
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-ligand sustains controlled generation of CF3 radicals by well-defined copper complex†
Jérémy
Jacquet
,
Sébastien
Blanchard
,
Etienne
Derat
,
Marine
Desage-El Murr
* and
Louis
Fensterbank
*
Institut Parisien de Chimie Moléculaire, UMR CNRS 8232, Sorbonne Universités UPMC Univ Paris 06, 4 Place Jussieu, CC 229, 75252 Paris Cedex 05, France. E-mail: marine.desage-el_murr@upmc.fr; louis.fensterbank@upmc.fr
First published on 3rd December 2015
Abstract
A well-defined copper complex bearing iminosemiquinone ligands performs single electron reduction of an electrophilic CF3+ source into CF˙3 radicals. This redox behavior is enabled by the ligand which shuttles through two different redox states (iminosemiquinone and iminobenzoquinone) while the copper center is preserved as a Cu(II). This system was used in the trifluoromethylation of silyl enol ethers, heteroaromatics and in the hydrotrifluoromethylation of alkynes. This is the first example of cooperative redox catalysis for the controlled generation of CF˙3 radicals.
Introduction
Cooperative redox catalysis steps away from the established organometallic paradigm involving redox events occurring at the metal center by introducing ligand-based electronic participation. Drawing inspiration from the redox relays involved in enzymatic processes,1 this area of catalysis hinges upon a participative role of the non-innocent ligand in catalytic elementary steps in order to achieve multielectronic transformations with base metals.2–8While redox non-innocence in biological settings has long been familiar to bioinorganic chemists, the use in a broader context of “synthetic” redox ligands as surrogates of the original biological radicals is emerging as a catalytic tool of its own. These privileged molecular scaffolds have been shown to actively participate in catalytic events through reversible delocalization of spin density.4 This redox interplay between ligand and metal can provide attractive alternative mechanistic venues for catalyst development. Among expected benefits, the enabling potential of these ligands towards first-row transition metals or redox silent metals could enlarge their chemistry and unravel unprecedented reactivities. This area of catalysis is currently on the fast track for the development of alternative catalytic approaches circumventing the use of precious noble metals.5
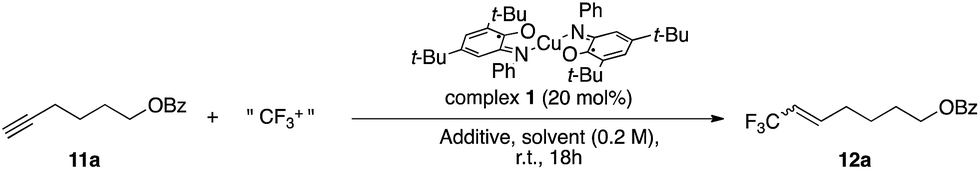
Seeking to enlarge the scope of applications, we have been focusing on a well-known family of metal complexes exhibiting non-innocent behavior: copper complexes with iminosemiquinone ligands. Originally developed as enzyme mimics,9 these complexes are of particular interest due to the versatile ligand scaffold which can accommodate two successive monoelectronic oxidation steps through a redox chemical interplay between three oxidation states: amidophenolate (AP), iminosemiquinone (SQ) and iminobenzoquinone (BQ). These systems are now being developed in broader (catalytic) contexts10 with several metals including iridium,11 cobalt,12,13 palladium14,15 and copper.16,17 Previous work allowed us to establish that the interaction of complex 1 [Cu(II)(LSQ)2] with an electrophilic source of CF3 triggers CF3 uptake by the complex through ligand-based bis-electronic redox participation while preserving the metal Cu(II) oxidation state.18 The resulting [Cu(II)(LBQ)2CF3]+ complex 2 exhibits a nucleophilic intramolecular CF3 reactivity, thus suggesting that redox involvement of ligands can sustain formal umpolung of the CF3 moiety (Fig. 1).
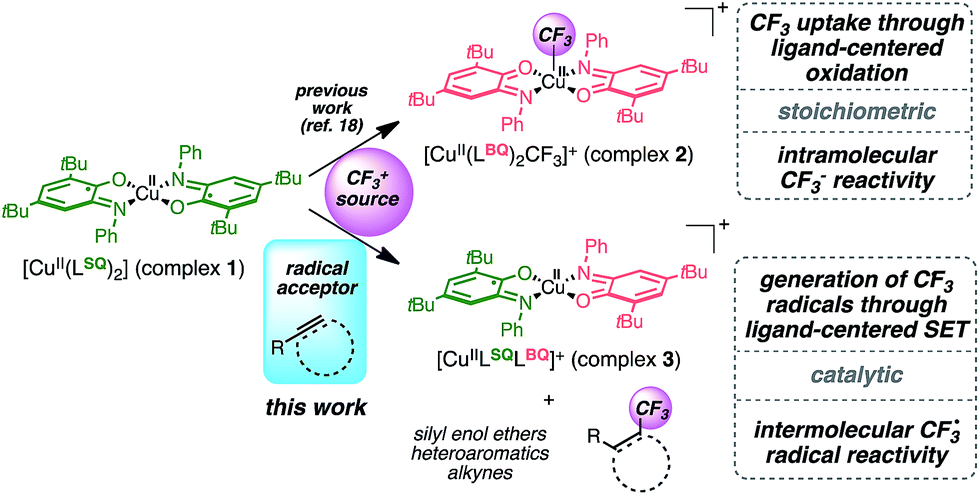 | ||
| Fig. 1 Ligand-based redox reactivity of complex 1 with an electrophilic CF3+ source. | ||
Transfer of trifluoromethyl groups through metal-catalyzed processes is currently a topic of acute interest as this group belongs to the privileged moieties in synthetic chemistry. Its introduction in a biologically active scaffold enhances metabolic stability and favors permeation of drugs through the blood brain barrier among other upsides.19–23 The widespread benefits of the introduction of trifluoromethyl groups also pervade through materials chemistry and agrochemistry. Accordingly, numerous strategies have been devised by chemists for the introduction of this chemical functionality.24–26 The wide range of available nucleophilic, electrophilic and radical trifluoromethylating sources provides varied catalytic manifolds opportunities, which plays no mean part in the success of this flourishing area. The wealth of CF3 sources available has allowed flexibility in the development of a variety of approaches differing from the electronics involved.27–30 In this matter, several metals have proven valuable to the chemists among which copper31 and palladium.32–34 Strategies involving the addition of CF3 moieties across multiple bonds have also been developed and these efficient approaches allow building of molecular complexity.35–37
Recently, among these strategies, major focus has been placed on synthetic application of systems implying CF˙3 radicals38 and recent elegant contributions rely on photoredox catalytic manifolds as means to generate such radicals in a clean and controlled fashion.39–44 Radical trifluoromethylation of unsaturated moieties is inherently challenging because of competing radical-atom transfer and several methods have sought to take advantage of this reactivity in order to develop hetero- and carbotrifluoromethylation.45–50 However, direct hydrotrifluoromethylation of multiple bonds, thus generating simple trifluoromethylated alkenes and alkanes, has been less explored and is an emerging application.51–56 While these methods are an elegant and efficient way to produce CF˙3 radicals in a controlled fashion, they mostly imply expensive and less sustainable noble metals such as ruthenium and iridium and/or also often imply the use of additives playing the role of sacrificial electron donors and redox relays.
