
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneration of artificial sequence-specific nucleases via a preassembled inert-template†
Xianjin
Xiao
,
Tongbo
Wu
,
Feidan
Gu
and
Meiping
Zhao
*
Beijing National Laboratory for Molecular Sciences and MOE Key Laboratory of Bioorganic Chemistry and Molecular Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: mpzhao@pku.edu.cn
First published on 7th December 2015
Abstract
Sequence specific nucleases are important tools for processing nucleic acids in a predictable way. Herein, we demonstrate a conceptually new approach for generating sequence-specific nucleases via a preassembled inert-template (PAIT). A fairly stable DNase I/inert-DNA complex was prepared with a customized sequence specificity for a target DNA which contains a sequence complementary to the inert-DNA template. The complex could efficiently cleave the targeted sequence within either a long double-stranded DNA or a single-stranded DNA without affecting other unrelated DNA strands. The discrimination factor against single-base mismatch strands within a 14 nt target region was as high as 25.3. The strategy was also successfully applied to RNase A. Our findings may hold great potential for the development of a number of new powerful enzymatic tools.
Introduction
Sequence specific nucleases are important enzymatic tools for processing nucleic acid molecules in a predictable and reproducible way.1–4 Restriction enzymes are the most commonly used sequence specific nucleases. However, the available restriction enzymes can only recognize and cut a limited number of specific sequences.5,6 Chemical nucleases targeted to a specific sequence guided by RNA7 or PNA8 have been developed. However, they suffer from poor biocompatibility and low efficiency. The rational engineering of restriction endonucleases9,10 and other rare-cutting endonucleases such as homing endonucleases11,12 through predictable mutagenisis13,14 and reassortment of target recognition domains15–17 has created hundreds of new sequence-specific enzymes. However, the range of predictability and customization is still quite limited. The recombination of restriction enzymes and sequence-specific DNA binding proteins such as zinc fingers18 and TALEN19 have proven to be more general approaches. The targeting range (the precision at which efficient cleavages can be introduced in a DNA target) of ZFN could reach one site in every 200 bp20 and there is no apparent limit for the targeting range of TALEN.21,22 However, the procedures for constructing ZFNs and TALENs are time consuming and labour intensive,23 and the overall success rate is low. The more recently developed CRISPR technology has provided a brand new tool for gene editing. This technology is based on the discovery of Cas-9/RNA complexes in prokaryote.24 It is much more convenient than others and the targeting range is theoretically unlimited.25 However, CRISPR technology requires the construction of gRNAs, which is not a very fast process (including 3 to 4 steps that may take several hours), especially when large-scale or high-throughput in vitro applications are in demand.Results
Herein we demonstrate a more facile and conceptually new approach for generating sequence-specific nucleases via a chemically preassembled inert-template (PAIT). The approach is based on a thermodynamically tight-binding state between deoxyribonuclease I (DNase I) and phosphorothioated single-stranded DNA (S-DNA) that we recently observed in our experiments. Under optimized reaction conditions, DNase I and S-DNA would assemble into a tight-binding complex, within which the S-DNA would block DNase I and only DNA strands complementary to the S-DNA template could be cleaved (Fig. 1a). This finding offers a fascinating method for the facile generation of sequence specificity of nucleases.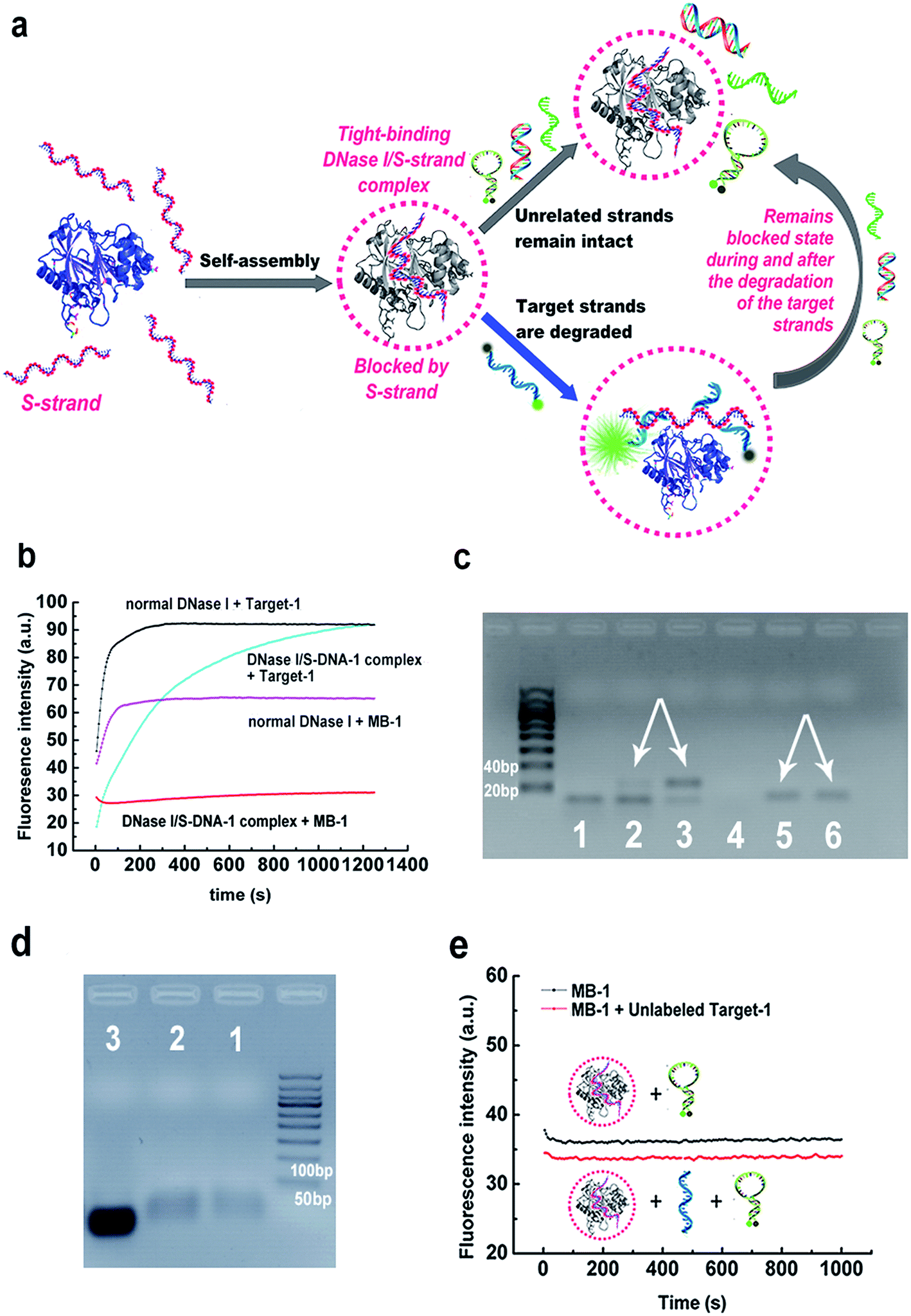 | ||
