailed characterization is provided in the ESI.†Heterogeneous Diels–Alder reactions
The catalytic reaction of MVK and isoprene is described. Reaction was carried out under an inert atmosphere using standard Schlenk line techniques or an Ar atmosphere dry box. Freshly prepared solutions of MVK (0.493 M) and Iso (0.500 M) in dry and degassed CH2Cl2 were used. Inside the glovebox, [(Fp–L)0.6(TEA)2.4][In3(BTC)4] (38 mg, 1.9% w/w Fe from ICP, 0.013 mmol Fe) was added in a Schlenk tube. The Schlenk tube was mounted on the Schlenk line and MVK (0.25 mL = 0.123 mmol) and Iso (1.0 mL = 0.500 mmol) were added dropwise via a syringe. The tube was gently shaken for 96 h. Subsequently, the supernatant was decanted, filtered via a syringe filter (0.2 μm, Acrodisc® GHP) and analysed by GC. Major product's yield was determined after calibration with the commercially available compound. The major product was also characterized by GC-MS and 1H and 13C NMR after removing all the volatile compounds under vacuum and dissolving the organic residue in CDCl3.Acknowledgements
UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/K014706/1, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1). A. G. would like to acknowledge Dr Marco Zanella (SEM) and Dr Xiaofeng Wu (CO experiments). We are grateful to the Diamond Light Source (beamline I19) for beamtime.Notes and references
- J. M. Thomas and W. J. Thomas, Principles and practice of heterogeneous catalysis, VCH, Weinheim, 1997 Search PubMed.
- J. M. Thomas, Design and applications of single-site heterogeneous catalysts, Imperial College Press, London, 2012 Search PubMed.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC.
- U. Diaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083 RSC.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589 CrossRef CAS PubMed.
- C. A. McNamara, M. J. Dixon and M. Bradley, Chem. Rev., 2002, 102, 3275 CrossRef CAS PubMed.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef CAS PubMed.
- P. Barbaro and F. Liguori, Chem. Rev., 2009, 109, 515 CrossRef CAS PubMed.
- C. E. Song and S.-G. Lee, Chem. Rev., 2002, 102, 3495 CrossRef CAS PubMed.
- D. E. de Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615 CrossRef CAS PubMed.
- A. Corma and H. García, Chem. Rev., 2002, 102, 3837 CrossRef CAS PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed.
- A. Dhakshinamoorthy and H. Garcia, ChemSusChem, 2014, 7, 2392 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011 RSC.
- Z.-J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC.
- M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310 CrossRef CAS.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361 CrossRef CAS.
- P. Garcia-Garcia, M. Muller and A. Corma, Chem. Sci., 2014, 5, 2979 RSC.
- M. Zhang, Z.-Y. Gu, M. Bosch, Z. Perry and H.-C. Zhou, Coord. Chem. Rev., 2015, 293–294, 327 CrossRef CAS.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445 CrossRef CAS PubMed.
- C. Wang, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2012, 134, 19895 CrossRef CAS PubMed.
- K. Manna, T. Zhang, M. Carboni, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2014, 136, 13182 CrossRef CAS PubMed.
- B. Gui, K. K. Yee, Y. L. Wong, S. M. Yiu, M. Zeller, C. Wang and Z. Xu, Chem. Commun., 2015, 51, 6917 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498 RSC.
- L. Chen, H. Chen, R. Luque and Y. Li, Chem. Sci., 2014, 5, 3708 RSC.
- J. Canivet, S. Aguado, Y. Schuurman and D. Farrusseng, J. Am. Chem. Soc., 2013, 135, 4195 CrossRef CAS PubMed.
- H. Fei, J. Shin, Y. S. Meng, M. Adelhardt, J. Sutter, K. Meyer and S. M. Cohden, J. Am. Chem. Soc., 2014, 136, 4965 CrossRef CAS PubMed.
- C. Bai, S. Jian, X. Yao and Y. Li, Catal. Sci. Technol., 2014, 4, 3261 CAS.
- S. Wu, L. Chen, B. Yin and Y. Li, Chem. Commun., 2015, 51, 9884 RSC.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997 CrossRef CAS PubMed.
