
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modification of pea protein isolate for ultrasonic encapsulation of functional liquids
Qianyu
Ye
a,
Matthew
Biviano
b,
Srinivas
Mettu
 b,
Meifang
Zhou
a,
Raymond
Dagastine
b and
Muthupandian
Ashokkumar
*a
b,
Meifang
Zhou
a,
Raymond
Dagastine
b and
Muthupandian
Ashokkumar
*a
aSchool of Chemistry, The University of Melbourne, Parkville, Melbourne, Victoria 3010, Australia. E-mail: masho@unimelb.edu.au; Fax: +61 3 9347 5180
bDepartment of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, Melbourne, Victoria 3010, Australia
First published on 1st November 2016
Abstract
This study reports on the ultrasonic processing of pea protein isolate (PPI) in phosphate-buffered saline (PBS, pH 7.4) and Tris/HCl (pH 8) buffer systems in order to modify its properties for use in the encapsulation of functional liquids. Tetradecane-filled microspheres were synthesized using ultrasonically-modified PPI as a shell material under high intensity 20 kHz ultrasound irradiation. Tetradecane was used as a model liquid, which could in principle be replaced by functional liquids such as vitamins, fish oil, etc. The solubility of water-insoluble globulin present in PPI was significantly improved in the first sonication step, which was confirmed by solubility measurements and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The hydrodynamic diameter measurements indicated that the dissolved pea proteins formed soluble aggregates. The size, size distribution, shell thickness, mechanical strength and yield of PPI microspheres were controlled by the variation of ultrasonic parameters in the first step. In terms of stability, the microspheres maintained a core–shell structure and their size remained unchanged after one-month storage at 4 °C. Most of the microspheres had a spherical shape with a smooth surface morphology. The shell thickness varied with the surface activity and solubility of PPI, which in turn were affected by sonication time. Average stiffness ranging from 9.5 to 22 mN m−1 and average Young's modulus from 0.58 to 2.35 MPa were obtained by using atomic force microscopy (AFM). Disulphide crosslinking and noncovalent interactions played a role in the shell formation, also facilitating the storage stability of PPI microspheres.
1. Introduction
Core–shell microspheres have been widely studied due to the attractive double functionalities of cores and shells, which can be independently tuned. The core materials vary from air,1 liquids2 to solids.3 A variety of substances can work as shell materials such as proteins,1,2 polysaccharides4 and polymers.5 Animal-based proteins have been well utilized in microsphere synthesis. For example, bovine serum albumin (BSA) and lysozyme were used as shell materials to synthesize microbubbles1 and microspheres.2 However, the use of vegetable-based proteins as shell materials to encapsulate a liquid or air core has not been widely studied so far. Pea protein is a potential candidate that can be used to encapsulate valuable materials in processed foods and dairy products. The core materials can be nutrients, flavors or drugs. There is a growing interest in using pea (Pisum sativum L.) as an important source of vegetable protein and it is a promising alternative to soybean protein. The increase in acceptance of pea proteins is due to its high-protein value (20–30%),6 good functional properties,7,8 biological activity,9,10 many other qualities,11,12 and relatively lower cost.Generally, pea proteins are composed of water-soluble albumins (14–42%)13–15 and predominant water-insoluble globulins (75–80%),16 including legumin, vicilin and convicilin. The Pisum sativum albumin proteins (PA) consist of two major ingredients, PA1 and PA2. PA1 is composed of two dimeric proteins, ranging from 8 to 11 kDa. PA2 consists of two isomers of ∼53 kDa and ∼48 kDa.16 Both PA1 and PA2 have higher proportions of sulphur-containing amino acids (cysteine and methionine) than those of pea globulins, suggesting greater potential for nutrient delivery.13,17 In particular, the high content of cysteine in PA offers the possibility of manipulating crosslinking ability in order to control the shell properties of microspheres. Based on such a crosslinking mechanism, several microstructures like lysozyme microspheres2 and bovine serum albumin (BSA) microbubbles1 have been synthesized.
With regard to water-insoluble globulins, legumin contains a hexamer of 330–410 kDa, held together by noncovalent bonding. Each pair of subunits is constituted of an acidic (∼40 kDa) and a basic (∼20 kDa) polypeptide joined by one disulphide bridge.18 Vicilin and convicilin are trimeric proteins in structures of 150 and 210–280 kDa, respectively. Vicilin subunits (∼50 kDa) are shown to have two potential processing sites, at which cleavage is responsible for the numerous and small fragments on SDS-PAGE.19,20 Convicilin subunits (∼70 kDa) are highly homologous with vicilin along the core of amino acid sequence, except for the N-terminus.21,22 Compared to pea albumin proteins, the globulins are deficient in sulfur-containing amino acids but rich in nitrogen-containing ones (amide, arginine and lysine). This is because of the inherent characteristics of storage proteins to provide amino acids and nitrogen to the growing seedling after hydrolysis on seed germination. The storage proteins keep inert and insoluble in aqueous media for the purpose of deposition. Due to inertness and insolubility issues, there are not many studies existing in the literature that prepared pea protein-shelled microspheres encapsulating liquid or air core, apart from spray-drying23–25 and complex coacervation.26,27
Since Osborne et al. reported that the solubility of the globulins can be adjusted by pH and ionic strength in 1989,28 researchers have tried various methods to extract them.29–32 However, some of the methods29,32 involved the use of a reducing agent (dithiothreitol) or denaturation agent (urea), which are banned in food and pharmaceutical industry. Without these agents, low working temperature and long dissolution time were employed in extraction and purification.30 Under mild conditions, the final concentration of globulins is relatively low.31 In addition to chemical denaturation, thermal denaturation was also performed in pea globulin processing. Heat treatment above the denaturation temperature commonly resulted in partial unfolding and subsequent aggregation of protein.33 Mession et al.34 reported that most of the subunits of pea globulins reassociated into soluble aggregates in high molecular weight of >700 kDa. However, this thermal denaturation was not only energy intensive but also heating-rate and protein-concentration dependent and salt sensitive.34,35
Apart from chemical and thermal denaturation, ultrasound has been used to alter protein conformation. Guzey et al. reported that high-intensity ultrasonic processing enhanced the emulsifying properties of whey protein isolates.36 Jambrak et al. demonstrated that ultrasonic treatment effectively improved the solubility, foaming property and foaming stability of whey proteins.37 Based on the effect of sonication on the physicochemical properties of water-soluble whey proteins, there is a high possibility that high-intensity ultrasonic treatment can also be used for the dissolution of water-insoluble pea globulin proteins and even the cleavage of disulphide bonds in albumin or legumin. Although the chemistry of pea proteins is complex, it is still likely for pea proteins to hold a microstructure with good stability, based on the potential of crosslinkability, surface activity and solubility.