| Fig. 1 (a) Schematic illustration of the principle of the preassembled inert-template (PAIT) strategy for generating sequence-specific nucleases. For DNase I, a phosphorothioated single-stranded DNA (S-DNA) is used as the inert template, while for RNase A an S-RNA template is used. (b and c) Verification of the sequence specificity of the prepared DNase I/S-DNA-1 complex toward Target-1 by (b) fluorescence measurement and (c) agarose gel electrophoresis. Lane 1: normal DNase I + Target-1. Lane 2: the DNase I/S-DNA-1 complex + Target-1. Lane 3: Target-1 alone. Lane 4: normal DNase I + MB-1. Lane 5: the DNase I/S-DNA-1 complex + MB-1. Lane 6: MB-1 alone. For lane 1 to 3, S-DNA was added to the product solution to form a Target-1/S-DNA duplex for better staining. (d) Gel electrophoresis results for demonstration of the DNase I/S-DNA-1 complex. Lane 1-2: DNase I/S-DNA-1 complex. Lane 3: S-DNA-1 alone. (e) Monitoring of the reactions between the DNase I/S-DNA-1 complex and MB-1 in the absence (black line) and presence (red line) of unlabeled Target-1. | ||
Previous studies have revealed that the binding between DNase I and DNA mainly relies on the ionic interactions between the DNA backbone phosphates and the positively charged amino acids on the surface of DNase I.26,27 As the bases on the DNA strands are not directly involved in the binding with the enzyme, we infer that the preassembly of an inert ssDNA analogue (referred to as a template) to DNase I might facilitate the enzyme to preferably bind and digest the template's complementary strand (referred to as a target strand) over other unrelated sequences. The primary key step was to obtain a tight-binding assembly, in which the inert template can inhibit DNase from degrading the unrelated sequences. We tried incubating a very high concentration of normal DNase I (0.1 mg mL−1, about 40-fold of that normally used in DNA degradation reactions) with synthesized uniformly phosphorothioated ssDNA (S-DNA-1, see ESI Table S1;† phosphorothioation can prevent the degradation by DNase I) at different S-DNA-1/DNase I ratios in buffer-1 (50 mM sodium phosphate, 0.15 M NaCl, 1 mM EDTA, pH 7.2). After incubation, the remaining free S-DNA-1 was removed by ultrafiltration. As desired, the results in ESI Fig. S1† showed obviously declined digestion rates of MB-1 (a sequence-unrelated reference probe, see Table S1†), which demonstrated that under the above conditions, the inert template S-DNA-1 has blocked the activity of DNase I. More importantly, the digestion rate of the target strand (Target-1) was not significantly affected. The highest sequence specificity was achieved when the ratio of S-DNA-1 to DNase I was 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, at which 95% of Target-1 was degraded whereas only 5% of MB-1 was degraded after 15 min (Fig. 1b and S1†). We also performed an agarose gel electrophoresis analysis of the digestion products in the above reaction solutions to further verify the sequence specificity (Fig. 1c). By comparison with the bands in lane 3 and lane 6, respectively, the bands in lane 2 and lane 5 clearly confirmed that the DNase I/S-DNA-1 complex could efficiently digest Target-1, while it had little effect on the unrelated sequence MB-1. Agarose gel electrophoresis shift assays were also conducted to prove the formation of the DNase I/S-DNA-1 complex (Fig. 1d).