- K. Sasan, Q. Lin, C. Mao and P. Feng, Chem. Commun., 2014, 50, 10390 RSC.
- K. Manna, T. Zhang and W. Lin, J. Am. Chem. Soc., 2014, 136, 6566 CrossRef CAS PubMed.
- M. I. Gonzalez, E. D. Bloch, J. A. Mason, S. J. Teat and J. R. Long, Inorg. Chem., 2015, 54, 2995 CrossRef CAS PubMed.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838 CrossRef CAS PubMed.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307 CrossRef CAS PubMed.
- M. H. Alkordi, Y. Liu, R. W. Larsen, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 12639 CrossRef CAS PubMed.
- C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren and Z.-M. Su, J. Am. Chem. Soc., 2009, 131, 1883 CrossRef CAS PubMed.
- F.-J. Ma, S.-X. Liu, C.-Y. Sun, D.-D. Liang, G.-J. Ren, F. Wei, Y.-G. Chen and Z.-M. Su, J. Am. Chem. Soc., 2011, 133, 4178 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 928 CrossRef CAS PubMed.
- L. Wang, W. Yang, Y. Li, Z. Xie, W. Zhu and Z.-M. Sun, Chem. Commun., 2014, 50, 11653 RSC.
- C. L. Whittington, L. Wojtas, W.-Y. Gao, S. Ma and R. W. Larsen, Dalton Trans., 2015, 44, 5331 RSC.
- A. Kozlov, A. Kozlova, K. Asakura and Y. Iwasawa, J. Mol. Catal. A: Chem., 1999, 137, 223 CrossRef CAS.
- N. A. Caplan, F. E. Hancock, P. C. Bulman Page and G. J. Hutchings, Angew. Chem., Int. Ed., 2004, 43, 1685 CrossRef CAS PubMed.
- K. Kervinen, P. C. A. Bruijnincx, A. M. Beale, J. G. Mesu, G. van Koten, R. J. M. Klein Gebbink and B. M. Weckhuysen, J. Am. Chem. Soc., 2006, 128, 3208 CrossRef CAS PubMed.
- J. Połtowicz, K. Pamin, E. Tabor, J. Haber, A. Adamski and Z. Sojka, Appl. Catal., A, 2006, 299, 235 CrossRef.
- R. J. Corrêa, G. C. Salomão, M. H. N. Olsen, L. C. Filho, V. Drago, C. Fernandes and O. A. C. Antunes, Appl. Catal., A, 2008, 336, 35 CrossRef.
- K. K. Bania, G. V. Karunakar, K. Goutham and R. C. Deka, Inorg. Chem., 2013, 52, 8017 CrossRef CAS PubMed.
- B. Li, Y. Zhang, D. Ma, T. Ma, Z. Shi and S. Ma, J. Am. Chem. Soc., 2014, 136, 1202 CrossRef CAS PubMed.
- Y. Chen, B. Fan, N. Lu and R. Li, Catal. Commun., 2015, 64, 91 CrossRef CAS.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746 CrossRef CAS PubMed.
- C. J. Brown, G. M. Miller, M. W. Johnson, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 11964 CrossRef CAS PubMed.
- Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 7358 CrossRef CAS PubMed.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100 CrossRef CAS PubMed.
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586 CrossRef CAS PubMed.
- S. Chen, J. Zhang, T. Wu, P. Feng and X. Bu, J. Am. Chem. Soc., 2009, 131, 16027 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- S.-T. Zheng, J. T. Bu, Y. Li, T. Wu, F. Zuo, P. Feng and X. Bu, J. Am. Chem. Soc., 2010, 132, 17062 CrossRef CAS PubMed.
- J. Yu, Y. Cui, C. Wu, Y. Yang, Z. Wang, M. O'Keeffe, B. Chen and G. Qian, Angew. Chem., Int. Ed., 2012, 51, 10542 CrossRef CAS PubMed.
- Z. Lin, F. Jiang, L. Chen, D. Yuan and M. Hong, Inorg. Chem., 2005, 44, 73 CrossRef CAS PubMed.