The aim of this study was to increase the solubility of pea protein isolates in two commonly used buffer systems, phosphate-buffered saline (PBS) and Tris buffer, by using a short-time ultrasonic processing in the absence of additional reducing or denaturing agents. In the second step, pea protein-shelled microspheres filled with tetradecane were synthesized using ultrasound. Tetradecane was used as a model core material. By tuning ultrasonic parameters in the first sonication step, the size, size distribution, shell thickness, mechanical strength and yield of pea protein microspheres were controlled.
2. Experimental details
2.1 Materials
Pea protein isolates (PPI) containing 88% w/w proteins were from Pisane. Tris(hydroxymethyl)amino methane was purchased from Chem-Supply. Phosphate buffered saline tablets (PBS buffer), poly-L-lysine (0.01% solution) and β-mercaptoethanol were from Sigma-Aldrich. Hydrochloric acid (analytical reagent, 36%) was from Univar and tetradecane (olefine free; >99%) from Fluka. All other chemicals were obtained from Bio-Rad. Milli-Q water was obtained from a Millipore system (18.2 MΩ cm−1 at 25 °C).2.2 Ultrasonic treatment
Pea protein isolate (PPI) was dispersed in 20 ml Tris–HCl buffer (50 mM, pH 8) and 20 ml PBS buffer (pH 7.4) solutions separately at 0.1% w/v. The PPI/Tris and PPI/PBS suspensions were stirred at room temperature for 2 h. The suspensions were centrifuged (8000 rpm, 15 min, 20 °C). The supernatants were named T-0 min and P-0 min and kept for further synthesis and analysis as the controls. Without stirring, the PPI/Tris and PPI/PBS suspensions were obtained by sonicating at 80 W for 1, 5, 10, 20, 30 and 40 min separately by placing a horn (microtip 1.2 cm in diameter; 20 kHz Branson sonifier) halfway down the suspensions. The supernatants were prepared by the centrifugation method mentioned above and were named T-1 min, T-5 min, T-10 min, T-20 min, T-30 min and T-40 min, P-1 min, P-5 min, P-10 min, P-20 min, P-30 min and P-40 min, respectively. The sonication glass cell used was surrounded by a water cooling jacket to maintain the temperature inside the cell at room temperature.2.3 Synthesis
Tetradecane saturated with a dye (Nile red) was layered on the surface of the supernatants (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v) prior to sonication. Tetradecane-filled PPI shelled microspheres were obtained by sonicating at 160 W for 30 s by placing a horn (microtip 3 mm in diameter; 20 kHz Branson sonifier) at the oil/supernatant interface. Then the microspheres were collected and washed 3 times with Milli-Q water and stored in aqueous solutions at pH 7 in the fridge, labelled: MT-0 min, MT-1 min, MT-5 min, MT-40 min, MP-0 min, MP-1 min, MP-5 min and MP-40 min.
:20 v/v) prior to sonication. Tetradecane-filled PPI shelled microspheres were obtained by sonicating at 160 W for 30 s by placing a horn (microtip 3 mm in diameter; 20 kHz Branson sonifier) at the oil/supernatant interface. Then the microspheres were collected and washed 3 times with Milli-Q water and stored in aqueous solutions at pH 7 in the fridge, labelled: MT-0 min, MT-1 min, MT-5 min, MT-40 min, MP-0 min, MP-1 min, MP-5 min and MP-40 min.
2.4 Characterization
The average microsphere size and size distribution were evaluated by measuring over 200 microspheres per system using optical microscopic images (Olympus). Fluorescence optical microscopy (Olympus) was employed to characterize the tetradecane core of the pea protein microspheres. Nile red was used as a fluorescence probe. The morphology of the microspheres was examined by scanning electron microscopy (SEM, FEI Quanta). The average shell thickness was determined by measuring the cross section of over 5 mechanically broken microspheres (cross section of each microsphere was measured over 5 times) using SEM images.38 The hydrodynamic diameter was carried out using Zetasizer (Malvern). The refractive index used was 1.52.392.5 Solubility and encapsulation efficiency measurements
After centrifugation (Section 2.2), the precipitates were collected and washed 3 times with Milli-Q water and oven-dried at 60 °C for 24 h. The dry samples were cooled to room temperature for 3 h and weighed. The solubility percentage was calculated as the percent ratio of the weight difference between the total PPI powder used and the precipitates on the total PPI powders. The encapsulation efficiency of PPI was determined by collecting the microsphere portions of 15 samples prepared in Section 2.2 and carrying out extensive dialysis against Milli-Q water, with a microsphere suspension-to-Milli-Q water 1:10 (4 changes for 24 h at 4 °C). Then the suspensions were oven-dried at 60 °C for 24 h and cooled to room temperature and washed with acetone 3 times to remove residual tetradecane, followed by similar drying and weighing procedures described above. The encapsulation efficiency was calculated as the percent ratio of microsphere shell weight to the total PPI powders added.
2.6 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis
The PPI compositions in the supernatants and microspheres were evaluated using SDS-PAGE (Criterion TGX, Bio-Rad). Samples were prepared by mixing the PPI supernatants described in Section 2.1 1:1 with Laemmli sample buffer and reducing buffer (50 μl of β-mercaptoethanol and 950 μl of Laemmli sample buffer) separately. Denaturation process was carried out by heating the mixed solutions at 95 °C for 5 min. The mixed solutions were cooled down to room temperature and 20 μl of them were applied to each SDS-PAGE well. The microsphere portions described in Section 2.2 were processed in a similar way as the supernatants. Unstained protein standards (broad range, 6.5–200 kDa) from Bio-Rad was mixed with the reducing buffer 1:20 and 20 μl of the mixed solutions was deposited on a separate strip. Gels were run at 200 V for 42 min. Bio-Safe Coomassie stain was employed. Image Lab and Gel Doc XR+ were used for imaging and analysis of the gels.