:1, at which 95% of Target-1 was degraded whereas only 5% of MB-1 was degraded after 15 min (Fig. 1b and S1†). We also performed an agarose gel electrophoresis analysis of the digestion products in the above reaction solutions to further verify the sequence specificity (Fig. 1c). By comparison with the bands in lane 3 and lane 6, respectively, the bands in lane 2 and lane 5 clearly confirmed that the DNase I/S-DNA-1 complex could efficiently digest Target-1, while it had little effect on the unrelated sequence MB-1. Agarose gel electrophoresis shift assays were also conducted to prove the formation of the DNase I/S-DNA-1 complex (Fig. 1d).
We further monitored the degradation rates of MB-1 by the DNase I/S-DNA-1 complex in the presence or absence of the target strands (unlabeled Target-1, see Table S1†). No evident increase of the fluorescence intensity in the presence of the target strands was observed, similar to the results in the absence of the target strands (Fig. 1e), demonstrating that the DNase I/S-DNA-1 complex would not dissociate even when it was digesting the complementary sequence of S-DNA. This data substantially suggested that the S-DNA templates have formed a very tight-binding complex with DNase I after incubation. Within the complex, the S-DNA had effectively blocked the active sites of DNase I. The target strands were able to hybridize with the preassembled S-DNA and thus, being recognized as a proper substrate, were digested by the enzyme. More importantly, DNase I remained blocked by the S-DNA during and after the degradation of the target strands.
To evaluate the discrimination ability of the DNase I/S-DNA-1 complex to different DNA strands, we tested six reference sequences. Fig. 2a showed that the complex had little activity on all of the six reference sequences. As a control, we used normal DNase I to react with the six tested sequences, and all of them were rapidly degraded (Fig. S2†). We then synthesized five analogues of S-DNA-1 with 1 to 5 mismatches at assorted locations (from an S-DNA-1/1 mismatch to an S-DNA-1/5 mismatch, see Table S1†), and prepared corresponding DNase I/S-DNA complexes, resulting in preassembled templates containing one to five mismatches with the target strand, respectively. From Fig. 2b, sequences containing more than two mismatches with S-DNA could be distinctly discriminated from the perfectly matched target strand, and strands containing five mismatches were almost unaffected by the complex.
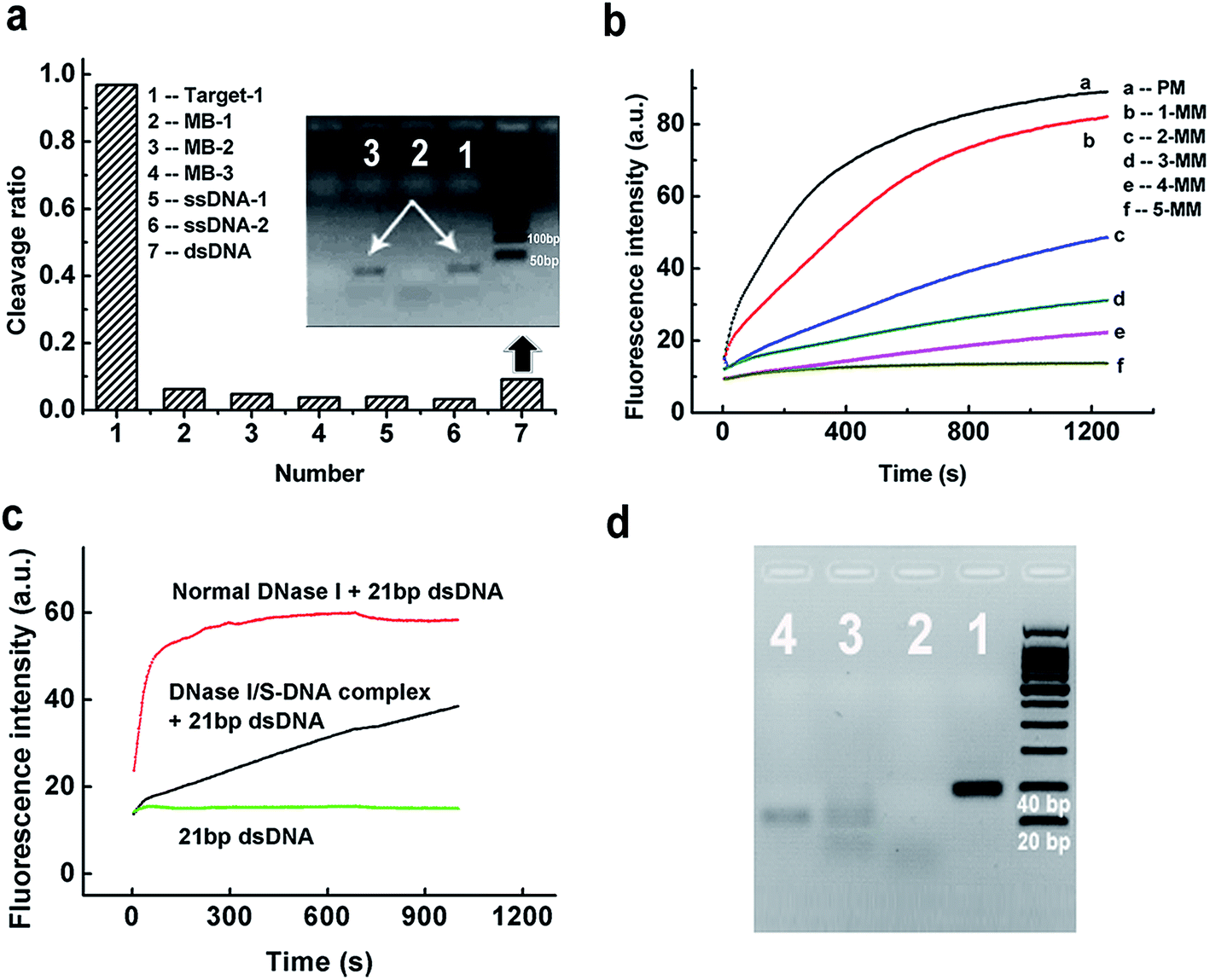 | ||
| Fig. 2 (a) Selectivity of the DNase I/S-DNA-1 complex to Target-1 over six unrelated ssDNA or dsDNA sequences. Inset: Lane 1: dsDNA. Lane 2: dsDNA + normal DNase I. Lane 3: dsDNA + the DNase I/S-DNA complex. (b) Discrimination capability of the DNase I/S-DNA complex against strands containing one to five mismatches with the S-DNA template. (c) Fluorescence experiments showing the cleavage of 21 bp dsDNA by the DNase I/S-DNA-1 complex. The 21 bp dsDNA is a Target-1/c-Target-1 duplex. (d) Gel electrophoresis results showing the cleavage of 41 bp dsDNA by the DNase I/S-DNA-1 complex within 2 h. The 41 bp dsDNA is a Target-3/c-Target-3 duplex. Lane 1: 41 bp dsDNA. Lane 2: 41 bp dsDNA + normal DNase I. Lane 3: 41 bp dsDNA + the DNase I/S-DNA-1 complex. Lane 4: c-Target-3. The gel was exposed to UV light and the image was captured at 595 nm. | ||