On the cheaper end of the metal spectrum, copper is a metal that has enjoyed recent exciting applications in trifluoromethylation of various organic substrates.31,57 Notably, an efficient Cu(I)-photocatalyzed trifluoromethylation of alkenes was recently reported.58 Another generation of CF˙3 radicals by Cu(I) salts from an electrophilic CF3+ source (5) in the trifluoromethylation of unactivated olefins to generate allylic trifluoromethylated products has been reported by Wang et al.59 using CuCl and by Buchwald et al.60 using [(MeCN)4Cu]PF6. A similar transformation was reported by Fu and Liu61 using electrophilic source 4 and CuTC ((thiophene-2-carbonyloxy)copper) and the authors postulated the involvement of a Cu(III)–CF3 species. The use of simple copper salts in these reactions is convenient but the lack of ligands that could stabilize the active copper species generates less controlled reactive intermediates and is a drawback for mechanistic elucidation. Moreover, these protocols often require between 10 and 20 mol% catalyst loading in order to provide good results.
Here, we show that well-defined complex 1 can be used in conjunction with an electrophilic CF3+ reagent as a catalytic source of CF˙3 radicals through SET sustained by the redox-active ligand scaffold. This cost-effective alternative way to generate CF˙3 radicals offers broad scope in transformations including trifluoromethylation of heteroaromatics, silyl enol ethers, and hydrotrifluoromethylation of alkynes.
Results and discussion
Initial experiments showed that reacting complex 1 with an electrophilic CF3+ source (Umemoto 4 or Togni II 5 reagents) in the presence of TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl radical, Scheme 1) led to the formation of the TEMPO–CF3 adduct, as evidenced by NMR 19F spectroscopy (δ = −55.7 ppm). This reactivity was evaluated in CH2Cl2 and NMP with two CF3+ sources (4 and 5) and was found to proceed in fairly good yields (up to 69%). These results suggest that complex 1 can promote the generation of CF˙3 radical from a CF3+ source in the reaction medium. An independent equimolar mixture of TEMPO and complex 1 was followed by UV-vis and no reaction was found to occur, thus ruling out the possibility of direct electronic transfer between complex 1 and TEMPO and subsequent ionic CF3 transfer. Also, no formation of the TEMPO–CF3 adduct occurs in the absence of complex 1, which is thus mandatory.49 This reactivity is in sharp contrast with that of the fully oxidized complex 2 [Cu(II)(LBQ)2CF3]+ which was found to be inert to reaction with TEMPO as no transfer of the CF3 moiety was observed.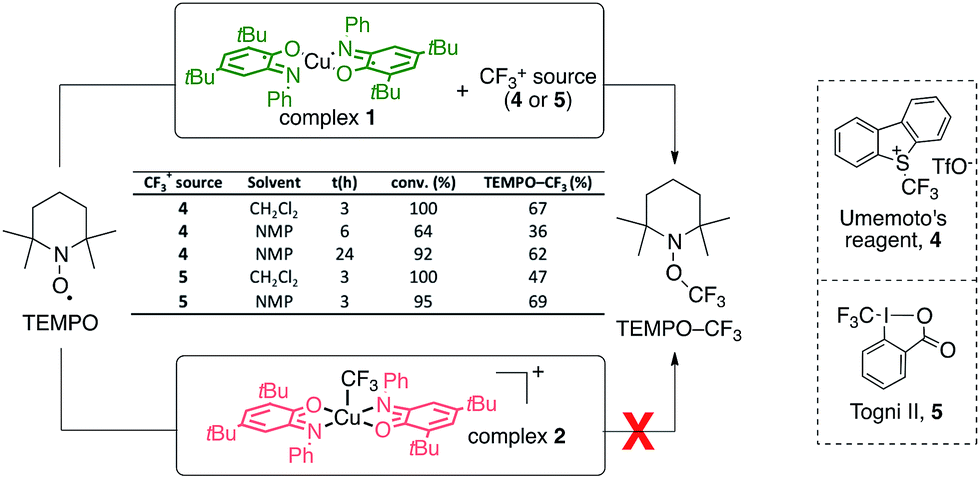 | ||
| Scheme 1 TEMPO trapping experiments for the generation of CF3 radicals with complexes 1 and 2. NMP: N-methylpyrrolidinone. | ||
During the trifluoromethylation of TEMPO, the reaction medium turns from dark green to dark purple (source 4) or dark orange (source 5). We thus decided to investigate the locus of the electronic transfer by UV-vis spectroscopy. The initial green colour of the solution mainly corresponds to that of [Cu(LSQ)2] complex 1, as attested by the presence on the spectra of its two characteristic bands around 300 nm and 800 nm,62 and as can be seen in Fig. 2 (green curve). At the end of the reaction, these two characteristic bands have disappeared, while broad new features appear with maxima at 425 nm, 525 nm and 725 nm (Fig. 2, brick red curve). Interestingly, very similar bands have been reported for the electrochemically synthesized [Cu(LSQ)(LBQ)]+ complex 3, arising from single electron oxidation of complex 1.62 We therefore independently synthesized complex 3via mediamutation of an equimolar mixture of [Cu(LSQ)2] complex 1 and its related dicationic complex 6 [Cu(LBQ)2]2+ (see ESI† for preparation) and recorded its absorption spectrum (Fig. 2, sky blue curve). Indeed, an excellent match between this reference spectrum and that of the reaction medium at the end of the reaction is observed. An almost perfect match is even obtained upon addition of one equivalent of dibenzothiophene, which is released in the reaction medium upon trifluoromethylation with Umemoto's reagent 4 (Fig. 2, purple curve).