2.7 AFM analysis
The stiffness and Young's modulus of the microspheres were measured by atomic force microscopy (AFM) (Asylum Research MFP-3D AFM in an acoustic isolation hood). The AFM measurements used a silica colloidal probe with diameters ranging from 32 to 39 μm, prepared using methods similar to those described by Ducker et al.40 AFM V-shaped MLCT cantilevers (Bruker) with cantilever spring constants ranged from 0.10 to 0.13 N m−1, measured by the thermal method of Hutter–Bechhoefer.41 The procedure for immobilization of microspheres onto glass slides was detailed in our previous paper,42 but briefly summarized here. The 35 mm glass slides used were treated by soaking in AJAX detergent for 1 h, and then in 10% w/w sodium hydroxide, washed in Milli-Q water, and dried with ultrapurified nitrogen gas. This is followed by a 20 min ozone treatment to remove any surface organic contaminants and increase the negative surface charge and hydrophobicity of the surface (UV/Ozone ProCleaner Plus, Bioforce Nanosciences). The slides are then soaked in a poly-L-lysine solution, (one drop of 0.01% poly-L-lysine solution diluted with Milli-Q water to 20 ml) for 1 hour to positively charge the surface, which will electrostatically hold the negatively charged PPI capsules at pH 7.4–8. Once the treatment is complete, the disk was centered on a plastic test tube with a 10 mm OD top, which was filled slightly above the brim with Milli-Q water holding a dispersed low concentration of microcapsules. Once the disk is placed on the tube, it is left for 12 hours at 4 °C to allow the capsules to rise to the surface, and adhere strongly to the surface.The diameter of immobilized individual microspheres (3–5 μm) was measured using inverted microscope (40× objective, Nikon Eclipse Ti-U). The AFM indentation experiments were conducted in a fluid cell with Milli-Q water and all force–distance curves were recorded at velocities of 100 nm s−1, with a maximum indentation force of approximately 10 nN. The experiment was repeated 10 times for each microsphere to evaluate the measurement reproducibility. No size variation of the microspheres was observed using the inverted microscope before and after the experiments. For each sample, 5 to 10 different individual microspheres were positioned on the glass substrates and the indentation experiment was carried out on each of them. Due to adhesion observations between the silica particle and microspheres observed in the retraction portion of the force–distance curve, only the approach portion of the indentation curve was analyzed to obtain the Young's modulus and elastic properties of the microspheres by using Reissner's theory.43,44
3. Results and discussion
As mentioned in the Introduction section, the main purpose of this study was to process PPI to increase its solubility in aqueous medium for the purpose of using it as a shell material for synthesizing core–shell microspheres.3.1 Solubility of PPI
Solubility in aqueous solution is a good index of whether the protein would be utilized or not by the ultrasonic encapsulation process to form a cross-linked shell around the encapsulated liquid. Solubility also indicates the availability of reactive hydrophilic groups to form a shell. It was evaluated by taking a ratio between the weight difference between the total PPI powder and the precipitates and the weight of total PPI powder used. The solubility was less than 40% in both buffer systems without ultrasonic treatment (Fig. 1). It increased to 51% in PBS buffer and 66% in Tris buffer when the sonication time increased to 5 min. Further increase in sonication time (40 min) caused a continuous increase in solubility, approaching a maximum percentage of 85% in PBS buffer and 94% in Tris buffer solutions. It is likely that high-intensity ultrasound promotes protein solubility by altering protein conformation and structure. The treatment may lead to an increase in hydrophilicity of the protein molecules.45–47 Another possibility is that ultrasonic irradiation may change the three-dimensional structures of globulins leading to an increase in the number of charged groups (NH4+, CH3COO−).37 The solubility of PPI in the pH range 7.4–8 is very similar.48 Therefore, the difference in solubility in PBS buffer and Tris buffer systems is presumably stemmed from ionic strength of each system.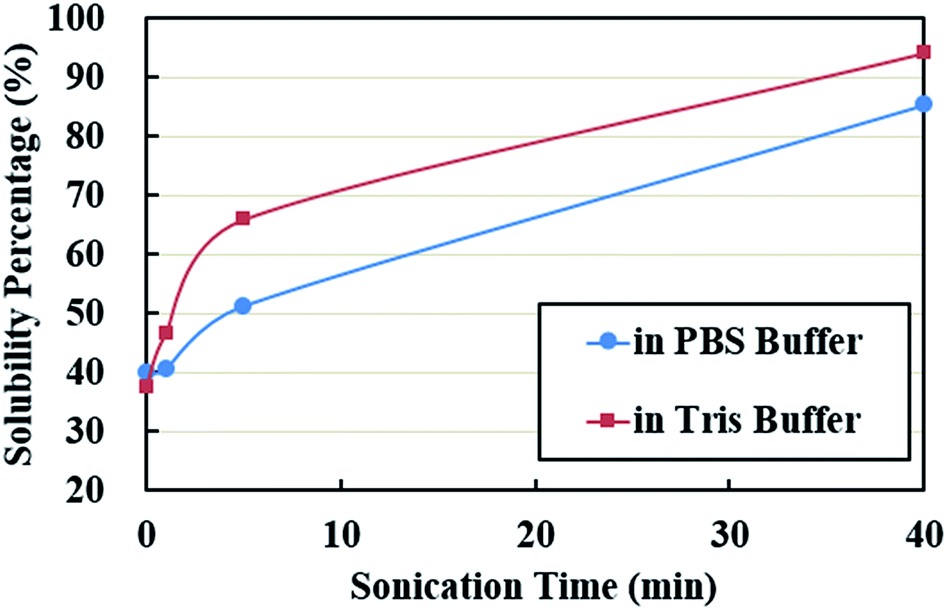 | ||
| Fig. 1 Solubility of the PPI suspensions in PBS buffer and Tris buffer solutions. | ||
3.2 Hydrodynamic diameter
The hydrodynamic diameter of PPI by number density is shown in Fig. 2 as a function of ultrasonic treatment time. The average hydrodynamic diameter in the supernatants, i.e., P-0 min and T-0 min, were about 5 and 9 nm, respectively, which are in the range of the hydrodynamic diameter of individual protein molecules.49 The hydrodynamic diameter increased up to 93 nm in PBS buffer and 44 nm in Tris buffer systems after 1 min treatment, followed by a gradual decrease as a function of sonication time. | ||
| Fig. 2 PPI hydrodynamic diameter by number in PBS buffer (A) and Tris buffer supernatants (B). | ||
This implies two possibilities. First, the ultrasonic denaturation may stretch the three-dimensional structures of proteins and raise the degree of asymmetry when exposing the hydrophilic parts of amino acids to aqueous systems, causing an increase in hydrodynamic diameter. The second possibility is that small proteins that are soluble are aggregating from the exposure to ultrasound. A similar observation was reported in thermal denaturation of PPI.34 In our experiment, the hydrodynamic diameter was higher in PBS buffer system than in Tris buffer system. This could be due to the higher ionic strength of PBS buffer solution, which leads to the nonspecific charge-shielding effect between charged groups of proteins. The ions enhance intramolecular interactions, hence enhancing salting-out effect and aggregation.50
Considering the results of solubility and hydrodynamic diameter, at short sonication times (0–5 min), the hydrodynamic diameter increased with a simultaneous increase in solubility of the protein. However, at long sonication times (5–40 min) there was a decrease in hydrodynamic diameter even though the solubility increased. To further understand the effect of sonication, SDS-PAGE analysis was carried out.
