
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailor-made preparation of Co–C, Co–B, and Co catalytic thin films using magnetron sputtering: insights into structure–composition and activation effects for catalyzed NaBH4 hydrolysis†
M. Paladini,
V. Godinho *,
G. M. Arzac,
M. C. Jiménez de Haro,
A. M. Beltrán and
A. Fernández*
*,
G. M. Arzac,
M. C. Jiménez de Haro,
A. M. Beltrán and
A. Fernández*
Instituto de Ciencia de Materiales de Sevilla (CSIC-Univ. Sevilla), Avda. Américo Vespucio 49, 41092-Sevilla, Spain. E-mail: asuncion@icmse.csic.es; godinho@icmse.csic.es
First published on 7th November 2016
Abstract
The magnetron sputtering (MS) methodology is a powerful tool for tailor-made fabrication of Co-based thin film catalysts with controlled microstructures and compositions for sodium borohydride (SBH) hydrolysis. In particular, Co–C catalysts were tested in this reaction and compared to Co–B and Co catalyst coatings. The microstructural and chemical analyses by X-ray diffraction (XRD), scanning and transmission electron microscopy (SEM and TEM), Rutherford back scattering (RBS) and X-ray photoelectron spectroscopy (XPS) were used to characterize a complete library of thin film catalysts. Pure Co materials were characterized by their nanocrystalline microstructure, and grain refinement was achieved via an increase in the deposition pressure. The incorporation of boron or carbon via co-deposition results in amorphization and dispersion of the active metallic Co phase. The composition can be tuned while keeping a controlled microstructure, and a comparison of activity at 25 °C was performed on catalysts deposited on Ni foam substrates. A comparison of the initial activities showed that the Co–B samples were more active than the Co–C samples because of electronic effects. However, a strong activation was found for the Co–C catalysts after the first use. This effect was dependent upon the incorporation of cobalt boride (CoxB) species on the catalysts' surface, as shown by XPS. After the first several uses, the activity of the Co–C samples (values up to 2495 mL min−1 gcatalyst−1) were as high as that of fresh Co–B, and the surface composition of both the catalysts was similar. This activation was not observed for the pure Co and was very weak for the Co–B catalysts. The use of polymeric (PTFE) substrates (flexible membranes) illustrated the versatility of the methodology to obtain catalytic membranes and allowed for a TEM microstructural analysis at the nanoscale. Catalytic activities at 60 °C were as high as 16.7 and 20 L min−1 gCo−1 for the Co–C and Co–B membranes, respectively. We determined the optimized conditions to increase the catalytic activity of Co-based coatings prepared via magnetron sputtering.
1. Introduction
The search for applicable hydrogen storage materials is important because of the advantages of hydrogen energy.1 The catalyzed hydrolysis of lithium and sodium borohydride, ammonia borane, hydrazine, and formic acid have been extensively investigated as promising hydrogen storage materials based on their relatively high hydrogen contents and low molecular weights. In particular, the catalyzed hydrolysis of sodium borohydride (SBH)2–4 is a safe reaction that produces hydrogen according to eqn (1):| BH4− (aq) + 4H2O (l) → 4H2 (g) + B(OH)4− (aq) | (1) |
Co-based catalysts have been widely investigated for this reaction, and the ammonia borane hydrolysis, as cost-effective materials5–8 with the aim of replacing the more expensive and scarce noble metals catalysts. Pure Co, Co–B, Co–B–O, Co–P and bimetallic Co-based materials have been widely investigated.5–8 Recently, metal-carbide catalysts have been proposed as alternative non-precious materials for fuel cells or electrolyzers in electrochemical energy conversion devices.9,10 A volcano plot was presented for the different metals and metal carbides for their use in hydrogen evolution reactions (HER).9
For technological/practical applications, i.e., systems and reactor designs, it is highly desirable to have catalysts in a supported form.3 In a previous paper,11 we proposed the magnetron sputtering (MS) deposition technique as a method to prepare pure Co catalysts supported on different substrates for the catalyzed hydrolysis of SBH and ammonia borane. MS is a technique currently used in industry that permits the deposition of a wide range of materials with varied compositions, microstructures, crystallinities or porosities on varied supports by varying the target composition and deposition conditions (nature and pressure of discharge gas, deposition power, temperature of substrate, etc.).11–14 Supported Co catalysts have mainly been prepared using the impregnation-chemical reduction method. In most cases, SBH is employed as a reducing agent to form the supported Co–B materials.15,16 Pure Co catalysts could be obtained for fundamental studies with boron-free targets using MS. Electrodeposition has also been presented as a convenient scale-up method for the production of supported Co catalysts.17,18 However, the use of this method was limited to metallic (or bimetallic) films and can only be used for conductive substrates. Pulsed laser deposition has also been used for the preparation of supported catalysts for SBH and ammonia borane hydrolysis. Co–B thin films and Co nanoparticles on a B film have been prepared using this method.19,20 MS deposition has been used to fabricate pure Co catalysts supported by non-conductive substrates for water oxidation21 and for the SBH hydrolysis reaction.11 In the present work, we studied Co–C catalysts and compared them with Co and Co–B catalysts, and all the catalysts were prepared using MS. The superior adhesion of these coatings on porous substrates (Ni foam and polymeric membranes) is an additional advantage of the MS deposition in this work.
There is still controversy regarding the nature of the active phase(s) and the role of boron and/or microstructure in the activity and durability of these Co-based catalysts.5–7,22–24 Even though the CoxB phase has been widely proposed as active phase (also by its in situ formation at the surface in the presence of SBH23); a Co core with a (poly)borate shell was proposed as well.6,18 The tailor-made catalytic coatings presented here were used in a set of experiments to expand the knowledge on these issues. Supported catalysts with a controlled microstructure and/or composition were all prepared using the same experimental set-up and were investigated and compared to other reported catalysts in the literature.