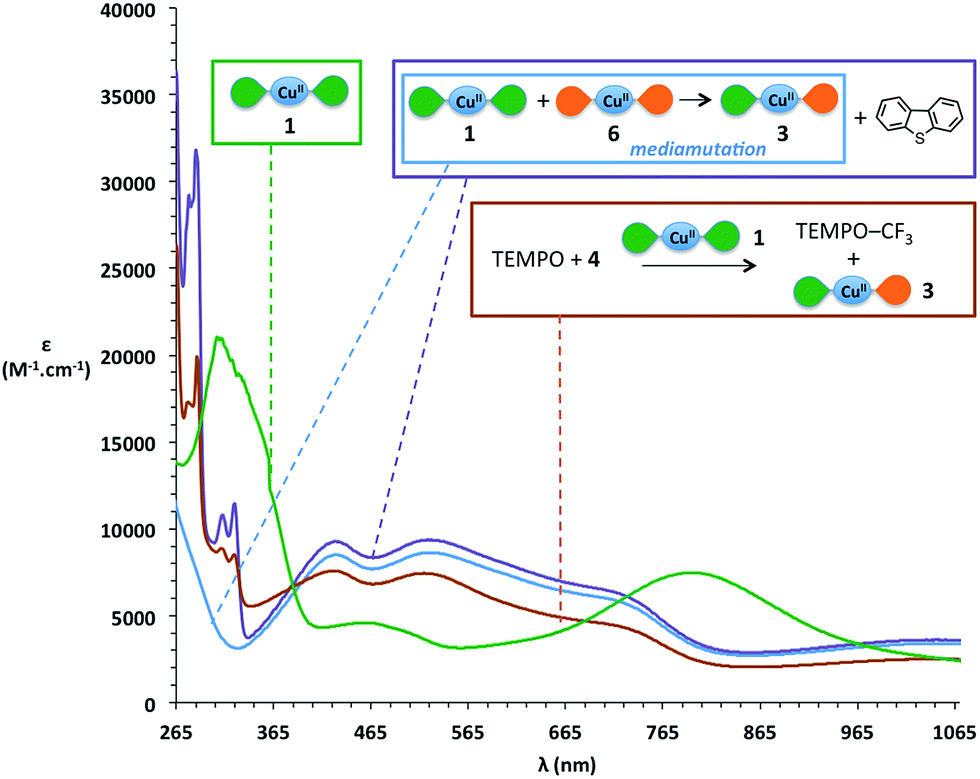 | ||
| Fig. 2 Comparison of UV-vis spectra in CH2Cl2 of: green: [Cu(LSQ)2] 1; brick red: reaction with Umemoto's source 4; sky blue: in situ generated [Cu(LSQ)(LBQ)]+3; purple: in situ generated [Cu(LSQ)(LBQ)]+3 with added dibenzothiophene. Color code for ligands: green petal is SQ ligand and orange petal is BQ ligand. | ||
These results strongly suggest that [Cu(LSQ)(LBQ)]+ complex 3, the monooxidized product of [Cu(LSQ)2] complex 1, is formed during the reaction and point towards a SET process involved in the reaction and sustained by the redox ligand.
Since complex 3 seems to be a cornerstone for the reactivity examined here, its electronic structure was evaluated through DFT calculations. These calculations were performed with Turbomole v6.4, using the B3LYP functional complemented by the D3 dispersion scheme and with the Def2-SV(P) basis set. In Fig. 3 can be seen the optimized structure together with the total spin density. It appears that one unpaired electron remains on the metallic center (in the dx2 − y2 orbital) while the second one is dispatched over the two ligands, with an antiferromagnetic coupling between the two. Thus, while we formally write that complex 3 is bearing two different ligands (one SQ and one BQ), the picture emerging from DFT calculations shows that the unpaired electron is fully delocalized on both ligands. The corresponding UV-vis spectrum of complex 3 was calculated using the same DFT level (Fig. 4). In the 400–800 nm range, the experimental and calculated spectra appear to be very similar. Two groups of transitions, centered around 425 nm and 600 nm, can be associated with the 425 nm and 525 nm bands, while an additional band around 790 nm may be associated with the 725 nm shoulder. Overall, the calculated spectrum fits well with the experimental data. Thus, all the data collected allow us to confirm the nature of complex 3 as [Cu(LSQ)(LBQ)]+.