To further demonstrate the generality of the developed PAIT strategy, we designed another target strand (Target-2, see Table S1†) which had a totally different sequence from that of Target-1. The prepared DNase I/S-DNA-2 complex showed expected sequence specificity toward Target-2 (Fig. S3†). We then tested a third target strand (Target-3) with a 15 nt longer sequence than Target-1. As shown in Fig. S4,† Target-3 was degraded as fast as Target-1, indicating that the DNase I/S-DNA complex could also be used to degrade longer target strands that contained a sequence complementary to the template S-strand. The above results demonstrated that we can easily prepare an artificial sequence-specific nuclease whose target sequence is predetermined by assembling its complementary S-DNA strands with DNase I.
Considering the fact that the genome of humans and other species comes as a double strand, we further tested the capability of the DNase I/S-DNA complex to cleave targeted sequences within double-stranded DNA (dsDNA). DNase I/S-DNA-1 was used to react with three different DNA duplexes containing the 21 nt target sequence, including a Target-1/c-Target-1 duplex (21 bp dsDNA), a Target-3/c-Target-3 duplex (41 bp dsDNA) and a 59 bp dsDNA. Fig. 2c and S5† showed that within 15 min, the DNase I/S-DNA-1 complex could digest nearly half of the targeted dsDNA. Fig. 2d showed that after 2 h of cleavage, nearly all the 41 bp dsDNA was degraded. The resultant band of c-Target-3 in lane 3 indicated that the DNase I/S-DNA-3 complex successfully cleaved the Target-3 sequence within the duplex. The above results confirmed that the obtained DNase I/S-DNA complex could efficiently cleave dsDNA containing the targeted sequences.
We then tried to find out the key factors to generate such a unique DNase I/S-DNA complex with desired sequence specificity. Fig. 3a showed that at least 60 min were needed for the formation of the DNase I/S-DNA complex. In comparison with the normal binding between DNase I and DNA that leads to the degradation of DNA, the above binding process was rather slow. Then we investigated the effect of incubation temperature. Interestingly, there is a sudden drop of the residual nonspecific activity of the DNase I/S-DNA complex from 23 °C to 29 °C (Fig. 3b). This distinct change strongly suggested that the DNase I/S-DNA complex might have to overcome a potential barrier to turn into a final thermodynamically stable state. From Fig. 3c, the complex could not form or remain stable in solution at low reaction concentration levels when the molar ratio of S-DNA-1 to DNase I and the incubation time were fixed at 4.5:1 and 60 min, respectively. By contrast, when the concentration of DNase I increased to higher than 0.1 mg mL−1, a stable and specific DNase I/S-DNA complex could be obtained under the above incubation conditions. The rigorous requirement of time, temperature and concentration for the formation of the DNase I/S-DNA complex strongly indicates a much more stable binding state between DNase I and S-DNA, in comparison with that between DNase I and normal DNA that leads to the degradation of DNA. Fig. 3d further confirmed that after incubation of DNase I with normal DNA strands or S-DNA-1 in buffer-1 without the addition of divalent ions, only S-DNA-1 formed a stable complex with DNase I that prevented DNase I from passing through the filter membrane (cut-off 30 kDa). The tight-binding between DNase I and S-DNA-1 was also proved by the fluorescence anisotropy and circular dichroism (CD) spectra measurement results (see Fig. S6 and S7†). We also measured the dissociation constant of the DNase I/S-DNA-1 complex under the optimized incubation conditions, which was found to be 22 nM (see Fig. S8†).