2. Experimental
2.1. Catalyst preparation and selection of materials and supports
On the basis of our previous work for pure Co in ref. 11, the deposition parameters selected for the coating fabrication in this paper are summarized in Table 1 with the deposition rates and thicknesses.| Catalyst | Ar pressure (×10−2 mbar) | Co power DC (W) | B or C power RF (W) | Thicknessa (nm) | Deposition rateb (nm min−1) |
|---|---|---|---|---|---|
| a Thickness measured from SEM cross-section on coatings grown on Si substrate.b Calculated from SEM thickness. | |||||
| Co50 | 2.8 | 50 | 1070 ± 10 | 4.2 | |
| Co50-hp | 4.5 | 50 | 440 ± 6 | 1.2 | |
| Co50B50 | 2.8 | 50 | 50 | 790 ± 7 | 2.4 |
| Co50B100 | 2.8 | 50 | 100 | 990 ± 12 | 4.8 |
| Co50C100 | 2.8 | 50 | 100 | 895 ± 13 | 3.6 |
| Co50C200 | 2.8 | 50 | 200 | 1045 ± 15 | 4.2 |
| Co100C300 | 2.8 | 100 | 300 | 1030 ± 17 | 10.8 |
| Co150C300 | 2.8 | 150 | 300 | 820 ± 16 | 16.2 |
The coatings were prepared via co-deposition, and two magnetron sources from AJA (N Scituate, MA, USA) were employed for 2′′ diameter targets. One was operated in a magnetic target configuration under DC power for a pure cobalt target (Kurt J. Lesker, 99.95% pure, 1 mm thick). The second one was operated when appropriate under an RF configuration for pure boron (Kurt J. Lesker, 99.95% pure) or carbon (Kurt J. Lesker, 99.99% pure) targets. The base pressure before deposition was 10−6 mbar, and the working pressure for Ar was 2.8 × 10−2 mbar for all the coatings, except for one Co sample grown at 4.5 × 10−2 mbar (named hp to indicate high pressure). Table 1 also summarizes the abbreviated names used for all the coatings, which were based on the target compositions, pressure and applied discharge power.
For the catalytic studies, the thin films were deposited on a commercial Ni foam (Goodfellow, 1.6 mm thick, 95% porosity, 20 pores per cm) and on PTFE (polytetrafluoroethylene) membranes (Pall Corporation, PTF002LH0A-SAMP, 0.02 μm pore size, polypropylene backed). For the chemical and microstructural characterization of the coatings, Si (100) pieces were employed as substrates in addition to the Ni foam and PTFE membranes. Prior to deposition, the Ni foam was cut into ca. 0.5 × 0.5 cm2 pieces and placed in groups to be used in a small reactor (see Section 2.3). Each group was weighed before and after deposition to obtain the total mass of the deposited catalyst. Before each synthesis, the Ni foam pieces were washed in an ultrasonic bath successively with distilled water, ethanol/acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 0.1 M HCl, distilled water, ethanol and acetone, and then it was dried in air for 24 hours. No previous treatment was performed on the PTFE membranes. The Si substrates were cleaned with acetone and dried in a nitrogen flow. When the support was different from the Ni foam, it will be clearly indicated in the text.
:1), 0.1 M HCl, distilled water, ethanol and acetone, and then it was dried in air for 24 hours. No previous treatment was performed on the PTFE membranes. The Si substrates were cleaned with acetone and dried in a nitrogen flow. When the support was different from the Ni foam, it will be clearly indicated in the text.
2.2. Catalyst characterization
X-ray diffraction measurements were performed using Cu Kα radiation in a Siemens D5000 diffractometer in a Bragg–Brentano configuration. The coatings grown on the PTFE membranes were used for these measurements.Scanning electron microscopy (SEM) was used to study the morphology and microstructure of the samples in a high resolution FEG microscope HITACHI S4800. The coatings were analyzed directly on the Ni foam and PTFE membranes for top view observations. For the cross section views, samples were cleaved from coatings grown on Si. The thickness of the coatings was evaluated using the SEM cross-section measurements.
TEM (Transmission Electron Microscopy) and ED (electron diffraction) studies were performed on a FEI Tecnai G2 F30 FEG (field emission gun) microscope equipped with a HAADF (High Angle Annular Dark Field) detector from Fischione Instruments. Images were obtained in STEM (scanning TEM) mode at 300 kV with HAADF detection. The thin film samples were grown on PTFE membranes. They were removed from the polypropylene back-support, fixed in a frame grid, and then submitted to ion thinning from the back side.
Proton-elastic back-scattering spectrometry (p-EBS) was used to determine the composition of the catalytic coatings grown on the Si substrates. This technique is well suited for characterization of the coatings as it provides simultaneous determination of Co and the light elements C, O and B.25 Measurements were carried out at the National Center for Accelerators (CNA, Sevilla, Spain) using a 3 MV tandem accelerator. The spectra were obtained using a 2 MeV H+ beam and passivated implanted planar silicon (PIPS) detector set at 165°. To obtain the thickness and composition of the films, the spectra were simulated using the SIMNRA code.26
XPS spectra were recorded using a SPECS electron spectrometer equipped with a PHOIBOS 150 9MCD analyzer using Al Kα radiation with a 35 eV pass energy at a normal emission take off angle. The spectra were calibrated to the position of the C (1s) signal (from contamination) at 284.6 eV and validated using the characteristic position of the surface oxidized Co (2p3/2) at 780.9 eV. Deconvolution was performed using the CasaXPS program provided by the SPECS Company. The coatings grown on the PTFE membranes were used for these measurements. The samples were investigated after deposition and after use for the SBH hydrolysis reaction.
2.3. Catalytic activity: hydrogen generation tests
The Ni foam pieces were grouped (10 to 15 pieces), cleaned and weighed before and after deposition, as described in Section 2.1. The pieces with the supported catalyst (1–6 mg) were placed at the bottom of a three necked heart-shaped flask. The flask was immersed in a water bath maintained at 25 ± 0.5 °C and connected to a 100 mL gas burette. The reaction (1) was started by injecting 38 mg of SBH dissolved in 1 mL of a 4.5 wt% NaOH solution. The amount of hydrogen generated was measured by reading the displacement of the piston in the gas burette (gas-tight with a mercury o-ring) as a function of time. No additional stirring was used for the experiments, except for the stirring created by the evolving hydrogen. The HGR (Hydrogen Generation Rate, mL min−1) was obtained from the slope of a plot of the volume of hydrogen evolved vs. time in a linear regime. In this paper, the catalytic activity (expressed in mL min−1 gcat−1) was the slope from the plot of HGR (mL min−1) as a function of the mass of the supported catalyst. The activity was also expressed per gram of cobalt considering the composition of the catalysts determined using p-EBS spectrometry. Finally, the catalytic activities were measured for selected samples at different temperatures, and the Arrhenius plots were analyzed to determine the apparent activation energies. Cycling experiments were also conducted. After each test, the supported catalyst was extracted from the reaction medium, washed with distilled water and ethanol and then dried for one day under atmospheric conditions before the activity test was repeated.The activities of the samples supported on the PTFE membrane were tested at 60 °C (in a thermostatic bath) using the same experimental set-up described for the Ni foam samples. Typically, 4 pieces of 1 cm2 each were cut into small pieces and placed in the reaction flask. The procedure was similar to the one described above except the mass of the catalyst was not determined by weight. Activity was expressed per gram of cobalt. For Co quantification in the membrane deposited catalysts, the colorimetric nitroso-R salt (NRS) method27 was used. A 1 cm2 piece of the supported catalyst was treated with 1 M HCl for complete dissolution of the Co-based catalysts. The amount of Co determined using the photometric method was evaluated by comparing the results with the results obtained using plasma ICP measurements for selected samples. We found that the colorimetric method agreed with the ICP results with less than 12% error.