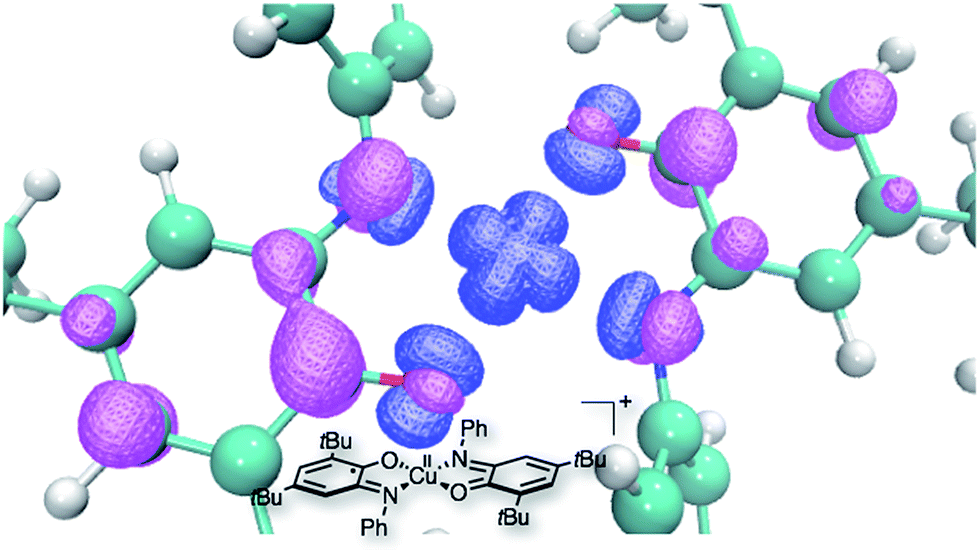 | ||
| Fig. 3 DFT optimized structure for complex 3, together with a spin density isosurface (plotted at 0.005e− Å−3). | ||
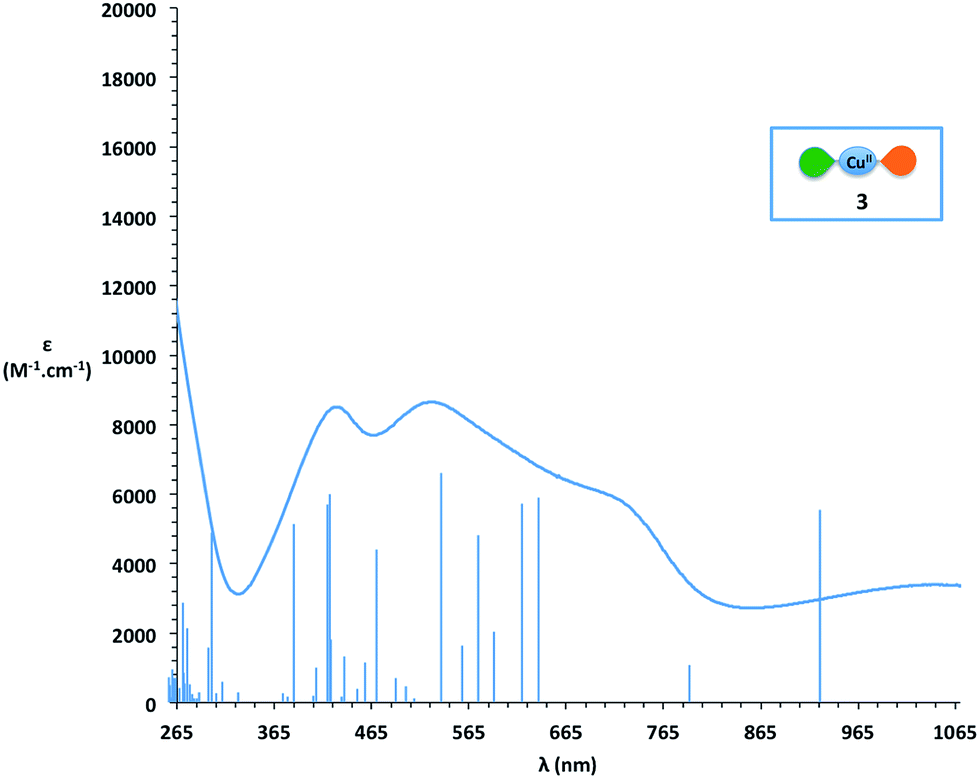 | ||
| Fig. 4 Comparison of DFT calculated peak positions (blue peaks) and experimental (blue curve) UV-vis spectrum of complex 3. | ||
When using Togni's reagent 5, the spectrum at the end of the reaction displays maxima around 300 nm, 390 nm and 500 nm (Fig. 5, brick red curve), which differs notably from the spectrum of [Cu(LSQ)(LBQ)]+ complex 3. We wondered if the iodobenzoate released from Togni's reagent might interact with the final copper complex and to our delight, adding the corresponding carboxylate to independently generated [Cu(LSQ)(LBQ)]+ complex 3 induces a change in its UV-vis spectrum, which then displays a very good match with the spectrum observed at the end of the reaction (Fig. 5, purple curve).