3.3 Effect of ultrasonic denaturation on protein composition
Protein composition in the supernatants was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as shown in Fig. 3. In order to investigate the disaggregation and crosslinkage of disulphide bonds in legumin and pea albumin (PA) as a function of sonication time, each sample was treated under non-reducing conditions and reducing conditions with β-mercaptoethanol, separately. The non-denatured samples (P-0 min and T-0 min) in lanes 9 and 13 shown in Fig. 3 are mainly composed of convicilin subunits (∼70 kDa), traces of legumin L main subunits (∼60 kDa), pea albumin PA2 (53–48 kDa), dissociated vicilin fragments α:β or β:γ (30–36 kDa or 25–30 kDa), vicilin fragments β or γ (13 kDa or 12–16 kDa), and pea albumin PA1 (8–11 kDa). This was due to water-soluble property of PA and good emulsifying capacity of vicilin in comparison to legumin.51,52 The samples after 1 min treatment (T-1 min and P-1 min in lanes 10 and 14, respectively) showed a similar protein composition and band intensity with the non-denaturing ones, in agreement with the solubility measurements discussed above. With an increase in sonication time, polymerized proteins with high molecular weight (>200 kDa) were observed at the top of lanes 10, 11, 12, 15 and 16 in Fig. 3, highlighted by red dotted circles. The diffusive and unresolved bands of very low electrophoretic mobility indicated protein denaturation,53,54 mainly caused by noncovalent interactions,53 including hydrophobic interaction55–57 and minor hydrogen bonding.58 With 5 min and 40 min treatments, band intensity of water-insoluble pea globulins increased. The supernatants were comprised of denatured proteins (>200 kDa), lipoxygenase Lip (∼89 kDa),59 convicilin monomer (∼70 kDa), legumin L main subunits (∼60 kDa), dissociated vicilin polypeptides, and PA. Legumin L main subunits were disrupted in the presence of β-mercaptoethanol into acidic Lα (38–40 kDa) and basic Lβ (20–22 kDa) fragments, initially bridged via disulphide bonds (indicated by solid circles in Fig. 3). The double bands at ∼40 kDa were presumably ascribed to the heterogeneity of legumin.29,30 Some acidic Lα and basic Lβ fragments were observed with long-time ultrasonic treatment without addition of reducing agent, shown by white half-filled circles in Fig. 3. This implies that legumin L main subunits were partially disrupted by ultrasonic irradiation and sulfhydryl groups were formed. A weak band at ∼40 kDa was observed in the samples with non- and short-time denaturation treatment (lane 9, 10, 13 and 14) under non-reducing conditions. This impurity was possibly unprocessed legumin subunits, dissociated with Lβin vivo.60,61 Finally, an intensive band was observed at the bottom of each lane, ascribing to the small fragments of PA and other denatured polypeptides of low MW (<6 kDa). | ||
| Fig. 3 SDS-PAGE profile of PPI in PBS buffer and Tris buffer supernatants as a function of sonication time. The standards (STD) were deposited in the first strip. Lanes applied by proteins in PBS buffer systems are labelled in normal font and lanes in Tris buffer systems in italic font. | ||
The water-insoluble PPI possessed relatively high solubility after short ultrasonic treatment in both buffer systems. The high solubility facilitates material delivery and shell formation during encapsulation of oil, which is of importance for the next step of microsphere preparation.
3.4 Size and size distribution of pea protein isolate (PPI) microspheres
In order to further study the effect of ultrasonic denaturation on the physical and functional properties of PPI in the two buffer systems, tetradecane-filled PPI microspheres were synthesized as a function of sonication pretreatment time. Nile red was dissolved in tetradecane as a fluorescent probe to demonstrate the core formation within the PPI microspheres. The optical and fluorescence microscopic images of the microspheres (MT-1 min) are shown in Fig. 4. The optical microscopic images of the eight samples are shown in Fig. 5. | ||
| Fig. 4 Optical (A) and fluorescence (B) microscopic images of tetradecane (labelled with Nile red) filled PPI microspheres (MT-1 min). Scale: the images reported have a size of 110 μm × 147 μm. | ||
 | ||
| Fig. 5 Comparison of PPI microspheres obtained within different buffer solutions at different sonication times ((A) MP-0 min, (B) MP-1 min, (C) MP-5 min, (D) MP-40 min, (E) MT-0 min, (F) MT-1 min, (G) MT-5 min, (H) MT-40 min). Scale: all images displayed have a size of 110 μm × 147 μm. | ||
In the PBS buffer solutions, the microspheres of MP-0 min, had an average size of 2.9 ± 1.0 μm (Table 1). When the ultrasonic denaturation was introduced for 1 min, the mean size of the microspheres (MP-1 min) increased to 4.1 ± 2.2 μm. Further increase in sonication time resulted in a decrease in microsphere size, with a minimum size of 3.0 ± 1.3 μm observed at 40 min. In Tris buffer system, the microsphere size stayed almost constant around 2.9 ± 1.0 μm. The microsphere size distributions are summarized in Fig. 6. There exists a primary peak ranging from 2 to 4 μm and a tail up to 9 μm in the size distribution in PBS buffer systems and a relatively mono-modal distribution from 2 to 3 μm in Tris buffer solutions. The observed trend will be discussed in the following section. Before discussion the size distribution data, it is important to consider the shell thickness of the microspheres.