The catalysts supported on the PTFE membranes were more convenient for XPS analysis (thinner and flatter samples compared to the Ni foam substrate). The samples were investigated after deposition and after use. In the latter case, the catalytic surface of the membranes was kept in contact with a 19 wt% SBH solution in a 4.5 wt% NaOH solution for 1.5 or 24 hours. The samples were rinsed with water and dried under atmospheric conditions prior to XPS analysis.
3. Results and discussions
3.1. The supported catalysts and their microstructural and chemical characterization
The following samples (see Table 1) were prepared starting with 50 W of DC power at the Co target: two pure Co catalysts at medium (2.8 × 10−2 mbar) and high (4.5 × 10−2 mbar) pressures of the process gas (Ar), and two Co–B catalysts using 50 and 100 W RF power at the metalloid (B) target with medium process gas pressure. Four samples were prepared with different combinations of power at the Co and C targets for the newly investigated Co–C catalysts. The medium Ar pressure was used when applying the DC and RF power at the Co and C targets in all of the samples.The first characterization of the samples was performed using XRD, and the data are shown in Fig. 1 for the catalysts supported on the PTFE membranes. The pure Co50 materials show small and broad peaks that can be assigned to a nanocrystalline hcp Co phase (ICDD 00-005-0727), but the cubic phase (ICDD 00-015-0806) cannot be disregarded.17 As we demonstrated in our previous publication,11 higher deposition powers are needed for higher crystallization of the cobalt phase. The incorporation of B and C results in amorphization of the catalysts at similar deposition powers. Based on research with powder materials, the presence of boron is generally associated with the formation of amorphous black solids.5,6 In previous work, nanocrystalline and amorphous phases appeared to be more active catalytically.5,6 In the present work, the addition of carbon produces a similar amorphization effect on the microstructure. Only when the highest deposition powers were used was a certain degree of nanocrystallization observed in Co150C300. Data obtained from the electron diffraction (ED) analyses are shown in Section 3.3.
 | ||
| Fig. 1 X-ray diffraction patterns for the as prepared Co, Co–B and Co–C coatings. The PTFE substrate is included as a reference. | ||
To gain additional microstructural information, Fig. 2 and 3 show the SEM top and cross sectional views of the coatings grown on Si substrates for all of the investigated samples. The SEM analysis also permits determination of the thin film thickness and deposition rate (Table 1). In general, all catalysts have a columnar growth, which is typical of magnetron sputtered deposited thin films under low adatom mobility conditions, and surface shadowing governs the film growth.28 The top views and details of the columns, presented in Fig. 2 and 3, reveal a microstructure of mesocolumns (between nano and micro), which are typically formed via the association of nanocolumns.29 A certain degree of intra- and inter-columnar porosity is also observed. The mesocolumn sizes were evaluated based on the direct observation of the columns in the cross sectional views and the grain sizes measured from the top view images. The results are summarized in Table 2. In the case of the two nanocrystalline, pure Co samples (Co50 and Co50-hp), a reduction in the mesocolumns/grain sizes was found for the sample growth under high pressure (clearly shown in the top view images). For Co–B and Co–C, the mesocolumns/grain sizes were similar to the ones for Co50-hp. It was concluded that Co50-hp, Co–B and Co–C samples have a similar microstructure and grain and mesocolumn sizes at the selected deposition conditions. Only the Co50 sample showed a clear increase in the grain/mesocolumn size. The use of high pressure during the magnetron sputtering for pure Co deposition produced grain refinement, as described in our previous paper.11 The introduction of B and C produces the same effect through enhanced amorphization. The Co–B and Co–C samples are directly comparable as they have a similar amorphization degree (XRD data) and similar thicknesses and mesocolumn sizes (Tables 1 and 2). This allows for a new fundamental understanding of the role of metalloid addition through comparison to pure Co samples with similar and controlled microstructures.
 | ||
| Fig. 2 SEM images of the planar (right) and cross sectional (left) views of Co and Co–B coatings. Images are labeled according to their corresponding sample. | ||
 | ||
| Fig. 3 SEM images of the planar (right) and cross sectional (left) views of Co–C coatings. Images are labeled according to their corresponding sample. | ||
| Sample | Composition by p-EBS (at%) | Mesocolumn size (nm) | ||||
|---|---|---|---|---|---|---|
| Co | B | C | O | SEM cross section | SEM top view | |
| Co50 | 87 | 13 | 30–250 | 20–375 | ||
| Co50-hp | 87 | 13 | 30–80 | 10–90 | ||
| Co50B50 | 75 | 15 | 10 | 50–80 | 10–100 | |
| Co50B100 | 65 | 23 | — | 12 | 70–120 | 40–100 |
| Co50C100 | 79 | — | 13 | 8 | 60–100 | 50–110 |
| Co50C200 | 64 | — | 27 | 9 | 50–120 | 40–140 |
| Co100C300 | 59 | — | 31 | 10 | 50–100 | 30–140 |
| Co150C300 | 70 | — | 23 | 7 | 50–100 | 30–150 |
The composition determination for the Co, Co–B and Co–C coatings is crucial to quantify the incorporation of B and C in the samples and the oxidation degree. Proton-EBS is well established for these analyses of thin films and coatings and for simultaneous quantification of light elements.25 Table 2 summarizes the results obtained for the samples grown on Si substrates. For the Co–C catalysts, the samples studied cover Co/C atomic ratios ranging from 6.1 to 1.9. The measured values for Co50B100 are compatible with the typical stoichiometries previously reported for Co–B powders (i.e., Co3B).30 An additional composition is given for a lower B content. The presence of a certain amount of oxygen in the Co deposited coatings is unavoidable because of the presence of oxygen and water in the residual vacuum.31 It is also characteristic for columnar coatings to have a certain degree of inter-columnar porosity, which also favors a post-deposition oxidation.32 Nevertheless, the presence of oxygen is a characteristic of powdery Co-based catalysts for this reaction.5,6,23 In conclusion, the tailored-made coatings can be used as a representative library for these types of catalysts.