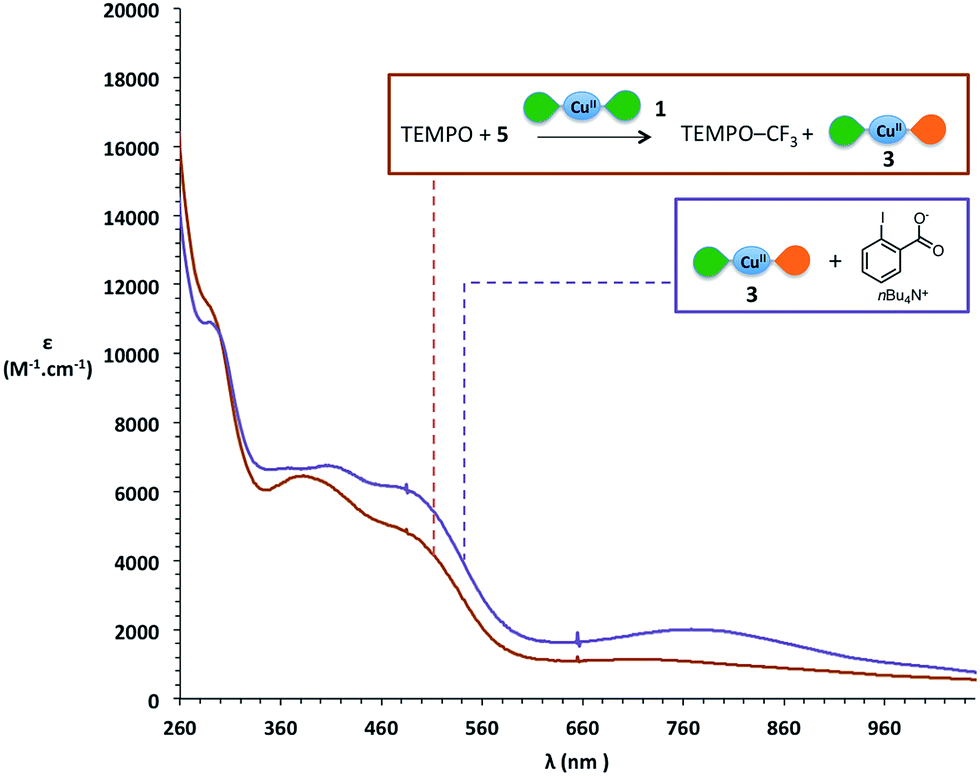 | ||
| Fig. 5 Comparison of UV-vis spectra in CH2Cl2 of: brick red: reaction with Togni II's source 5; purple: in situ generated [Cu(LSQ)(LBQ)]+3 with added o-iodobenzocarboxylate. Color code for ligands: green petal is SQ ligand and orange petal is BQ ligand. | ||
Thus, in the case of Togni's reagent 5, a SET from one of the iminosemiquinone ligands of [Cu(LSQ)2] complex 1 to form [Cu(LSQ)(LBQ)]+ complex 3 also appears very likely. These experiments strongly support a SET occurring between complex 1 and the CF3+ source, in which the CF3+ source is reduced to form a CF˙3 radical while the concomitant oxidation of complex 1 into complex 3 is ligand-based and sustained by oxidation of an iminosemiquinone ligand into an iminobenzoquinone ligand.
These observations led us to probe the reactivity of this system and evaluate the possibility of CF3 uptake in various families of radical acceptors. Photoredox63 and copper-catalyzed64 catalytic trifluoromethylation of silyl enol ethers to generate α-trifluoromethyl ketones have been reported and this family of substrates was used as benchmark reactivity for our system.
We were pleased to see that reaction of silyl enol ethers 7a–d with reagent 5 in the presence of 5 mol% of complex 1 yielded the corresponding trifluoromethylated adducts 8a–d in promising to good yields from 47 to 94% (Table 1). However, substituting source 4 for 5 provided lower yields. The reaction proceeds within 18 hours at room temperature, which compares well with reported conditions (24 h at rt63 and 12 h at rt to 50 °C64).
Yields were determined by 19F NMR analysis, reaction conditions: 1 equiv. silyl enol ether, 1.5 equiv. reagent 5. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was performed with 4 instead of 5. Isolated yields in brackets. Reaction was performed with 4 instead of 5. Isolated yields in brackets. |
|---|

|
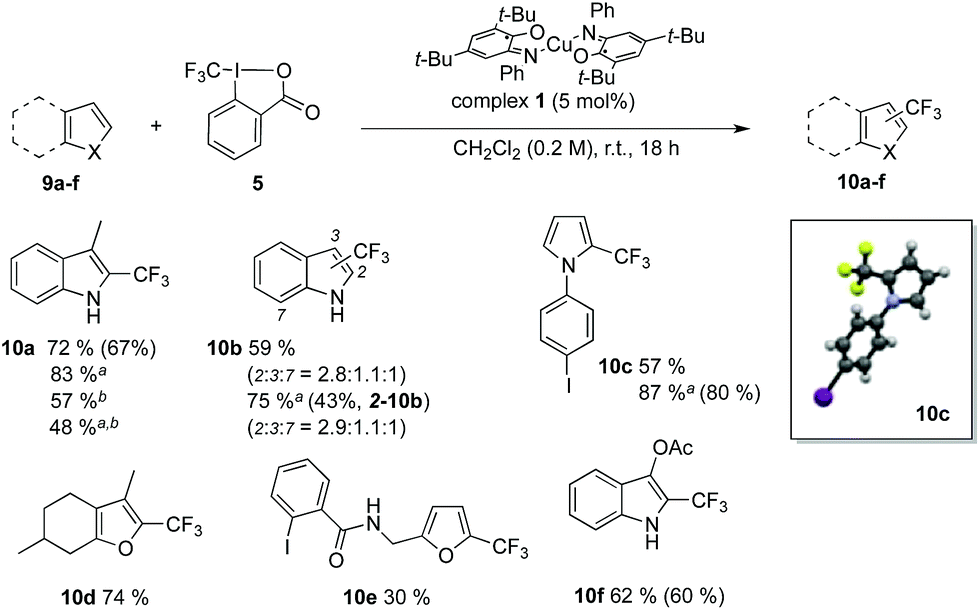
Introduction of CF3 motifs in heteroaromatics is also a topic of interest and several radical-based methods have been reported.40,65,66 Efficiency of the redox catalyst 1 was probed with indole, pyrrole and furane derivatives (Table 2, 9a–f). The yields range from 59 to 87% and the regioselectivity obtained for 10b is consistent with reported radical trifluoromethylation conditions.40 Lowering the catalyst loading to 2 mol% resulted in decreased yields. Furthermore, the structure of compound 10c was confirmed by X-ray crystallography. Successful preparation of 10c and 10e also demonstrates the mildness of our reaction conditions towards electrophiles such as aryl iodides, which are typically reactive under photoredox conditions67,68 and could in our approach be used as synthetic handles for further functionalization by classic methods.