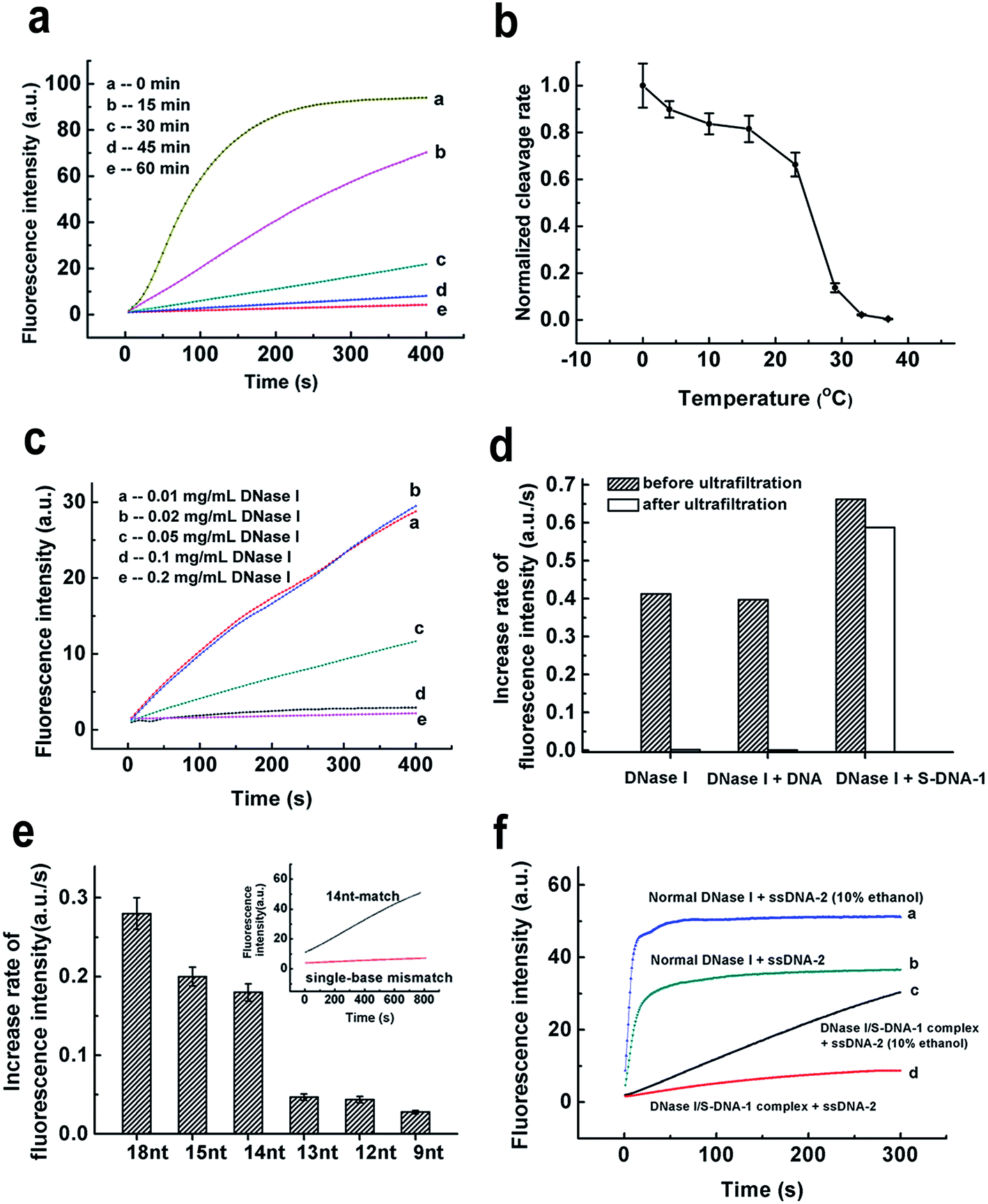 | ||
| Fig. 3 Mechanism studies of the acquired sequence specificity. (a–c) Key factors for the formation of the DNase I/S-DNA complex with the desired sequence specificity. The effects of (a) time, (b) temperature, and (c) initial concentrations of DNase I and S-DNA on the formation of the stable DNase I/S-DNA-1 complex. Unrelated sequences (MB-2) were used to monitor the activity of free DNase I, which provided a much larger increase of the fluorescence intensity than MB-1 and thus showed the changes more clearly. The sharp drop of the cleavage rate from 23 °C to 29 °C (b) indicates that the tight-binding state between DNase I and S-DNA-1 may need to overcome a potential barrier. The molar ratio of S-DNA-1 to DNase I was fixed at 4.5:1. Error bars show the standard deviation of three independent experiments. (d) The ultrafiltration experiments for the direct demonstration of the tight-binding state between DNase I and S-DNA. The concentration of DNase I was 0.1 mg mL−1, and the molar ratio of DNA to DNase I or S-DNA-1 to DNase I was 4.5:1. The reaction solutions were incubated at 37 °C for 1 h before ultrafiltration. Target-1 was used to monitor the activity of DNase I before and after ultrafiltration. (e) The effect of the length of the target region on the cleavage rate of the DNase I/S-DNA complex. S-DNAs with different lengths of the complementary region to Target-1 were used to prepare the DNase I/S-DNA complex. (f) The non-specific activity of DNase I in the formed DNase I/S-DNA-1 complex may be partially recovered in the presence of 10% ethanol. | ||
We then changed the length of the complementary region of Target-1. Fig. 3e showed the minimum length needed for effective degradation was 14 nt. We also observed effective discrimination toward single-base mismatch within the 14 nt target region with a discrimination factor (the ratio of the cleavage rate of the perfect-match target to that of the single-base mismatched target) of 25.3, indicating that the DNase I/S-DNA complex holds great potential in identifying small genetic changes. Importantly, the cleavage rate is strongly dependent on the length and the local structure of the target region (Fig. 3e and S9†), demonstrating that the hybridization to the S-DNA template was the primary step of subsequent degradation.