Fig. 4 shows the XPS surface analysis performed on selected, representative, fresh samples deposited on the PTFE membranes (Co50-hp, Co50B100 and Co50C100). The Co (2p) binding energies at 777.8 (Co 2p3/2) and 792.9 eV (Co 2p1/2) are consistent with those of metallic cobalt,5,23,33,34 cobalt borides (CoxB)35,36 or cobalt carbides,37 which cannot be clearly differentiated using XPS analysis. Oxidized species are also detected on the surface, corresponding to Co (2p) peaks at higher binding energies of 780.9 (Co 2p3/2) and 796.4 eV (Co 2p1/2).5,23,34 The shape of the peaks and satellites are compatible with the CoO phase.23,34 The B (1s) peak in the Co–B sample shows two peaks at binding energies of 187.6 and 191.6 eV. The former peak at 187.6 eV can be assigned to elemental boron or boron in CoxB compounds, which is in agreement with literature data.33,36,38,39 The second peak at 191.6 eV can be assigned to oxidized boron species at the surface.5,22 For the Co–C sample, the analysis of the C (1s) peak is not presented because of strong overlapping with the adventitious carbon peak, which was a result of contamination and air exposure. For oxygen, the O (1s) peak corresponded to the characteristic Co–OH and Co-oxide species at the surface.40
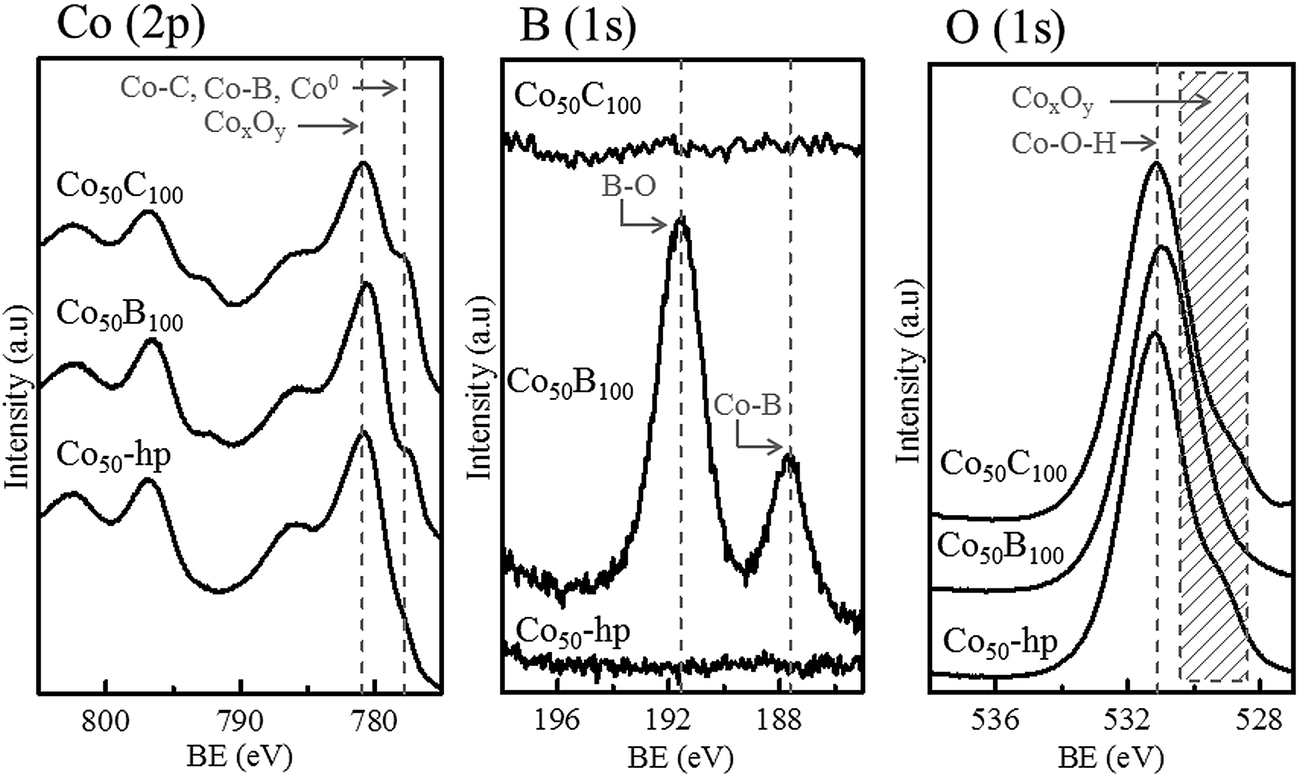 | ||
| Fig. 4 XPS data for Co (2p), B (1s) and O (1s) for the representative samples indicated in the figure. | ||
The versatility of magnetron sputtering has been demonstrated for catalyst design. Composition and microstructure can be controlled while the catalysts grow directly on the substrates of interest.
3.2. Catalytic activity: a comparative study
The activity for the SBH hydrolysis reaction was measured for each prepared Ni foam supported catalyst in 4.5 wt% NaOH and 3.8 wt% SBH solutions. The results are summarized in Table 3. Fig. 5 shows the hydrogen evolution curves (Fig. 5a), and hydrogen generation rate plot (Fig. 5b) as a function of the catalyst mass for the Co50C100 sample measured at 25 °C. For all of the tested catalysts, the hydrogen evolution curves followed a straight line, which indicated zero-order kinetics for the SBH concentration. No induction periods were detected in the hydrogen evolution experiments. The linear plot of the hydrogen generation rate as a function of the catalyst mass indicated the kinetic regime for all of the measurements. The activities in the range from 15 to 35 °C were measured for the as prepared samples. A characteristic Arrhenius plot is shown in Fig. 5c for the Co50C100 catalyst.| Samplea (Ni foam substrate) | Co at% | Co wt% | Activity maximum first 5 cycles (mL min−1 gCo−1) | Activity initial (mL min−1 gCo−1) | Activity maximum first 5 cycles (mL min−1 gcat−1) | Activity initial (mL min−1 gcat−1) |
|---|---|---|---|---|---|---|
| a Thicknesses as indicated in Table 1. | ||||||
| Co50 | 87 | 96 | 1667 | 1667 | 1600 | 1600 |
| Co50-hp | 87 | 96 | 2083 | 2083 | 2000 | 2000 |
| Co50B50 | 75 | 93 | 2204 | 2005 | 2050 | 1865 |
| Co50B100 | 65 | 90 | 2983 | 2674 | 2685 | 2407 |
| Co50C100 | 79 | 94 | 1648 | 1222 | 1594 | 1149 |
| Co50C200 | 64 | 92 | 2229 | 1179 | 1984 | 1050 |
| Co100C300 | 59 | 86 | 2901 | 1337 | 2495 | 1150 |
| Co150C300 | 70 | 91 | 2186 | 978 | 1990 | 890 |
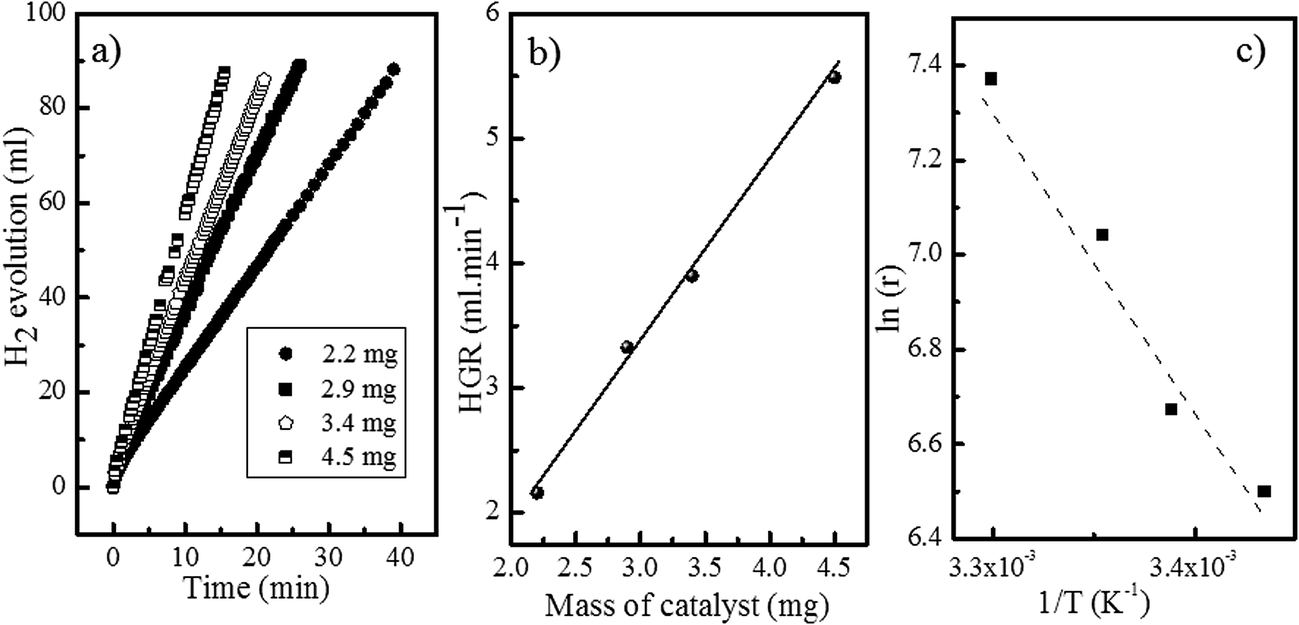 | ||
| Fig. 5 Catalytic tests for the Co50C100 sample: (a) hydrogen evolution curves for the catalyzed SBH hydrolysis. (b) Hydrogen generation rate as a function of the mass of the catalyst. (c) Arrhenius plot. | ||