| Yields were determined by 19F NMR analysis, reaction conditions: 1 equiv. heteroaromatic, 1.5 equiv. reagent 5. aReaction conditions: 5 mol% complex 1, 4 equiv. heteroaromatic, 1 equiv. reagent 5. b2 mol% catalyst loading. Isolated yields in brackets. |
|---|
|
Having established the catalytic activity of complex 1 with well-known radical acceptors, we turned our attention towards hydrotrifluoromethylation of alkynes or alkenes. Since a seminal study from Kitazume in 1985 reporting the ultrasound-promoted hydrotrifluoromethylation of alkynes with trifluoroalkylcuprates, generated in situ from CF3I or CF3Br, zinc powder and substoichiometric amounts of CuI,69 only a handful of strategies for selective hydrotrifluoromethylation have been reported.70 These involve inorganic electride,51 silver-catalyzed52 or organic53,54 and organometallic55,56 photoredox systems.
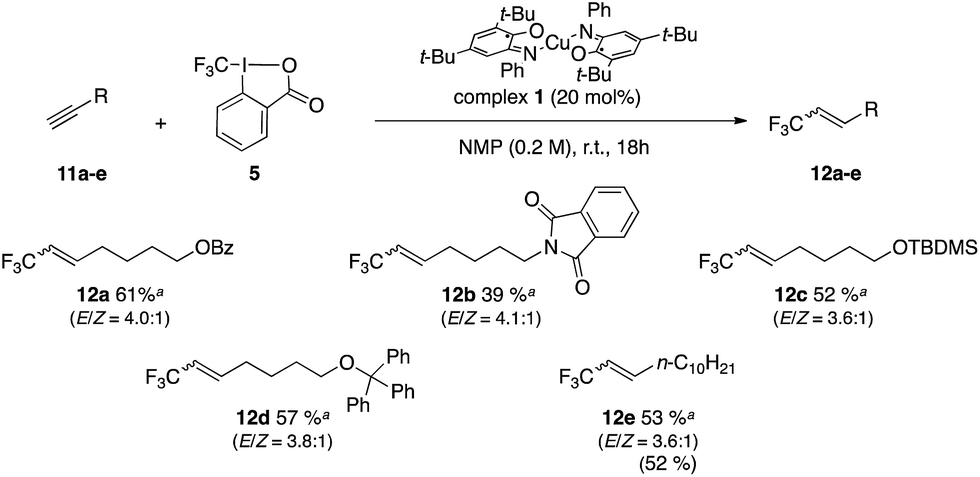
Cho and co-workers have reported the controlled trifluoromethylation of alkynes with CF3I, fac-[Ir(ppy)3] as catalyst and DBU (10 equiv.) as reductive quench and H source.56 The Gouverneur group has reported a method for alkynes and alkenes using photoredox catalyst [Ru(bpy)3]2+ in conjunction with Umemoto reagent 4 as CF3+ source and methanol as H source.55 An organic photoredox system was reported for alkynes by Scaiano and co-workers using reagent 5 along with organic photocatalyst Methylene Blue and DBU (2 equiv.).54 We selected alkyne 11a as a model for optimization studies and established that the best conditions involved the use of complex 1 and reagent 5 in NMP (Table 3, entry 2). Introduction of additive such as CHD (1,4-cyclohexadiene) as H atom donor did not improve the yields (entries 3 and 4). The scope of the reaction was evaluated and provided the corresponding trifluoromethylated alkenes in yields between 39 and 61%. These results prove competitive towards the literature with 68% yield reported for alkene 12a (conditions: 10 mol% [Ru(bpy)3Cl2·6H2O], visible light, 1.8 equiv. of 4 in methanol at 25 °C for 24 h).55
|
|
||||
|---|---|---|---|---|
| Entry | CF3+ source | Solvent | Additive | 12a a |
| CHD: 1,4-cyclohexadiene. Reaction conditions: alkyne (4 equiv.), CF3+ source (1 equiv.), solvent, additive, rt, 18 h. aYields were determined by 19F NMR analysis. Isolated yields in brackets. |
||||
| 1 | 4 | NMP | — | 11% |
| 2 | 5 | NMP | — | 61% |
| 3 | 5 | NMP | CHD (1 equiv.) | 35% |
| 4 | 5 | CH2Cl2 | CHD (1 equiv.) | 25% |
|
||||
Hydrotrifluoromethylation of alkene 13 (Table 4) was attempted but failed to deliver any expected hydrotrifluoromethylated product 14a. Vinyl–CF314b and allyl–CF314c compounds were obtained instead. This could be due to the fact that the alkyl radical generated upon addition of CF˙3 onto the alkene (Scheme 2) is easier to oxidize than its vinylic counterpart – generated upon addition of CF˙3 on the alkyne – and thus undergoes oxidation to the alkyl cation followed by proton loss to generate the allylic CF3 product more quickly than H transfer. This observation also extends to allene 15 which was found to undergo oxytrifluoromethylation to yield product 16 presumably arising from trapping of the cationic intermediate by 2-iodobenzoate generated in the reaction upon reduction of reagent 5.71
|
|
||||
|---|---|---|---|---|
| Entry | CF3+ source | 14a | 14b | 14c |
| 1,4-CHD = 1,4-cyclohexadiene. Reaction conditions: alkene (4 equiv.), CF3+ source (1 equiv.), 1,4-CHD (1 equiv.), NMP, rt, 18 h. Yields were determined by 19F NMR analysis. aReaction conditions: 5 mol% complex 1, 1.5 equiv. reagent 5, CH2Cl2, r.t., 18 h. |
||||
| 1 | 4 | Traces | Traces | Traces |
| 2 | 5 | — | 19% | 29% |

|
||||

 | ||
| Scheme 2 Proposed mechanism for the controlled generation of CF˙3 radicals sustained by redox ligand. [NMP–H] corresponds to the species resulting from H atom abstraction, more likely at the CH2 position adjacent to the nitrogen.76 | ||
Based on literature precedents, mechanistic possibilities for this transformation include organometallic, ionic or radical pathways. An organometallic route was proposed by Beller and co-workers72 and by Sanford and co-workers41 for the copper-catalyzed trifluoromethylation of vinyl boronic acids with CF˙3 radicals generated by t-BuOOH and CF3SO2Na or CF3I and photocatalyst Ru(bpy)3Cl2·6H2O respectively. Both involved a CuIII–CF3 intermediate releasing the final product through reductive elimination. Investigating the copper-catalyzed allylic trifluoromethylation of alkenes with reagent 4, Fu and Liu postulated a similar mechanism based on DFT calculations.61 This mechanism can be ruled out in our system since the corresponding Cu–CF3 complex 2 resulting from CF3 uptake by complex 1 has been isolated and cannot transfer its CF3 moiety.18 An ionic pathway seems unlikely as the presence of the catalyst is mandatory and this route would be in contradiction with the TEMPO trapping experiments (Scheme 1). Indeed, adding TEMPO to a solution of complex 1 does not change the UV-vis profile, therefore suggesting that no electronic transfer occurs between TEMPO and complex 1.73