| Sample label | Average microsphere diameter (μm) (1 week) | Average microsphere diameter (μm) (4 week) | Average shell thickness (nm) (1 week) | Average shell thickness (nm) (3 week) | Average oil core diameter (μm) (1 week) | Average stiffness (mN m−1) (1 week) | Average modulus ES (MPa) (1 week) |
|---|---|---|---|---|---|---|---|
| MP-0 min | 2.9 ± 1.0 | 3.9 ± 1.5 | 77 ± 9 | — | 2.7 ± 1.0 | 9.5 ± 6.0 | 1.20 ± 0.74 |
| MP-1 min | 4.1 ± 2.2 | 4.3 ± 1.6 | 131 ± 21 | 107 ± 6 | 3.8 ± 2.2 | 22.0 ± 8.1 | 1.02 ± 0.37 |
| MP-5 min | 3.5 ± 2.2 | 3.9 ± 1.6 | 117 ± 12 | — | 3.3 ± 2.2 | 10.8 ± 3.4 | 0.58 ± 0.18 |
| MP-10 min | 3.3 ± 1.2 | — | — | — | — | — | — |
| MP-20 min | 3.2 ± 1.2 | — | — | — | — | — | — |
| MP-30 min | 3.1 ± 1.2 | — | — | — | — | — | — |
| MP-40 min | 3.0 ± 1.3 | 3.2 ± 1.1 | 127 ± 10 | 130 ± 20 | 2.7 ± 1.3 | 13.5 ± 2.2 | 0.62 ± 0.11 |
| MT-0 min | 2.9 ± 1.0 | 2.8 ± 0.8 | 59 ± 8 | — | 2.8 ± 1.0 | 11.2 ± 3.1 | 2.35 ± 0.61 |
| MT-1 min | 2.9 ± 1.3 | 3.4 ± 1.6 | 108 ± 5 | 100 ± 11 | 2.7 ± 1.3 | 18.2 ± 7.8 | 1.19 ± 0.55 |
| MT-5 min | 2.7 ± 0.9 | 3.3 ± 1.2 | 61 ± 6 | — | 2.6 ± 0.9 | 9.9 ± 1.5 | 2.00 ± 0.30 |
| MT-10 min | 3.1 ± 1.1 | — | — | — | — | — | — |
| MT-20 min | 2.9 ± 1.1 | — | — | — | — | — | — |
| MT-30 min | 2.7 ± 1.1 | — | — | — | — | — | — |
| MT-40 min | 2.9 ± 0.9 | 2.9 ± 1.0 | 78 ± 3 | 80 ± 3 | 2.7 ± 0.9 | 14.2 ± 2.3 | 1.70 ± 0.32 |
 | ||
| Fig. 6 Size distributions of tetradecane filled PPI microspheres prepared in PBS buffer (A) and Tris buffer solutions (B). | ||
3.5 Morphology and shell thickness of microspheres
The shell thickness was measured using SEM images by mechanically breaking the microspheres. SEM images (Fig. 7) of broken microspheres was for this purpose.38 The surface morphology is shown with hollow interiors and thin shells ranging from 60 to 130 nm. Most of the microspheres were devoid of surface features and the surface of sample MP-40 min seemed to possess a larger degree of nanoscale structure. In PBS buffer systems, the shell thickness increased from about 77 nm in non-denatured samples to 131 nm after 1 min treatment and it was about 120 nm with further increase in sonication time. The shell thickness in Tris buffer solutions was less than that in PBS buffer. However, the trend observed (as a function of sonication treatment) is similar in both buffer solutions. The core diameters are calculated by subtracting 2 times shell thickness (h) from the diameter of the microspheres (D), in Table 1. It can be seen that the core size trend followed the microsphere size trend. | ||
| Fig. 7 SEM images of mechanically broken tetradecane-filled PPI microspheres ((A) MP-1 min, (B) MP-40 min, (C) MT-1 min, (D) MT-40 min). | ||
Solution viscosity will increase with the continuous dissolution of macromolecules, which leads to a decrease in the strength of shear forces generated by acoustic cavitation due to large power dissipation. Because of the lack of efficient shear forces, larger microspheres are expected to form. As shown in Table 1, there is only slight increase in average microsphere diameter at short-time treatment, indicating similar levels of viscosity. On the other hand, ultrasonic treatment improved the solubility and hence an increase in shell thickness was observed with sonication time. Compared with PBS buffer systems, Tris buffer system has lower ionic strength, leading to less aggregation, smaller hydrodynamic diameter of proteins, and thinner shell thickness of microspheres.
3.6 Encapsulation efficiency and protein shell composition
Encapsulation efficiency was examined as the weight percent ratio of the microsphere portions collected from the suspensions on the total PPI powder added. The encapsulation efficiency of the shell material is shown as a function of sonication time in Fig. 8. Due to low solubility of proteins in non-denatured samples, the corresponding encapsulation efficiency was about 23% in both buffer solutions. With an increase in solubility, the encapsulation efficiency at 5 min treatment became twice higher than that of the non-denatured samples in Tris buffer systems, about 57%. The final encapsulation efficiency at 40 min was up to 78% in PBS buffer and 88% in Tris buffer solutions. | ||
| Fig. 8 Encapsulation efficiency of the PPI microspheres in PBS buffer and Tris buffer solutions. | ||
The composition of microsphere shell as a function of sonication treatment time is given in Fig. 9. Considering that the concentrations of microspheres used were unknown quantitative comparison of the band intensities is difficult. Large amount of denatured proteins with various MW were involved in the shell formation, reflected by the high intensity of bands spreading over lanes 4, 7, 8, 11, 12, 15, and 16. These proteins did not dissociate in the presence of SDS and β-mercaptoethanol showing the possibility of noncovalent interactions (hydrophobic bonding and hydrogen bonding). The band around 24–25 kDa in the reduced microsphere shells in lanes 4, 7, and 8 is attributed to the subunits of cysteine-rich pea albumin PA2, highlighted by solid circles in Fig. 9. Theoretically, in the absence of β-mercaptoethanol, a band at 48–53 kDa should be observed in corresponding non-reduced samples due to uncleaved PA2. However, only a weak band is found in lanes 15 and 16, shown by white half-filled circles. It implies that most of the PA2 subunits were crosslinked with polypeptides containing free sulfhydryl groups such as PA fragments, legumin acidic Lα and basic Lβ fragments. Relatively large protein aggregates were generated during the shell formation, indicated by red dotted circles. The intensive bands at the bottom of each lane show that the protein fragments of low MW observed in the supernatants by SDS-PAGE probably took part in the shell formation as well. The disulphide bond crosslinking and noncovalent strong interactions may play a role in controlling the stability and mechanical strength of the microspheres prepared using solutions sonicated for longer time (5–40 min).