In previous work with Co and Ni based solids,6,41 a certain activation of the catalysts upon first uses (approximately 10% from the initial activity) was found. In addition, a repetitive cycling produced a progressive deactivation at the end.6,42 For these reasons, each catalyst was cycled up to 7 times to determine the initial and maximum catalytic activity. Table 3 summarizes these values expressed in mL min−1 per gram of catalysts and per gram of cobalt (according to the stoichiometry given by p-EBS) for all of the investigated samples. Long life durability experiments will be presented in a future study. The data corresponding to the first 7 cycles are included as ESI (Fig. S1†), and the data show the initial activation effects and tendency towards deactivation after cycling. Fig. 6 represents the catalytic activity data (both initial and maximum in mL min−1 gCo−1) as a function of the Co content (at%) for the investigated materials indicated in the figure.
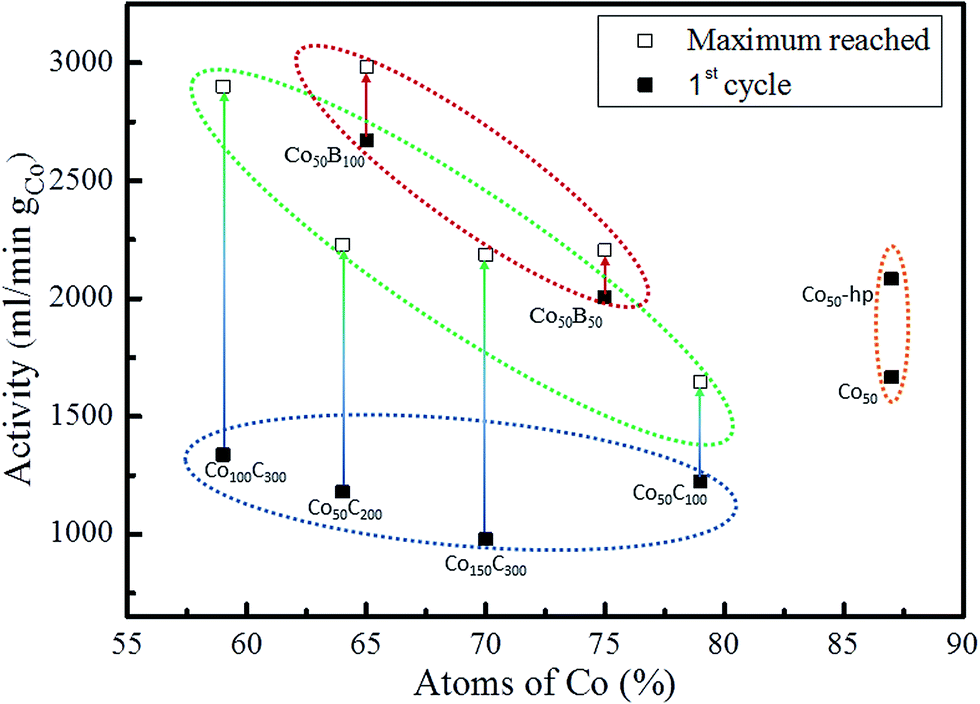 | ||
| Fig. 6 Initial (■) catalytic activity values for all of the investigated samples plotted against the Co content for each sample (according to p-EBS data). Maximum (□) activity reached during the first 7 cycles when reactivation was observed. | ||
As discussed above, the prepared Co50-hp, Co–B and Co–C samples are comparable in terms of microstructure and grain and mesocolumn size. We found that the initial activity of the Co50-hp sample is on the order of the Co50B50 activity, as shown in Table 3. An increase in the activity is observed if the amount of boron is increased, which occurs with the Co50B100. This is consistent with the idea that the presence of boron is crucial for the high activity of the Co catalysts.5,6 In the presence of boron, cobalt is electron-enriched and benefits catalytic activity apart from increasing the dispersion of the metallic phase.5,6 This hypothesis is confirmed because the Co–C catalysts show low initial activities with respect to the pure Co and Co–B samples. The addition of carbon increases the cobalt dispersion but no electronic effect was found in this case. However, a strong activation (up to a 220% increase in the activity with respect to the initial) is observed for the Co–C samples after the first uses (transition from points in a blue ellipse to points in a green ellipse in Fig. 6, see also Fig. S1†). The activity of the Co–C samples after use can be as high as that of Co–B (2495 vs. 2685 mL min−1 gcat−1 for Co100C300 and Co50B100, respectively). This effect is not observed for the pure Co catalysts and is very weak for the Co–B samples. Considering the maximum activities for the Co–C catalysts, there is a correlation between the activity and cobalt dispersion. An improvement in the activity was found when the dispersion of Co increased and was mediated by the higher incorporation of carbon (4 points inside the green ellipse in Fig. 6). A similar behavior is observed for the two Co–B samples (2 points inside the red ellipse) although the effect is not accompanied by a significant activation upon use. To understand the activation process occurring in the Co–C catalysts, an XPS analysis was conducted on a representative Co–C sample supported on PTFE membranes after use (see details in the Experimental section). The results can be found in Fig. 7 for the B (1s) region for as prepared samples and samples after use. The surface of the Co–C catalyst after use shows the two characteristics peaks of the B–O (191.6 eV) and B–Co (187.6 eV) species. In fact, the surface becomes similar to that of the Co–B catalysts (see Fig. 4), which explains the activation of the Co–C material during the first uses. The presence of borate reaction products on the surface of the catalysts after use is well known;5,6 however, the incorporation of boride was observed in our experiments for the case of the Co50C100 sample, which showed a clear activation effect upon first use. These results reinforce the idea that there is an electronic effect of the boride to improve catalytic activity in addition to a dispersion effect. An electronic effect coming from B incorporation as boride was also previously proposed for the Co–B black powder,5,43 which is generally obtained via the reduction of oxidized Co with SBH.