In light of these considerations, we propose a mechanism (Scheme 2) implying the generation of CF˙3 radicals by SET from complex 1 to the CF3+ source74 and subsequent addition of this radical onto the unsaturated moieties. The resulting alkyl–CF3 radical 17 (Scheme 2, black pathway) could then undergo another SET thereby regenerating complex 1 along with the oxidized cation 18 and closing the catalytic cycle. Proton loss provides the expected product 19. In the case of alkynes (Scheme 2, grey pathway), vinyl–CF3 radical 20 obtained presumably undergoes hydrogen transfer with NMP,52 thereby providing trifluoromethylated alkene 21. The resulting NMP radical is then oxidized to the cation, thus regenerating complex 1. Also, performing the reaction of complex 3 instead of complex 1 with TEMPO in the presence of reagent 5 only provides TEMPO–CF3 adducts in trace amounts (<3%) thus indicating that only reduced complex 1 is capable of reducing the CF3+ source.75
Conclusions
We have shown that a well-defined copper(II) complex can promote generation of CF˙3 radicals by reduction of an electrophilic CF3+ source. This unusual behavior is enabled by a redox dialogue between the CF3+ source and the complex, mediated by the redox-active ligand which enables SET through ligand-centered oxidation. This strategy offers milder conditions and complementarity to photoredox catalytic manifolds as well-defined molecular complexes performing controlled generation of CF˙3 radicals. This method circumvents changes in the redox state of the copper center by promoting electronic transfer at the ligand instead, thus limiting uncontrolled reactivities at the metal center, and was successfully applied to the trifluoromethylation of silyl enol ethers, heteroaromatics and hydrotrifluoromethylation of alkynes. To the best of our knowledge, this is the first example of hydrotrifluoromethylation of alkynes catalyzed by a well-defined copper complex and overall of controlled generation of CF˙3 radicals by cooperative redox catalysis. This reactivity opens the way towards new developments in the field of redox ligand-based cooperative catalysis as possible mild alternatives to expensive noble metals such as iridium and ruthenium. Among other foreseeable broad catalytic perspectives, the participative nature of these ligands could allow them to stabilize high and/or disfavored metallic oxidation states, which is a prospect we are currently pursuing.Acknowledgements
The authors thank MENRT (JJ), UPMC, CNRS, ANR for funding (grant ANR-11-JS07-004-01), IUF (LF) and LabEx MiChem. The authors also wish to thank Lise-Marie Chamoreau for X-ray analysis.Notes and references
- W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580 CrossRef CAS.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 363 CrossRef CAS.
- S. Blanchard, E. Derat, M. Desage-El Murr, L. Fensterbank, M. Malacria and V. Mouriès-Mansuy, Eur. J. Inorg. Chem., 2012, 376 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC.
- V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228 CrossRef CAS PubMed.
- H. Grützmacher, Angew. Chem., Int. Ed., 2008, 47, 1814 CrossRef PubMed.
- P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599 CrossRef CAS.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886 RSC.
- M. R. Ringenberg, S. L. Kokatam, Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 788 CrossRef CAS PubMed.
- A. L. Smith, K. I. Hardcastle and J. D. Soper, J. Am. Chem. Soc., 2010, 132, 14358 CrossRef CAS PubMed.
- For a highlight, see: W. I. Dzik, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2011, 50, 3356 CrossRef CAS PubMed.
- D. L. J. Broere, B. de Bruin, J. N. H. Reek, M. Lutz, S. Dechert and J. I. van der Vlugt, J. Am. Chem. Soc., 2014, 136, 11574 CrossRef CAS PubMed.
- D. L. J. Broere, L. L. Metz, B. de Bruin, J. N. H. Reek, M. A. Siegler and J. I. van der Vlugt, Angew. Chem., Int. Ed., 2015, 54, 1516 CrossRef CAS PubMed.
- M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2013, 52, 12329 CrossRef CAS PubMed.
- Z. Alaji, E. Safaei, L. Chiang, R. M. Clarke, C. Mu and T. Storr, Eur. J. Inorg. Chem., 2014, 6066 CrossRef CAS.
- J. Jacquet, E. Salanouve, M. Orio, H. Vezin, S. Blanchard, E. Derat, M. Desage-El Murr and L. Fensterbank, Chem. Commun., 2014, 50, 10394 RSC.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS PubMed.