 | ||
| Fig. 9 SDS-PAGE profile of the PPI microspheres in PBS buffer and Tris buffer systems with sonication time. The standards (STD) were deposited in the first lane. Lanes applied by proteins in PBS buffer systems are labelled in normal font and lanes in Tris buffer systems in italic font. | ||
3.7 Stability of PPI microspheres
The storage stability of the PPI microspheres in two buffer systems was monitored for 1 month. The size variation of microspheres with different sonication times is shown in Fig. 10 and 11(a) and Table 1. | ||
| Fig. 10 Cross-linked PPI microsphere size and size distribution as a function of storage time and sonication time, in PBS buffer (A) and Tris buffer systems (B). | ||
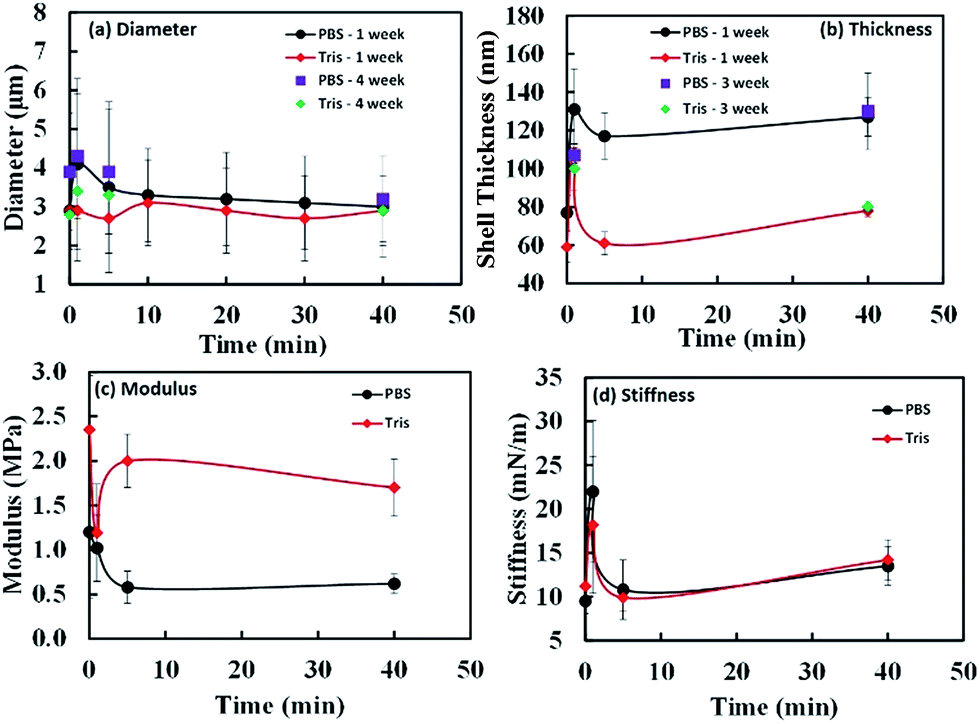 | ||
| Fig. 11 The effect of sonication times on diameter (a), shell thickness (b), modulus (c) and shell stiffness (d) in PBS and Tris buffers. The data for (c) and (d) are obtained using the data presented in Fig. 12. | ||
The oil core remained intact and no leakage was noticed. After 1 month, the mean size of the samples fluctuated in a small range (≤1 μm) without any trend within experimental errors. We have also plotted the comparison of diameters and thickness for fresh and stored samples in Fig. 11(a) and (b). The observed diameters for weeks 1 and 4 samples do not change significantly within experimental error limits. The data shown in Fig. 11(a) clearly indicates that there is not much difference between the two data sets. As observed from Fig. 11(b) and Table 1, except for thickness of shell for MP-1 min sample, all other measured samples did not show much variation of thickness over 1 month period. The microspheres prepared with long-time ultrasonic denaturation exhibited greater storage stability than those with short-time treatment. We found that when sonication time is longer than 1 minute, the microspheres were stable for at least 5 months when stored at 4 °C.
3.8 AFM indentation experiments on individual PPI microspheres
Stiffness and Young's modulus of individual microspheres were measured by AFM indentation experiments. The stiffness of a microsphere is described as the capsule's resistance to deformation when a force is applied. Typical force–indentation curves obtained by AFM on ultrasonically synthesized PPI microspheres (∼4 μm) are shown in Fig. 12 for varying sonication times. The nearly vertical black filled circles represent force–indentation curves in case that a given load is applied to a rigid glass surface so that no indentation can be detected. However, indentation increases linearly with applied force when deformable microspheres are compressed in a small deformation regime (indentation ∼ crossover deformation εcrossover).43,44,62,63 In this regime, the microspheres show elastic response with no instabilities (buckling and inflection of shell). The applied load versus indentation curve is linear and the slope of the force curves is proportional to the stiffness of the microsphere given the relation:43,44,62,63| F = kshellδ | (1) |
 | (2) |
 | (3) |
 | ||
Fig. 12 Force–indentation curves of microspheres prepared at various sonication times ((a) MP-0 min, (e) MT-0 min, (b) MP-1 min, (f) MT-1 min, (c) MP-5 min, (g) MT-5 min, (d) MP-40 min, and (h) MT-40 min). The black closed circles are the force–indentation curves against a rigid glass reference substrate. The dashed vertical lines represent the crossover deformation 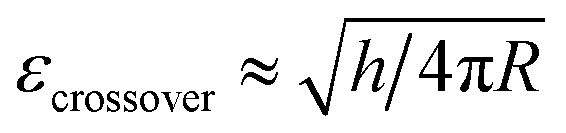 . The four-coloured curves in each graph represent consecutive measurements on the same sample to verify the reproducibility of the experiments (red curves: run 1, pink curves: run 2, blue curves: run 3 and green curves: run 4). . The four-coloured curves in each graph represent consecutive measurements on the same sample to verify the reproducibility of the experiments (red curves: run 1, pink curves: run 2, blue curves: run 3 and green curves: run 4). | ||
In Table 1 and Fig. 11, we show the average stiffness and Young's modulus of microspheres with different sonication times. The average stiffness of microspheres changed from 9.5 to 22.0 mN m−1 and the Young's modulus (ES) from 0.58 to 2.35 MPa. According to the eqn (2), the Young's modulus is inversely proportional to quadratic power of shell thickness so that the largest shell thickness has the lowest modulus. The Young's modulus can be related to the extent of crosslinking and non-covalent interactions of the shell molecules. Control experiments were performed with microspheres made of non-denatured proteins (MP-0 min and MT-0 min) that exhibited highest Es in each buffer system. The Young's modulus increased from 1.20 to 2.35 MPa when the buffer system was varied from PBS to Tris (a decrease in ionic strength). The image analysis of Fig. 9 indicates that various vicilin fragments and aggregates with high MW dominated the shell composition of MP-0 min (∼47% and ∼33% band intensity, respectively), while MT-0 min was mainly composed of aggregates with high MW (∼40%) and cysteine-rich PA fragments (∼27%). It suggests that proteins that initially existed in their inherent structure may exhibit higher mechanical strength due to their compact molecular state. And the ionic strength of system can significantly affect the preference of shell composition. In comparison to vicilin with good emulsifying capacity, aggregates made of PA fragments show greater crosslinkability and hence the shell material MT-0 min shows higher Young's modulus.