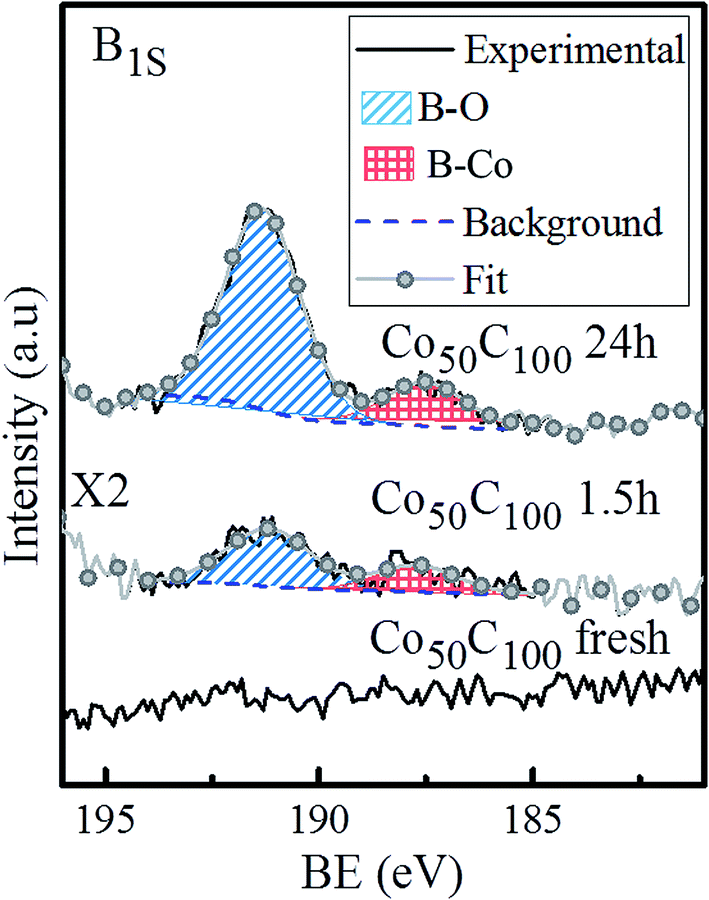 | ||
| Fig. 7 B (1s) XPS spectra for the Co50C100 sample: as prepared and after use for 1.5 and 24 hours. Deconvolution of the two peaks is also shown. | ||
The nanocrystalline pure cobalt samples are also highly active (orange ellipse in Fig. 6). However, the lower dispersion does not seem to favor incorporation of B in the form of cobalt borides in this case, and the samples do not show strong activation during the first uses (see data in Table 3 and Fig. S1†). In agreement with our previous paper, ref. 11, we found that an increase in the Ar deposition pressure produced an increase in catalytic activity for the Co coatings prepared at the same DC deposition power of 50 W. This effect is associated11 with a higher exposed surface due to a smaller mesocolumn size. It was then assumed that a further increase in the Ar pressure during MS deposition of pure Co coatings would improve the catalytic activity. However, the use of higher pressures produced powdery coatings44 with a loss of adhesion. Based on this, the use of dispersant agents such as B and C is suitable for achieving grain refinement without a loss of adhesion.
Our results identified two catalytic phases: the amorphous cobalt boride for the Co–B and Co–C (in situ formed) catalysts and the nanocrystalline Co particles (with their corresponding surface oxide layer) in the case of pure Co. In the pure Co catalysts, there was no evidence of the formation of CoxB species on the surface. However, the formation of surface cobalt borides upon operation cannot be completely disregarded. Cobalt borides are unstable and would transform into metallic cobalt (which would then become oxidized) and borates in aqueous solutions during reaction quenching. One possible interpretation is that cobalt borides do form when in contact with SBH on the surface of any cobalt precursor (even in the form of oxides) and then decompose during the reaction quenching.45 This would explain the small amounts of borides in comparison to the borates detected using XPS. The catalytic activity would correlate with the amount of cobalt boride phase formed in situ.45 The highly dispersed (and thus more reactive) Co–C should incorporate enough surface cobalt boride to be detected after quenching. This result shows that a high dispersion of cobalt is required for high catalytic activity, and the nature of the dispersant atom (B or C) is indistinct after the first uses. We suggest that in operando studies would help clarify these points.
The apparent activation energies (in the 15–35 °C range) were 54, 44, 57 and 53 kJ mol−1, for the Co50, Co50-hp, Co50C100 and Co50B100 samples, respectively, which correlated with the activity trend at 25 °C.
The activity of the prepared samples can be compared to reference materials in the literature. For the Co–B powdery materials, the typically reported activities (at 20–25 °C) are in the range of 400–3350 mL min−1 gcat−1, depending on the composition and microstructure.7 Our results (i.e., 2685 mL min−1 gcat−1 for the Co50B100 sample) are in the same order of magnitude. However, new advances in the dispersion of Co–B nanoparticles on silica substrates46 or in the design of new alloys, including P and Cr or Mo, as well as B,7,47 have resulted in higher activities (up to 30800 mL min−1 gcat−1). Some of these alternative nanostructures and compositions could be implemented using MS in multi-target chambers in future work and possibly prove the dispersion and electronic effect mechanism proposed here.