- T. Yamazaki, T. Taguchi and I. Ojima, Unique Properties of Fluorine and Their Relevance to Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, 2009 Search PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed.
- C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed.
- C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC.
- O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 1744 CrossRef CAS PubMed.
- J.-A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975 CrossRef CAS.
- S.-M. Wang, J.-B. Han, C.-P. Zhang, H.-L. Qin and J.-C. Xiao, Tetrahedron, 2015, 71, 7949 CrossRef CAS.
- T. Liu and Q. Shen, Eur. J. Org. Chem., 2012, 6679 CrossRef CAS.
- E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679 CrossRef CAS PubMed.
- R. J. Lundgren and M. Stradiotto, Angew. Chem., Int. Ed., 2010, 49, 9322 CrossRef CAS PubMed.
- Y. Ye and M. S. Sanford, Synlett, 2012, 23, 1696 CrossRef.
- E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC.
- G. Han, Q. Wang, Y. Liu and Q. Wang, Org. Lett., 2014, 16, 5914 CrossRef CAS PubMed.
- Y. Li, Y. Lu, G. Qiu and Q. Ding, Org. Lett., 2014, 16, 4240 CrossRef CAS PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS PubMed.
- For a review: T. Koike and M. Akita, Top. Catal., 2014, 57, 967 CrossRef CAS.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed.
- Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034 CrossRef CAS PubMed.
- S. Park, J. M. Joo and E. J. Cho, Eur. J. Org. Chem., 2015, 4093 CrossRef CAS.
- J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS PubMed.
- W. J. Choi, S. Choi, K. Ohkubo, S. Fukuzumi, E. J. Cho and Y. You, Chem. Sci., 2015, 6, 1454 RSC.
- A. Carboni, G. Dagousset, E. Magnier and G. Masson, Chem. Commun., 2014, 50, 14197 RSC.
- G. Dagousset, A. Carboni, E. Magnier and G. Masson, Org. Lett., 2014, 16, 4340 CrossRef CAS PubMed.
- W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef CAS PubMed.
- Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221 CrossRef CAS PubMed.
- E. Kim, S. Choi, H. Kim and E. J. Cho, Chem.–Eur. J., 2013, 19, 6209 CrossRef CAS PubMed.
- S. Choi, Y. J. Kim, S. M. Kim, J. W. Yang, S. W. Kim and E. J. Cho, Nat. Commun., 2014, 5, 4881 CrossRef CAS PubMed.
- X. Wu, L. Chu and F.-L. Qing, Angew. Chem., 2013, 125, 2254 CrossRef.
- D. J. Wilger, N. J. Gesmundo and D. A. Nicewicz, Chem. Sci., 2013, 4, 3160 RSC.
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, ACS Catal., 2014, 4, 2530 CrossRef CAS.
- S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Médebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505 CrossRef CAS PubMed.
- N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS PubMed.
- H. Wang and D. Vicic, Synlett, 2013, 24, 1887 CrossRef CAS.
- R. Beniazza, F. Molton, C. Duboc, A. Tron, N. D. McClenaghan, D. Lastécouères and J.-M. Vincent, Chem. Commun., 2015, 51, 9571 RSC.
- X. Wang, Y. Ye, S. Zhang, J. Feng, Y. Xu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 16410 CrossRef CAS PubMed.
- A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120 CrossRef CAS PubMed.
- J. Xu, Y. Fu, D.-F. Luo, Y.-Y. Jiang, B. Xiao, Z.-J. Liu, T.-J. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 15300 CrossRef CAS PubMed.
- P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 2213 CrossRef CAS PubMed.
- P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119 CrossRef CAS PubMed.
- L. Li, Q.-Y. Chen and Y. Guo, J. Org. Chem., 2014, 79, 5145 CrossRef CAS PubMed.
- Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411 CrossRef CAS PubMed.
- E. Mejía and A. Togni, ACS Catal., 2012, 2, 521 CrossRef.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed.
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed.
- T. Kitazume and N. Ishikawa, J. Am. Chem. Soc., 1985, 107, 5186 CrossRef CAS.
- P. Gao, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Chem.–Eur. J., 2015, 21, 7648 CrossRef CAS PubMed.
- Y. Wang, M. Jiang and J.-T. Liu, Adv. Synth. Catal., 2014, 356, 2907 CrossRef CAS.
- Y. Li, L. Wu, H. Neumann and M. Beller, Chem. Commun., 2013, 49, 2628 RSC.
- J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357 CrossRef CAS PubMed.
- L. Ling, K. Liu, X. Li and Y. Li, ACS Catal., 2015, 5, 2458 CrossRef CAS.
- Possible pathways for catalyst deactivation could arise from conversion of complex 1 into complex 2 (Fig. 1) upon CF˙3 uptake, which would result in a catalytic dead-end as complex 2 does not perform CF3 transfer (ref. 18). Deactivation of a photoredox catalyst by ligand functionalization with the generated radical has recently been reported ( J. J. Devery III, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers II and C. R. J. Stephenson, Chem. Sci., 2015, 6, 537 RSC ). This type of deactivation pathway could also be at work in our system. We thank one referee for this suggestion.
- Hydrogen abstraction from NMP has been reported and can occur at two sites: the methyl group on the nitrogen or the CH2 position adjacent to the nitrogen ( S. M. Aschmann and R. Atkinson, Atmos. Environ., 1999, 33, 591 CrossRef CAS; C. Li, T. Takanohashi, I. Saito and M. Iino, Energy Fuels, 2003, 17, 1399 CrossRef; G. Solignac, I. Magneron, A. Mellouki, A. Muñoz, M. Martin Reviejo and K. Wirtz, J. Atmos. Chem., 2006, 54, 89 CrossRef ).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1413274. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03636d |
| This journal is © The Royal Society of Chemistry 2016 |