Next, we examined the stability of the DNase I/S-DNA complex under harsher conditions. As shown in Fig. S10–S16,† the complex remained stable at a low temperature (4 °C for 24 h), high temperature (50 °C), low or high salt concentrations (5 mM and 150 mM), high pH (8.8), ultrasonication, and 10% DMSO. We also observed an interesting phenomenon that the DNase I/S-DNA complex might partially dissociate in 10% ethanol and release part of its activity toward reference probes (Fig. 3f). These results proved that the DNase I/S-DNA complex can serve as a reliable sequence-specific enzyme in various biological samples. If necessary, the non-specific activity of DNase I towards other DNA strands can be readily recovered by 10% ethanol, which offers a flexible tool for regulation of the sequence specificity of nucleases as desired.
Encouraged by the above results, we further applied our strategy to regulate the sequence specificity of RNase A. By incubating 0.04 mg mL−1 RNase A with 31 μM S-RNA strand in buffer-1 at 37 °C for 1 h, a very stable RNase A/S-RNA complex was obtained and showed the desired sequence specificity toward a target RNA strand that is complementary to the S-RNA strand (Fig. 4). In Fig. 4b, the position and density of the band in lane 2 was almost the same as that in lane 3, demonstrating that the RNase A/S-RNA complex hardly digested the unrelated RNA strand. In Fig. 4c, the target RNA strand (Table S1†) was labelled with FAM and BHQ-1 at its two ends. If not cleaved, the FAM would be quenched by BHQ-1 through FRET. Therefore, no obvious bands could be seen in lane 1. In lane 2 and 3, the target RNA strands were cleaved, thus emitting strong fluorescence signals. Normal RNase A degraded the target RNA strands more deeply, so the resultant shorter fragments migrated slightly faster than the fragments in lane 3. These results demonstrated that our strategy was applicable for regulating the sequence specificity of RNase A, as well.
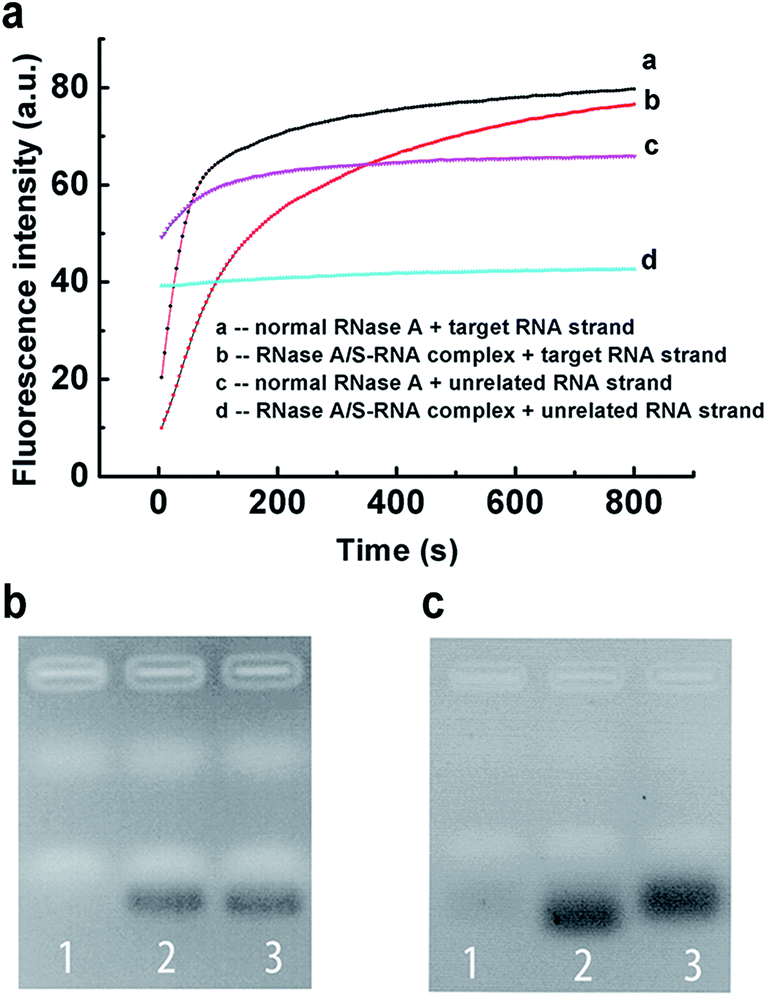 | ||
| Fig. 4 Application of the PAIT strategy for regulation of the sequence specificity of RNase A. The sequence specificity of the RNase A/S-RNA complex toward a target RNA strand is demonstrated by (a) fluorescence experiments and (b and c) gel electrophoresis. (b) Lane 1: unrelated RNA strand + normal RNase A. Lane 2: unrelated RNA strand + RNase A/S-RNA complex. Lane 3: unrelated RNA strand. The gel was stained by Gelsafe and the detection wavelength is 595 nm. (c) Lane 1: target RNA strand. Lane 2: target RNA strand + normal RNase A. Lane 3: target RNA strand + RNase A/S-RNA complex. The target RNA strand is labeled with FAM and BHQ-1 at the 5′ end and 3′ end. The gel was not stained and the signal is produced by FAM and detected at 535 nm. | ||
Discussion
Previous studies on the crystal structure of DNase I have revealed that DNase I has a sandwich-type structure with two 6-stranded β-sheets surrounded by α-helices and loop regions.26,27 The two β-sheets form a hydrophobic core where the catalytic center is located. The binding between DNase I and dsDNA normally comprises three types of interactions: ionic interactions between the DNA backbone phosphates and positively charged amino acids, further ionic interactions between phosphates and negatively charged side chains mediated by divalent cations, and van der Waals' interactions. In our experiments, the initial binding of S-DNA to DNase I might occur very quickly through random hydrophobic and ionic interactions. But such a primary binding was not stable enough, so the resultant solution could still rapidly digest MB-1 (Fig. 3a, curve b). With the increase of the incubation time and the concentration, the binding between S-DNA and DNase I turned out to be very tight and the activity of DNase I was completely blocked, indicating a successful assembly of the inert template on the enzyme. In our work, the optimum conditions for preparation of the DNase I/S-DNA complex were determined by first fixing the initial concentration of DNase I at 0.1 mg mL−1, and the incubation time and temperature at 1 h and 37 °C, respectively, and then testing different molar ratios of