3.3. Supported catalysts on flexible polymeric membranes
To illustrate the versatility of the deposition methodology, we studied polymeric (PTFE) membranes as substrates for the catalytic coatings. Flexible Teflon membranes (commercially available) consist of expanded fibers, as shown in the SEM image in Fig. 8. Catalyst depositions were conducted under the conditions reported in Table 1 but with shorter deposition times to reduce the coating thickness (Data are included in Table 4.). As shown in the SEM image in Fig. 8 for the Co50C100 sample, a good coverage of the polymer fibers was achieved while keeping the characteristic open pores of the membrane structure. A good adhesion while also maintaining the membrane flexibility was also obtained for all of the investigated Co-based catalysts. | ||
| Fig. 8 SEM images of the PTFE membrane before (left) and after (right) deposition of the Co50C100 coating. | ||
The TEM microstructural analysis at the nanoscale is shown in Fig. 9 (STEM-HAADF images) for samples Co-hp, Co50B100 and Co50C100. At this magnification, we were able to visualize the nanocolumns that compose the mesocolumns. The sizes of the nanocolumns range from 4 to 12 nm, and no significant differences were found among the three samples, as expected from the SEM analysis. The electron diffraction patterns (insets in Fig. 9) clearly show the formation of nanocrystals in the pure Co sample, whereas alloying with B or C produces amorphization. For the Co-hp sample, rings were identified that corresponded to the interplanar distances of 2.17, 2.06 and 1.93 Å, which were assigned to the (100), (002) and (101) planes of metallic hcp Co, respectively (ICDD 00-05-0727) (The cubic phase cannot be completely disregarded.). In addition, rings at spacings of 2.46 and 1.51 Å were measured and assigned to the (111) and (220) planes of the CoO phase (ICDD 00-0481719) (The Co3O4 phase can also form.). The surface oxidation of metallic Co is expected and is more important in the thin samples used for TEM analysis because additional crystallization can occur under the 300 kV electron beam.
 | ||
| Fig. 9 STEM-HAADF images for selected coatings (Co50-hp, Co50B100 and Co50C100) deposited on the PTFE membrane fibers. The corresponding electron diffraction patterns are included as insets. | ||
The catalytic activities at 60 °C are presented (in liters per minute and per gram of Co) for selected coatings in Table 4. For practical applications, taking into account the exothermic character of the SBH hydrolysis reaction, it is common in reference papers to report the activity at 60 °C. For the Co–C and Co–B catalytic coatings on porous polymeric membranes, activities as high as 16.7 and 20 L min−1 gCo−1 were measured, respectively. The nanostructured character of the coatings associated with the dispersion and electronic effect of the boron lead to very thin films (100–600 nm) with activities in the range of the highest reported values for catalysts with similar compositions.7
4. Conclusions
The magnetron sputtering deposition methodology is presented here as a versatile tool for the fabrication of Co-based supported catalysts for hydrolytic hydrogen generation from SBH. In addition to the adaptability of the technique to support catalysts on porous, metallic or polymeric substrates, we have shown its capacity to create tailor-made catalysts by controlling both the microstructure and composition. Novel Co–C catalytic coatings were investigated and compared to Co–B and pure Co. The composition can be controlled by using a dual head MS deposition chamber and tuning the power applied to the Co and the metalloid (B or C) targets. The microstructure can be controlled by increasing the deposition pressure, which produces grain refinement for the nanocrystalline pure Co coating. The co-deposition of C or B also induced grain refinement through amorphization and dispersion of the Co active phase.A library of Co–C, Co–B and Co catalysts deposited onto Ni foam were tested for their SBH catalytic hydrolysis reaction activity. We determined the optimized conditions to increase the catalytic activity of Co-based coatings prepared using magnetron sputtering. We were able to prepare a series of samples with comparable microstructures, grain and crystal sizes to study the effect of the addition of carbon and boron to pure Co samples. We found a correlation between the degree of dispersion in the active Co phase and the activity for the Co–C and Co–B catalysts, as well as an electronic effect of B in the Co–B catalyst that improved catalytic activity. The Co–C catalysts showed evidence of in situ formation of CoxB on their surfaces, which explains the increase in activity after the first cycles (up to 220% with respect to the initial). Nanocrystalline pure Co and amorphous CoxB were proposed as catalytic phases.
The versatility and potential of this methodology was shown by the fabrication of highly active catalytic membranes, which was performed by depositing thin films on flexible polymeric substrates.
Acknowledgements
Financial support is acknowledged from the Spanish MINECO (grants CTQ2012-32519 and CTQ2015-65918-R, EU co-financed by FEDER), the Junta de Andalucía (grant PE2012-TEP862, EU co-financed by FEDER) and the CSIC (PIE-201460E018). AMB thanks to Talent-Hub Program funded by the Junta de Andalucía and the European Commission under the Co-funding of the 7th Framework Program in the People Program (Marie Curie Special Action). TEM measurements were performed at the Laboratory of Nanoscopies and Spectroscopies LANE at the ICMS. Authors thank I. Rosa, O. Montes and M. R. García for their very valuable technical support.Notes and references
- Hydrogen as a Future Energy Carrier, ed. A. Züttel, A. Borgschulte and L. Schlapbach, Wiley-VCH, 2008 Search PubMed.
- U. B. Demirci, O. Akdim, J. Andrieux, J. Hannauer, R. Chamoun and P. Miele, Fuel Cells, 2010, 10, 335–350 CrossRef CAS.
- S. S. Muir and X. Yao, Int. J. Hydrogen Energy, 2011, 36, 5983–5997 CrossRef CAS.
- B. H. Liu and Z. P. Li, J. Power Sources, 2009, 187, 527–534 CrossRef CAS.
- U. B. Demirci and P. Miele, Phys. Chem. Chem. Phys., 2010, 12, 14651–14665 RSC.
- U. B. Demirci and P. Miele, Phys. Chem. Chem. Phys., 2014, 16, 6872–6885 RSC.
- N. Patel and A. Miotello, Int. J. Hydrogen Energy, 2015, 40, 1429–1464 CrossRef CAS.
- J. Liao, H. Li, X. Zhang, K. Feng and Y. Yao, Catal. Sci. Technol., 2016, 11, 3893–3899 Search PubMed.
- R. Michalsky, Y.-J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274–1278 CrossRef CAS.
- N. A. Fadil, G. Saravanan, G. V. Ramesh, F. Matsumoto, H. Yoshikawa, S. Ueda, T. Tanabe, T. Hara, S. Ishihara, H. Murakami, K. Ariga and H. Abe, Chem. Commun., 2014, 50, 6451–6453 RSC.
- M. Paladini, G. M. Arzac, V. Godinho, M. C. Jiménez De Haro and A. Fernández, Appl. Catal., B, 2014, 158–159, 400–409 CrossRef CAS.
- V. Godinho, C. López-Santos, T. C. Rojas, D. Philippon, M. C. Jiménez de Haro, S. Lucas and A. Fernández, J. Alloys Compd., 2012, 536(S1), S398–S406 CrossRef CAS.
- V. Godinho, J. Caballero-Hernández, D. Jamon, T. C. Rojas, R. Schierholz, J. García-López, F. J. Ferrer and A. Fernández, Nanotechnology, 2013, 24, 275604 CrossRef CAS PubMed.
- S. Lucas, J.-F. Colomer, C. Bittencourt, P. Moskovkin and N. Moreau, Appl. Phys. A: Mater. Sci. Process., 2010, 99, 125–138 CrossRef CAS.
- H. Tian, Q. Guo and D. Xu, J. Power Sources, 2010, 195, 2136–2142 CrossRef CAS.
- G. M. Arzac, D. Hufschmidt, M. C. Jiménez De Haro, A. Fernández, B. Sarmiento, M. A. Jiménez and M. M. Jiménez, Int. J. Hydrogen Energy, 2012, 37, 14373–14381 CrossRef CAS.
- O. Akdim, R. Chamoun, U. B. Demirci, Y. Zaatar, A. Khoury and P. Miele, Int. J. Hydrogen Energy, 2011, 36, 14527–14533 CrossRef CAS.
- O. Akdim, U. B. Demirci and P. Miele, Int. J. Hydrogen Energy, 2011, 36, 13669–13675 CrossRef CAS.
- N. Patel, A. Miotello and V. Bello, Appl. Catal., B, 2011, 103, 31–38 CrossRef CAS.
- N. Patel, R. Fernandes, A. Santini and A. Miotello, Int. J. Hydrogen Energy, 2012, 37, 2007–2013 CrossRef CAS.
- E. R. Young, D. G. Nocera and V. Bulović, Energy Environ. Sci., 2010, 3, 1726–1728 CAS.
- Z.-L. Wang, J.-M. Yan, H.-L. Wang and Q. Jiang, J. Power Sources, 2013, 243, 431–435 CrossRef CAS.
- J. Andrieux, D. Swierczynski, L. Laversenne, A. Garron, S. Bennici, C. Goutaudier, P. Miele, A. Auroux and B. Bonnetot, Int. J. Hydrogen Energy, 2009, 34, 938–951 CrossRef CAS.
- A. M. Ozerova, V. I. Simagina, O. V. Komova, O. V. Netskina, G. V. Odegova, O. A. Bulavchenko and N. A. Rudina, J. Alloys Compd., 2012, 513, 266–272 CrossRef CAS.
- F. J. Ferrer, M. Alcaire, J. Caballero-Hernández, F. J. Garcia-Garcia, J. Gil-Rostra, A. Terriza, V. Godinho, J. García-López, A. Barranco and A. Fernández-Camacho, Nucl. Instrum. Methods Phys. Res., Sect. B, 2014, 332, 449–453 CrossRef CAS.
- M. Mayer, AIP Conf. Proc., 1999, 475, 541–544 CrossRef CAS.
- G. Wünsch, Talanta, 1979, 26, 177–179 CrossRef.
- J. A. Thornton, Annu. Rev. Mater. Res., 1977, 7, 239–260 CrossRef CAS.
- R. Messier, A. P. Giri and R. A. Roy, J. Vac. Sci. Technol., A, 1984, 2, 500–503 CAS.
- J. C. Walter, A. Zurawski, D. Montgomery, M. Thornburg and S. Revankar, J. Power Sources, 2008, 179, 335–339 CrossRef CAS.
- P. Pokorný, J. Musil, P. Fitl, M. Novotný, J. Lančok and J. Bulíř, Plasma Processes Polym., 2015, 12, 416–421 CrossRef.
- J. E. Castle, J. Adhes., 2008, 84, 368–388 CrossRef CAS.
- A. B. Dávila-Ibáñez, J. L. Legido-Soto, J. Rivas and V. Salgueirino, Phys. Chem. Chem. Phys., 2011, 13, 20146–20154 RSC.
- Y. P. Wang, Y. J. Wang, Q. L. Ren, L. Li, L. F. Jiao, D. W. Song, G. Lui, Y. Han and H. T. Yuan, Fuel Cells, 2010, 10, 132–138 CAS.
- G. M. Arzac, T. C. Rojas and A. Fernández, ChemCatChem, 2011, 3, 1305–1313 CrossRef CAS.
- G. Mavel, J. Escard, P. Costa and J. Castaign, Surf. Sci., 1973, 35, 109–116 CrossRef CAS.
- A. V. Syugaev, N. V. Lyalina, S. F. Lomayeva and A. N. Maratkanova, J. Solid State Electrochem., 2015, 19, 2933–2941 CrossRef CAS.
- J. Geng, D. A. Jefferson and B. F. G. Johnson, Chem.–Eur. J., 2009, 15, 1134–1143 CrossRef CAS PubMed.
- H. Wang, Z. Yu, H. Chen, J. Yang and J. Deng, Appl. Catal., A, 1995, 129, L143–L149 CrossRef CAS.
- N. S. McIntyre and M. G. Cook, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- A. M. F. R. Pinto, D. S. Falcão, R. A. Silva and C. M. Rangel, Int. J. Hydrogen Energy, 2006, 31, 1341–1347 CrossRef CAS.
- M. Rakap and S. Özkar, Appl. Catal., B, 2009, 91, 21–29 CrossRef CAS.
- U. B. Demirci, O. Akdim, J. Hannauer, R. Chamoun and P. Miele, Sci. China: Chem., 2010, 53, 1870–1879 CrossRef CAS.
- H. Hahn and R. S. Averback, J. Appl. Phys., 1990, 67, 1113–1115 CrossRef CAS.
- V. I. Simagina, O. V. Komova, A. M. Ozerova, O. V. Netskina, G. V. Odegova, D. G. Kellerman, O. A. Bulavchenko and A. V. Ishchenko, Appl. Catal., A, 2011, 394, 86–92 CrossRef CAS.
- C.-C. Yang, M.-S. Chen and Y.-W. Chen, Int. J. Hydrogen Energy, 2011, 36, 1418–1423 CrossRef CAS.
- D.-W. Zhuang, Q. Kang, S. S. Muir, X. Yao, H.-B. Dai, G.-L. Ma and P. Wang, J. Power Sources, 2013, 224, 304–311 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23171c |
| This journal is © The Royal Society of Chemistry 2016 |