- Y. Liu, V. C. Kravtsov, R. Larsen and M. Eddaoudi, Chem. Commun., 2006, 1488 RSC.
- Y. Liu, V. C. Kravtsov and M. Eddaoudi, Angew. Chem., Int. Ed., 2008, 47, 8446 CrossRef CAS PubMed.
- S. Yang, X. Lin, A. J. Blake, K. M. Thomas, P. Hubberstey, N. R. Champness and M. Schroder, Chem. Commun., 2008, 6108 RSC.
- S. Yang, X. Lin, A. J. Blake, G. S. Walker, P. Hubberstey, N. R. Champness and M. Schröder, Nat. Chem., 2009, 1, 487 CrossRef CAS PubMed.
- S. Yang, S. K. Callear, A. J. Ramirez-Cuesta, W. I. F. David, J. Sun, A. J. Blake, N. R. Champness and M. Schroder, Faraday Discuss., 2011, 151, 19 RSC.
- S. Huh, T.-H. Kwon, N. Park, S.-J. Kim and Y. Kim, Chem. Commun., 2009, 4953 RSC.
- L. Wang, T. Song, C. Li, J. Xia, S. Wang, L. Wang and J. Xu, J. Solid State Chem., 2012, 190, 208 CrossRef CAS.
- C. Zhu, G. Yuan, X. Chen, Z. Yang and Y. Cui, J. Am. Chem. Soc., 2012, 134, 8058 CrossRef CAS PubMed.
- Z.-J. Lin, T.-F. Liu, Y.-B. Huang, J. Lü and R. Cao, Chem.–Eur. J., 2012, 18, 7896 CrossRef CAS PubMed.
- Z.-J. Lin, Y.-B. Huang, T.-F. Liu, X.-Y. Li and R. Cao, Inorg. Chem., 2013, 52, 3127 CrossRef CAS PubMed.
- Y. Huang, Z. Lin, H. Fu, F. Wang, M. Shen, X. Wang and R. Cao, ChemSusChem, 2014, 7, 2647 CrossRef CAS PubMed.
- G. Zhou, Y. Yang, R. Fan, W. Cao and B. Yang, CrystEngComm, 2012, 14, 193 RSC.
- J.-M. Gu, S.-J. Kim, Y. Kim and S. Huh, CrystEngComm, 2012, 14, 1819 RSC.
- S.-T. Zheng, C. Mao, T. Wu, S. Lee, P. Feng and X. Bu, J. Am. Chem. Soc., 2012, 134, 11936 CrossRef CAS PubMed.
- E.-Y. Cho, J.-M. Gu, I.-H. Choi, W.-S. Kim, Y.-K. Hwang, S. Huh, S.-J. Kim and Y. Kim, Cryst. Growth Des., 2014, 14, 5026 CAS.
- S. N. Zhao, X. Z. Song, M. Zhu, X. Meng, L. L. Wu, J. Feng, S. Y. Song and H. J. Zhang, Chem.–Eur. J., 2015, 21, 9748 CrossRef CAS PubMed.
- I. R. Butler, in Organometallic Chemistry, ed. M. Green, The Royal Society of Chemistry, Cambridge, 2002, 30, 385 Search PubMed.
- K. Akhbari and A. Morsali, Dalton Trans., 2013, 42, 4786 RSC.
- B. Cheng, F. Zare Karizi, M.-L. Hu and A. Morsali, Mater. Lett., 2014, 137, 88 CrossRef CAS.
- D. Braga, L. Scaccianoce, F. Grepioni and S. M. Draper, Organometallics, 1996, 15, 4675 CrossRef CAS.
- D. Braga, O. Benedi, L. Maini and F. Grepioni, J. Chem. Soc., Dalton Trans., 1999, 2611 RSC.
- M. Zhang, M. Bosch and H.-C. Zhou, CrystEngComm, 2015, 17, 996 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- D. J. Casper, A. V. Sklyarov, S. Hardcastle, T. L. Barr, F. H. Försterling, K. F. Surerus and M. M. Hossain, Inorg. Chim. Acta, 2006, 359, 3129 CrossRef CAS.
- E. P. Kündig and C. M. Saudan, Lewis Acids in Organic Synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed.
- M. Redlich, M. F. Mayer and M. M. Hossain, Aldrichimica Acta, 2003, 36, 3 CAS.
- D. Bézier, G. T. Venkanna, L. C. M. Castro, J. Zheng, T. Roisnel, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2012, 354, 1879 CrossRef.
- J. Zheng, L. C. Misal Castro, T. Roisnel, C. Darcel and J.-B. Sortais, Inorg. Chim. Acta, 2012, 380, 301 CrossRef CAS.
- W. J. Seitz, A. K. Saha and M. M. Hossain, Organometallics, 1993, 12, 2604 CrossRef CAS.
- M. F. Mayer and M. M. Hossain, J. Org. Chem., 1998, 63, 6839 CrossRef CAS PubMed.
- B. D. Heuss, M. F. Mayer, S. Dennis and M. M. Hossain, Inorg. Chim. Acta, 2003, 342, 301 CrossRef CAS.
- S. J. Mahmood, A. K. Saha and M. M. Hossain, Tetrahedron, 1998, 54, 349 CrossRef CAS.
- S. J. Mahmood and M. M. Hossain, J. Org. Chem., 1998, 63, 3333 CrossRef CAS.
- P. V. Bonnesen, C. L. Puckett, R. V. Honeychuck and W. H. Hersh, J. Am. Chem. Soc., 1989, 111, 6070 CrossRef CAS.
- M. E. Bruin and P. E. Kundig, Chem. Commun., 1998, 2635 RSC.
- D. L. Reger and C. Coleman, J. Organomet. Chem., 1977, 131, 153 CrossRef CAS.
- P. Boudjouk, J. B. Woell, L. J. Radonovich and M. W. Eyring, Organometallics, 1982, 1, 582 CrossRef CAS.
- T. Kückmann, H.-W. Lerner and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m966 Search PubMed.
- D. L. Reger, C. J. Coleman and P. J. McElligott, J. Organomet. Chem., 1979, 171, 73 CrossRef CAS.
- S. E. Gibson, H. Ibrahim, C. Pasquier, M. A. Peplow, J. M. Rushton, J. W. Steed and S. Sur, Chem.–Eur. J., 2002, 8, 269 CrossRef CAS.
- C. Volkringer, H. Leclerc, J.-C. Lavalley, T. Loiseau, G. Férey, M. Daturi and A. Vimont, J. Phys. Chem. C, 2012, 116, 5710 CAS.
- E. Alvarez, N. Guillou, C. Martineau, B. Bueken, B. van de Voorde, C. le Guillouzer, P. Fabry, F. Nouar, F. Taulelle, D. de Vos, J.-S. Chang, K. H. Cho, N. Ramsahye, T. Devic, M. Daturi, G. Maurin and C. Serre, Angew. Chem., Int. Ed., 2015, 54, 3664 CrossRef CAS PubMed.
- N. J. Coville and L. Cheng, J. Organomet. Chem., 1998, 571, 149 CrossRef CAS.
- S. D. Pike and A. S. Weller, Philos. Trans. R. Soc. London, Ser. A, 2015, 373, 20140187 CrossRef PubMed.
- J. Rickerby, M. Vallet, G. Bernardinelli, F. Viton and E. P. Kündig, Chem.–Eur. J., 2007, 13, 3354 CrossRef CAS PubMed.
- S. Kobayashi, T. Tsuchiya, I. Komoto and J.-I. Matsuo, J. Organomet. Chem., 2001, 624, 392 CrossRef CAS.
- D. Carmona, F. Viguri, A. Asenjo, F. J. Lahoz, P. García-Orduña and L. A. Oro, Organometallics, 2012, 31, 4551 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251 CrossRef CAS PubMed.
- K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877 RSC.
- D. Feng, Z.-Y. Gu, Y.-P. Chen, J. Park, Z. Wei, Y. Sun, M. Bosch, S. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 17714 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and methods, additional figures and tables describing materials characterisation and catalytic activity, crystallographic details. CCDC 1417097 and 1417098. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03494a |
| This journal is © The Royal Society of Chemistry 2016 |