S-DNA to DNase I; the molar ratio was optimized to be 4.5:1. With this ratio fixed at 4.5:1, we verified the limiting values of other factors, including the initial DNase I concentration, the incubation time and temperature. As shown in Fig. 3, the initial DNase I concentration, the incubation time and temperature should be no less than 0.1 mg mL−1, 60 min and 37 °C, respectively. These results suggested that the above four factors cooperatively determined the formation of the DNase I/S-DNA complex with an expected sequence specificity. It should be pointed out that the initial concentrations of DNase I and S-DNA can be different if one uses a different incubation time and temperature. But relatively higher concentrations of DNase I and S-DNA are favorable for the rapid formation of a stable DNase I/S-DNA complex (Fig. 3c).
Within the tight-binding complex, S-DNA and DNase I were mainly held together by the interactions between the S-DNA backbone phosphates and charged side chains of the enzyme, leaving the bases of the template unoccupied and available for hybridization with a complementary sequence. Therefore, hybridization between the S-DNA and complementary target strand was the initiation step for the cleavage process. For efficient hybridization, the melting temperature of the target strand/S-DNA duplex should be at least higher than the degradation reaction temperature (37 °C). As shown in Table S1,† the melting temperatures of S-DNA-12 nt/Target-1 and S-DNA-13 nt/Target-1 were 35.2 °C and 42.3 °C, respectively. However, the DNase I/S-DNA-13 nt complex could not effectively degrade Target-1 at 37 °C (Fig. 3e), indicating that the hybridization between S-DNA-13 nt and Target-1 was hindered to some extent by the DNase I. This could be attributed to the ionic interactions between the backbone phosphates of the two strands and the positively charged amino acids on the surface of DNase I, which tend to dissociate the two strands. Based on our experimental results, the minimum length of the hybridization part needed for the effective degradation of Target-1 was 14 nt (Fig. 3e). For other sequences with a higher or lower GC content, the minimum length may be slightly different. It is also noteworthy to point out that the formation of secondary structures of the target strands can affect the performance of our strategy. If the secondary structure is so stable that S-DNA can not effectively hybridize with the target strand, the cleavage rate would be significantly reduced (Fig. S9†).
Regarding the mechanism for the selective degradation of dsDNA by the DNase I/S-DNA complex, the initial recognition steps may be very similar to that of normal DNase I, i.e., mainly through the ionic interactions between the positively charged amino acids on the surface of DNase I and the DNA backbone phosphates. Actually this binding to dsDNA was stronger than ssDNA because of more phosphate groups in the dsDNA.28 The positively charged side chains on DNase I interacted with the backbone phosphates of the two DNA strands, which would more or less bend the double helix and therefore partly deform the base pairs.28 Thus the S-DNA, in very close proximity, may have a chance to hybridize to the complementary part in dsDNA and lead to cleavage of the unprotected phosphodiester. This mechanism also explains why the cleavage rate of the targeted dsDNA was significant slower than that of the targeted ssDNA. For longer dsDNA, the degradation process was even slower, as can be seen in Fig. S5.†
To further prove the above mechanism, we synthesized a new S-DNA which contains two locked nucleic acid (LNA) nucleotides (denoted as S-DNA-LNA, see Table S1†). We inferred that the incorporation of LNA nucleotides in the S-DNA template would increase its binding affinity to the complementary strands and thus facilitate its hybridization to the target sequence within the dsDNA.29 From Fig. S17,† we observed a higher cleavage rate of the targeted dsDNA in comparison with that when using the S-DNA-1 template. These results substantially confirmed the above mechanism and also provided a very efficient way to enhance the cleavage rate of dsDNA.
Conclusions
In summary, we have proposed a very convenient and effective preassembled inert-template (PAIT) approach for the rapid preparation of sequence-specific nucleases. The strategy is applicable to both DNase I and RNase A, and the sequences of S-DNA/S-RNA can be flexibly designed according to the target strand of interest. The DNase I/S-DNA complex could efficiently cleave both dsDNA and ssDNA. The discrimination factor against single-base mismatch strands within a 14 nt target region was as high as 25.3. Since the strategy is very convenient to conduct, we anticipate that it will be a prevalent tool for the fast preparation of sequence-specific nucleases, especially in large-scale or high-throughput in vitro applications. Furthermore, it also holds great potential to be developed into an in vivo enzymatic tool.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21575008, 21375004).Notes and references
- F. Buchholz, Curr. Opin. Biotechnol., 2009, 20, 383–389 CrossRef CAS PubMed
.
- L. H. Lu, D. S. H. Chan, D. W. J. Kwong, H. Z. He, C. H. Leung and D. L. Ma, Chem. Sci., 2014, 5, 4561–4568 RSC
- Q. Wang, R. D. Li, B. C. Yin and B. C. Ye, Analyst, 2015, 140, 6306–6312 RSC
- T. Sprink, J. Metje and F. Hartung, Curr. Opin. Biotechnol., 2015, 32, 47–53 CrossRef CAS PubMed
- P. Doruker, O. Kurkcuoglu, A. Uyar and L. Nilsson, FEBS J., 2006, 273, 253 Search PubMed
- A. Pingoud and A. Jeltsch, Nucleic Acids Res., 2001, 29, 3705–3727 CrossRef CAS PubMed
- D. S. Sigman, C. B. Chen and M. B. Gorin, Nature, 1993, 363, 474–475 CrossRef CAS PubMed
- K. Ito and M. Komiyama, Methods Mol. Biol., 2014, 1050, 111–120 CAS
- Z. Y. Zhu, J. Zhou, A. M. Friedman and S. Y. Xu, J. Mol. Biol., 2003, 330, 359–372 CrossRef CAS PubMed
- J. C. Samuelson, R. D. Morgan, J. S. Benner, T. E. Claus, S. L. Packard and S. Y. Xu, Nucleic Acids Res., 2006, 34, 796–805 CrossRef CAS PubMed
- N. S. Jurica, R. J. Monnat and B. L. Stoddard, Mol. Cell, 1998, 2, 469–476 CrossRef PubMed
- P. Rouet, F. Smih and M. Jasin, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6064–6068 CrossRef CAS
- J. Ashworth, J. J. Havranek, C. M. Duarte, D. Sussman, R. J. Monnat, B. L. Stoddard and D. Baker, Nature, 2006, 441, 656–659 CrossRef CAS PubMed
- P. Redondo, J. Prieto, I. G. Munoz, A. Alibes, F. Stricher, L. Serrano, J. P. Cabaniols, F. Daboussi, S. Arnould, C. Perez, P. Duchateau, F. Paques, F. J. Blanco and G. Montoya, Nature, 2008, 456, 107–111 CrossRef CAS PubMed
- S. H. Chan, Y. M. Bao, E. Ciszak, S. Laget and S. Y. Xu, Nucleic Acids Res., 2007, 35, 6238–6248 CrossRef CAS PubMed
- S. Jurenaite-Urbanaviciene, J. Serksnaite, E. Kriukiene, J. Giedriene, C. Venclovas and A. Lubys, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10358–10363 CrossRef CAS PubMed
- R. D. Morgan and Y. A. Luyten, Nucleic Acids Res., 2009, 37, 5222–5233 CrossRef CAS PubMed
- Y. G. Kim, J. Cha and S. Chandrasegaran, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1156–1160 CrossRef CAS
- M. Christian, T. Cermak, E. L. Doyle, C. Schmidt, F. Zhang, A. Hummel, A. J. Bogdanove and D. F. Voytas, Genetics, 2010, 186, 757–761 CrossRef CAS PubMed
- J. A. Townsend, D. A. Wright, R. J. Winfrey, F. Fu, M. L. Maeder, J. K. Joung and D. F. Voytas, Nature, 2009, 459, 442–445 CrossRef CAS PubMed
- T. Cermak, E. L. Doyle, M. Christian, L. Wang, Y. Zhang, C. Schmidt, J. A. Baller, N. V. Somia, A. J. Bogdanove and D. F. Voytas, Nucleic Acids Res., 2011, 39, 7879 CrossRef CAS
- D. Reyon, S. Q. Tsai, C. Khayter, J. A. Foden, J. D. Sander and J. K. Joung, Nat. Biotechnol., 2012, 30, 460–465 CrossRef CAS PubMed
- D. F. Voytas, Annu. Rev. Plant Biol., 2013, 64, 327–350 CrossRef CAS PubMed
- B. Wiedenheft, S. H. Sternberg and J. A. Doudna, Nature, 2012, 482, 331–338 CrossRef CAS PubMed
- J. D. Sander and J. K. Joung, Nat. Biotechnol., 2014, 32, 347–355 CrossRef CAS PubMed
- D. Suck, A. Lahm and C. Oefner, Nature, 1988, 332, 464–468 CrossRef CAS PubMed
- D. Suck and C. Oefner, Nature, 1986, 321, 620–625 CrossRef CAS PubMed
- M. E. Hogan, M. W. Roberson and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9273–9277 CrossRef CAS
- L. Kvaerno and J. Wengel, Chem. Commun., 1999, 657–658 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary figures and tables. See DOI: 10.1039/c5sc04398k |
| This journal is © The Royal Society of Chemistry 2016 |