In terms of the ultrasonically denatured proteins, the Young's modulus decreased with increasing sonication time in PBS buffer system (Fig. 11(c)). This could be due to less compact protein structure after ultrasonic denaturation. Longer sonication time may counter against crosslinking between PA, Lα and Lβ fragments, caused by the nonspecific charge-shielding effect in PBS buffer system. In comparison, the microspheres prepared in Tris buffer system show relatively high Young's modulus and a different trend. For MT-5 min, an appropriate level of ultrasonic denaturation of PPI may have resulted in its low surface activity (the thin shell thickness ∼ 61 nm) but high solubility (∼66%). It could be due to high inter- and intrachain repulsion caused by a relatively large number of charged groups exposed (NH4+, CH3COO−). The highly charged protein mixtures exhibited the second highest Young's modulus (2 ± 0.3 MPa), presumably resulting from the noncovalent interactions and sulfhydryl group crosslinking (aggregates with high MW of band intensity ∼ 55%). Thus, ultrasonic irradiation affects the denaturation, adsorption and subsequent crosslinking of the PPI shell during treatment and synthesis in a complex manner. In general, sonication in the first step decreases the Young's modulus of the microspheres due to less compact molecular state of protein. In high ionic strength buffer system (PBS), microspheres with longer sonication time (≥5 min) tend to have smaller oil core, lower stiffness and modulus, due to low crosslinkability. In the low ionic strength buffer system (Tris) and longer sonication time (≥5 min) in the first step facilitates the synthesis of microspheres possessing thinner and more flexible shell with higher modulus because of high crosslinkability.
4. Conclusions
Ultrasonic treatment was carried out to promote the solubility of water-insoluble pea globulin proteins in PBS and Tris buffer systems. The ultrasonically treated pea proteins were used to prepare stable tetradecane-filled PPI microspheres. The size, size distribution, shell thickness, mechanical strength and yield of microspheres were controlled by altering of the ultrasonic parameters in the first step. Based on the SDS-PAGE and AFM indentation experiments, disulphide bond crosslinking and noncovalent interaction presumably affected the mechanical strength of PPI shell, leading to the generation of stable microspheres. The average stiffness of the microspheres first increased with increase in sonication time from 0 to 1 min. The stiffness decreased with further increase in sonication time (5–40 min). The shell thickness of microspheres also varied with sonication time. The strength of the microspheres can thus be tuned by sonication time. By using the two commonly used buffer solutions (PBS and Tris/HCl), this two-step method can be applied to synthesize PPI microspheres with desired size, stability and mechanical strength at an appropriate sonication time. Due to the increasing acceptance of PPI as a vegetable-based protein source, PPI microspheres could be used in encapsulation of aromas and oils in dairy and food products.Acknowledgements
Authors acknowledge financial support from the Australian Research Council (ARC-DP). Authors also acknowledge funding through ARC-ITRP grant IH120100053. AFM analysis was performed at the Materials Characterisation and Fabrication Platform (MCFP) at the University of Melbourne and the Victorian Node of the Australian National Fabrication Facility (ANFF).Notes and references
- F. Vong, Y. Son, S. Bhuiyan, M. Zhou, F. Cavalieri and M. Ashokkumar, Ultrason. Sonochem., 2014, 21, 23–28 CrossRef CAS PubMed.
- M. Zhou, T. S. H. Leong, S. Melino, F. Cavalieri, S. Kentish and M. Ashokkumar, Ultrason. Sonochem., 2010, 17, 333–337 CrossRef CAS PubMed.
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- E. Colombo, F. Cavalieri and M. Ashokkumar, ACS Appl. Mater. Interfaces, 2015, 7, 12972–12980 CAS.
- S. Freiberg and X. Zhu, Int. J. Pharm., 2004, 282, 1–18 CrossRef CAS PubMed.
- R. E. Aluko, O. A. Mofolasayo and B. M. Watts, J. Agric. Food Chem., 2009, 57, 9793–9800 CrossRef CAS PubMed.
- K. Maninder, K. S. Sandhu and N. Singh, Food Chem., 2007, 104, 259–267 CrossRef CAS.
- H. Koyoro and J. Powers, Cereal Chem., 1987, 64, 97 CAS.
- F. Roy, J. Boye and B. Simpson, Food Res. Int., 2010, 43, 432–442 CrossRef CAS.
- F. Gressent, I. Rahioui and Y. Rahbe, Eur. J. Biochem., 2003, 270, 2429–2435 CrossRef CAS PubMed.
- J. Bacon, N. Lambert, M. Phalp, G. Plumb and D. Wright, Anal. Biochem., 1987, 160, 202–210 CrossRef CAS PubMed.
- J. A. Gatehouse, R. R. Croy, H. Morton, M. Tyler and D. Boulter, Eur. J. Biochem., 1981, 118, 627–633 CrossRef CAS PubMed.
- D. R. Davies, Euphytica, 1976, 25, 717–724 CrossRef CAS.
- D. Grant, A. Sumner and J. Johnson, Can. Inst. Food Technol. J., 1976, 9, 84–91 CrossRef CAS.
- D. R. Murray, Plant, Cell Environ., 1979, 2, 221–226 CrossRef.
- R. R. Croy, M. S. Hoque, J. A. Gatehouse and D. Boulter, Biochem. J., 1984, 218, 795–803 CrossRef CAS PubMed.
- D. Boulter, Proc. Nutr. Soc., 1982, 41, 1–6 CrossRef.
- R. Croy, E. Derbyshire, T. Krishna and D. Boulter, New Phytol., 1979, 83, 29–35 CrossRef CAS.
- D. Spencer, P. M. Chandler, T. J. Higgins, A. S. Inglis and M. Rubira, Plant Mol. Biol., 1983, 2, 259–267 CrossRef CAS PubMed.
- J. A. Gatehouse, G. Lycett, R. Croy and D. Boulter, Biochem. J., 1982, 207, 629–632 CrossRef CAS PubMed.
- D. Bown, T. N. Ellis and J. Gatehouse, Biochem. J., 1988, 251, 717–726 CrossRef CAS PubMed.
- E. J. Newbigin, P. M. Chandler, A. Gould, R. J. Blagrove, J. F. March, A. A. Kortt and T. J. Higgins, Planta, 1990, 180, 461–470 CrossRef CAS PubMed.
- L. L. de Azevedo Bittencourt, C. Pedrosa, V. P. de Sousa, A. P. T. Pierucci and M. Citelli, Plant Foods Hum. Nutr., 2013, 68, 333–339 CrossRef PubMed.
- A. P. T. Pierucci, L. R. Andrade, E. B. Baptista, N. M. Volpato and M. H. M. Rocha-Leão, J. Microencapsulation, 2006, 23, 654–662 CrossRef CAS PubMed.
- A. P. T. Pierucci, L. R. Andrade, M. Farina, C. Pedrosa and M. H. M. Rocha-Leão, J. Microencapsulation, 2007, 24, 201–213 CrossRef CAS PubMed.
- V. Ducel, J. Richard, Y. Popineau and F. Boury, Biomacromolecules, 2004, 5, 2088–2093 CrossRef CAS PubMed.
- V. Ducel, J. Richard, P. Saulnier, Y. Popineau and F. Boury, Colloids Surf., A, 2004, 232, 239–247 CrossRef CAS.
- T. B. Osborne and G. F. Campbell, J. Am. Chem. Soc., 1898, 29, 348 CrossRef.
- R. Casey, Biochem. J., 1979, 177, 509–520 CrossRef CAS PubMed.
- J. A. Gatehouse, R. R. Croy and D. Boulter, Biochem. J., 1980, 185, 497–503 CrossRef CAS PubMed.
- F. E. O'Kane, R. P. Happe, J. M. Vereijken, H. Gruppen and M. A. van Boekel, J. Agric. Food Chem., 2004, 52, 3141–3148 CrossRef PubMed.
- H. Hirano, J. A. Gatehouse and D. Boulter, FEBS Lett., 1982, 145, 99–102 CrossRef CAS.
- J.-M. Wang, N. Xia, X.-Q. Yang, S.-W. Yin, J.-R. Qi, X.-T. He, D.-B. Yuan and L.-J. Wang, J. Agric. Food Chem., 2012, 60, 3302–3310 CrossRef CAS PubMed.
- J.-L. Mession, N. Sok, A. Assifaoui and R. M. Saurel, J. Agric. Food Chem., 2013, 61, 1196–1204 CrossRef CAS PubMed.
- Z. Cui, Y. Chen, X. Kong, C. Zhang and Y. Hua, J. Agric. Food Chem., 2014, 62, 1634–1642 CrossRef CAS PubMed.
- D. Guzey and J. Weiss, High-intensity ultrasonic processing improves emulsifying properties of proteins, Colloidal and Interfacial Food Science Laboratory, Department of Food Science and Technology, The University of Tennessee, 2001, vol. 2605, pp. 37996–31071 Search PubMed.
- A. R. Jambrak, T. J. Mason, V. Lelas, Z. Herceg and I. L. Herceg, J. Food Eng., 2008, 86, 281–287 CrossRef CAS.
- M. Zhou, F. Cavalieri and M. Ashokkumar, Soft Matter, 2011, 7, 623–630 RSC.
- A. Nesterenko, I. Alric, F. Silvestre and V. Durrieu, Food Res. Int., 2012, 48, 387–396 CrossRef CAS.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Nature, 1991, 353, 239–241 CrossRef CAS.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS.
- S. Mettu, M. Zhou, B. L. Tardy, M. Ashokkumar and R. R. Dagastine, Polymer, 2016, 102, 333–341 CrossRef CAS.
- E. Reissner, J. Math. Phys., 1946, 25, 279–300 CrossRef.
- E. Reissner, J. Math. Phys., 1946, 25, 80–85 CrossRef.
- M.-H. Morel, P. Dehlon, J. Autran, J. Leygue and C. Bar-L'Helgouac'h, Cereal Chem., 2000, 77, 685–691 CrossRef CAS.
- K. Moulton and L. Wang, J. Food Sci., 1982, 47, 1127–1129 CrossRef.
- L. Wang, J. Food Sci., 1975, 40, 549–551 CrossRef CAS.
- H.-N. Liang and C.-H. Tang, Food Hydrocolloids, 2013, 33, 309–319 CrossRef CAS.
- H. P. Erickson, Biol. Proced. Online, 2009, 11, 32 CrossRef CAS PubMed.
- S.-M. Choi and C.-Y. Ma, J. Agric. Food Chem., 2005, 53, 8046–8053 CrossRef CAS PubMed.
- C. Dagorn-Scaviner, J. Gueguen and J. Lefebvre, J. Food Sci., 1987, 52, 335–341 CrossRef CAS.
- A. Rangel, G. B. Domont, C. Pedrosa and S. T. Ferreira, J. Agric. Food Chem., 2003, 51, 5792–5797 CrossRef CAS PubMed.
- X. Li, Y. Li, Y. Hua, A. Qiu, C. Yang and S. Cui, Food Chem., 2007, 104, 1410–1417 CrossRef CAS.
- P. Shand, H. Ya, Z. Pietrasik and P. Wanasundara, Food Chem., 2007, 102, 1119–1130 CrossRef CAS.
- F. E. O'Kane, R. P. Happe, J. M. Vereijken, H. Gruppen and M. A. van Boekel, J. Agric. Food Chem., 2004, 52, 5071–5078 CrossRef PubMed.
- F. E. O'Kane, J. M. Vereijken, H. Gruppen and M. A. Boekel, J. Food Sci., 2005, 70, C132–C137 CrossRef.
- X. D. Sun and S. D. Arntfield, Food Hydrocolloids, 2012, 28, 325–332 CrossRef CAS.
- M. R. G. Tandang-Silvas, T. Fukuda, C. Fukuda, K. Prak, C. Cabanos, A. Kimura, T. Itoh, B. Mikami, S. Utsumi and N. Maruyama, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1432–1442 CrossRef CAS PubMed.
- X. D. Sun and S. D. Arntfield, Food Res. Int., 2010, 43, 509–515 CrossRef CAS.
- M. F. Marcone, Y. Kakuda and R. Y. Yada, Food Chem., 1998, 62, 27–47 CrossRef CAS.
- E. N. Tzitzikas, J.-P. Vincken, J. de Groot, H. Gruppen and R. G. Visser, J. Agric. Food Chem., 2006, 54, 425–433 CrossRef CAS PubMed.
- A. Fery, F. Dubreuil and H. Möhwald, New J. Phys., 2004, 6, 18 CrossRef.
- M. P. Neubauer, M. Poehlmann and A. Fery, Adv. Colloid Interface Sci., 2014, 207, 65–80 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |