
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Room temperature synthesis of CuInS2 nanocrystals†
Christine
Buchmaier
a,
Thomas
Rath
 *a,
Franz
Pirolt
a,
Astrid-Caroline
Knall
a,
Petra
Kaschnitz
a,
Otto
Glatter
a,
Karin
Wewerka
b,
Ferdinand
Hofer
b,
Birgit
Kunert
c,
Kurt
Krenn
d and
Gregor
Trimmel
a
*a,
Franz
Pirolt
a,
Astrid-Caroline
Knall
a,
Petra
Kaschnitz
a,
Otto
Glatter
a,
Karin
Wewerka
b,
Ferdinand
Hofer
b,
Birgit
Kunert
c,
Kurt
Krenn
d and
Gregor
Trimmel
a
aInstitute for Chemistry and Technology of Materials (ICTM), NAWI Graz, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: thomas.rath@tugraz.at
bInstitute for Electron Microscopy and Nanoanalysis, Graz University of Technology & Centre for Electron Microscopy Graz, Steyrergasse 17, 8010 Graz, Austria
cInstitute of Solid State Physics, Graz University of Technology, Petersgasse 16, 8010 Graz, Austria
dInstitute of Earth Sciences, University of Graz, Universitätsplatz 2, 8010 Graz, Austria
First published on 28th October 2016
Abstract
Herein, we investigate a synthetic approach to prepare copper indium sulfide nanocrystals at room temperature. The nanocrystals have a chalcopyrite crystal structure, a diameter of approximately 3 nm and are well soluble in organic solvents like toluene or chloroform. The synthesis is performed by dissolving metal xanthates as precursors together with oleylamine in toluene followed by stirring for several hours at room temperature leading to nanocrystals stabilized with oleylamine ligands. The nanoparticles are characterized in terms of inner structure by X-ray diffraction, transmission electron microscopy, Raman-, absorption- and photoluminescence spectroscopy. Their formation process is investigated by small angle X-ray scattering, UV-Vis absorption spectroscopy and NMR spectroscopy. The formation of the copper indium sulfide nanocrystals proceeds via a chemical reaction of the amine with the thiocarbonyl functionality of the xanthate. The presented method exemplifies a synthesis strategy, which can be easily expanded to other metal sulfide nanocrystals.
Introduction
Metal chalcogenide nanocrystals and nanomaterials have attracted a lot of attention during the last decade because of their interesting optical and electrical properties due to quantum confinement effects based on variations in size and shape and due to chemical tuning of the chemical composition and the ligand environment. The progress in synthetic routes, their properties as well as possible applications are discussed in several reviews (e.g.ref. 1–6).Copper indium sulfide (CuInS2) nanocrystals are an interesting alternative to cadmium or lead based II–VI materials. Copper indium sulfide is a non-toxic and environmentally stable material and has a high absorption coefficient and a direct band gap of 1.5 eV.7 Furthermore, its luminescent properties and a large Stokes shift make this semiconductor interesting for various applications.8 Therefore, CuInS2 nanocrystals find applications in numerous fields9 including bioimaging,10 photocatalysis,11 light emitting diodes12 and, in particular, solar energy conversion devices like semiconductor sensitized mesoscopic solar cells,13–15 quantum dot solar cells,16 as well as hybrid bulk heterojunction polymer/nanoparticle solar cells.17–19
For the synthesis of copper indium sulfide nanoparticles, numerous routes are reported, whereby a majority of them can be referred to as colloidal synthesis routes (heat up and hot injection syntheses), solvothermal routes as well as in situ syntheses in a polymeric matrix material.9,20–22 Almost all of these routes involve heat treatment at a certain stage in the synthetic procedure.
However, some protocols for room temperature synthesis have been also already developed, which is particularly interesting in terms of energy-efficiency and sustainability. In some of them, sulfur sources like hexamethyldisilathiane, which react at room temperature with the copper and indium salts, are used.23,24 In others, the energy needed for the reaction is introduced by UV-light. Examples for this are the synthesis of CuInS2 nanocrystals at room temperature by Nairn et al.,25 who prepared CuInS2 nanocrystals in an organic solvent by photochemical decomposition of a single-source metal–thiolate precursor, or the formation of nanocrystalline CuInS2 layers by UV-irradiation of a mixed copper and indium xanthate film under reduced pressure.26 In contrast to that, the formation of CuInS2 using xanthate precursors usually proceeds via a thermal decomposition at elevated temperatures. Examples are the preparation of CuInS2 nanoparticles from the corresponding copper and indium ethyl xanthates at 196 °C using glycol as solvent27 or at 210 °C using trioctylamine as solvent and oleylamine/trioctylphosphine as ligands.28 Alternatively, a mixture of Cu and In xanthates (named as CIX) were decomposed at a temperature of 110 °C using o-dichlorobenzene as solvent and oleylamine/trioctylphosphine as ligands29 or, in comparison to that, poly(3-hexylthiophene) as polymeric ligand.30 In this latter paper, the conversion to CuInS2 takes place in the presence of the polymer in solution. Alternatively, the precursors can be directly converted to metal sulfides in a thin film of a conjugated polymer, as we showed using dimethylpentyl xanthates of copper and indium for the fabrication of CuInS2–polymer hybrid solar cells.17,18 However, this approach requires temperatures above 160 °C, usually 200 °C. Recently, we have demonstrated that the addition of a small amount of hexylamine can reduce the conversion temperature in this approach to 140 °C.31 In this case, hexylamine induces the partial decomposition of the metal xanthates via a direct chemical reaction forming the corresponding thiocarbamate compound, whereas supposedly most of the metal xanthate still reacts via the thermally induced Chugaev reaction, i.e. the xanthate decomposes to a volatile alkene, COS and the metal sulfide. The Chugaev mechanism was also postulated for the overall reaction of the corresponding O-hexadecyl metal xanthates with long chain alkyl amines giving metal sulfide nanoparticles (CdS, ZnS, PbS, MnS, CuS, NiS, and HgS).32 Here, mild reaction temperatures and subsequent annealing at elevated temperatures yielded nanoparticles with high photoluminescence quantum yields. Amines were also investigated as catalyst and stabilizing ligand in solvothermal synthesis routes of metal sulfide nanoparticles using the chemically similar metal dithiocarbamate precursors, e.g. for the preparation of ZnS or SnS nanocrystals.33,34
In the present work, we investigate the synthesis of CuInS2 nanocrystals from the direct reaction of oleylamine with a mixture of copper and indium xanthates in solution at room temperature. Thereby we focus, besides the characterization of the properties of the formed nanocrystals, also on the investigation of the chemistry behind the conversion of the metal xanthates to the CuInS2 nanocrystals and their growth by time resolved small angle X-ray scattering (SAXS), UV-Vis and NMR spectroscopy.
Experimental
Materials and nanocrystal synthesis
For the synthesis of the CuInS2-nanocrystals metal dithiocarbonates, also referred to as metal xanthates, were used. Copper-O-2,2-dimethylpentan-3-yl-dithiocarbonate and indium-O-2,2-dimethylpentan-3-yl-dithiocarbonate were synthesized according to a previously published procedure.17 For the nanocrystal syntheses, which were carried out in nitrogen atmosphere in a glove box, 161 mg copper-O-2,2-dimethylpentan-3-yl-dithiocarbonate (0.63 mmol) and 430 mg indium-O-2,2-dimethylpentan-3-yl-dithiocarbonate (0.63 mmol) were dissolved in 10 mL of degassed toluene. To induce the nanoparticle formation, 1 mL (3.04 mmol, 1.2 equiv. to xanthate groups) degassed oleylamine was added to the yellow solution, which turned red immediately after the addition of the amine. The reaction solution was stirred at room temperature (25 °C) for 72 h and the progress of the nanoparticle formation was studied by UV-Vis and SAXS measurements in periodic intervals.The CdS nanocrystals were prepared analogously to the CuInS2 nanocrystals. Here, 308 mg cadmium-O-2,2-dimethylpentan-3-yl-dithiocarbonate (0.63 mmol), synthesized according to ref. 35, was dissolved in 10 mL degassed toluene followed by adding 1.6 mL oleylamine (4.92 mmol, 4 equiv. to xanthate groups). The solution was then stirred for 3 days in inert atmosphere at room temperature.
Characterization techniques
X-ray diffraction measurements were performed on a Siemens D 501 diffractometer in Bragg–Brentano geometry operated at 40 kV and 30 mA, using Cu Kα radiation and a graphite monochromator at the secondary side. Confocal Raman spectra have been obtained with a Raman microspectrometer HR-800 (HORIBA Jobin Yvon) using a 50 mW Ar+-laser with a wavelength of 532.12 nm (irradiation time 10 s; 5 times accumulation; pinhole 100 μm). Spectra were recorded unpolarized. Axially viewed inductively coupled plasma optical emission spectrometry (ICP-OES, Ciros Vision EOP, Spectro, Kleve, Germany) was used for the quantification of Cu (324.754 nm), In (230.606 nm) and S (180.731 nm) after microwave assisted digestion (Multiwave 3000, Anton Paar, Graz, Austria) of the sample in HNO3 (240 °C at 40 bar).For transmission electron microscopy (TEM), samples were prepared by dropping a nanoparticle solution (solvent: toluene) onto a nickel-TEM-grid (Quantifoil) with a carbon film and subsequent evaporation of the solvent at room temperature. TEM images as well as EDX spectra were acquired on a Tecnai F20 microscope (FEI Company) equipped with a Schottky emitter, an energy dispersive X-ray spectrometer and a high resolution Gatan imaging filter (HR-GIF) with an UltraScanCCD camera. Selected area electron diffraction (SAED) was performed on a FEI Tecnai 12 electron microscope.
The equipment for small angle X-ray scattering (SAXS) consisted of a high-flux SAXSess camera (Anton Paar, Graz, Austria) connected to a Debyeflex 3003 X-ray generator (GE-Electric, Germany), operating at 40 kV and 50 mA with a sealed-tube Cu anode. The Goebel-mirror focused and Kratky-slit collimated X-ray beam was line shaped (17 mm horizontal dimension at the sample). The scattered radiation was measured in the transmission mode and recorded by a one-dimensional MYTHEN-1k microstrip solid-state detector (Dectris Switzerland), within a q-range (with q being the magnitude of the scattering vector) of 0.1 to 5.0 nm−1. Using Cu Kα radiation with a wavelength of 0.154 nm and a sample-to-detector distance of 307 mm, this corresponds to a total 2θ region of 0.14° to 7°, applying the conversion q [nm−1] = 4π(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)/λ with 2θ being the scattering angle with respect to the incident beam and λ the wavelength of the X-rays.
θ)/λ with 2θ being the scattering angle with respect to the incident beam and λ the wavelength of the X-rays.
Samples were filled into a 1 mm (diameter) reusable quartz capillary (wall thickness 10 μm) with vacuum-tight sealing screw-caps at both ends. All measurements of the samples in the capillary were done in vacuum. The used SAXS curves for the evaluation were placed on an absolute scale with water as the secondary standard.36
The radius of gyration (RG) or Guinier radius, which is an estimate for the particle diameter, was directly approximated using the linear region (low q) from the SAXS data via the Guinier plot (lnI(q) vs. q2). The volume or intensity weighted size distribution functions DV(R) and DI(R) were calculated from the scattering data using the indirect Fourier transformation method.37
UV-Vis absorption spectra were measured on a Shimadzu 1800 spectrophotometer. Photoluminescence (PL) studies were performed on a F-7000 fluorescence spectrophotometer from Hitachi.
FT-IR spectra were acquired using a Bruker Alpha FT-IR spectrometer. All FT-IR spectra of the samples were recorded in transmission mode (films on silicon wafers, spectral range between 4000 and 800 cm−1).
Thermal gravimetric analyses (TGA) were performed on a Netzsch Jupiter STA 449C. All measurements were carried out in helium atmosphere and a heating rate of 10 °C min−1 from room temperature to 550 °C.
NMR spectroscopy (1H, 13C) was carried out on a Bruker Avance 300 MHz spectrometer. Deuterated solvents (CDCl3) were obtained from Cambridge Isotope Laboratories Inc. The NMR spectra were referenced to solvent residual peaks according to values given in the literature.38
Results and discussion
Nanocrystal synthesis
The synthesis of the CuInS2 nanoparticles was carried out at room temperature using copper and indium xanthates as precursors. Their chemical structures are depicted in Fig. 1. These metal xanthates bear branched alkyl chains, which enable their solubility in non-polar organic solvents such as chloroform, toluene or chlorobenzene. The formed solutions are stable over weeks when kept at room temperature. However, by addition of alkyl amines, such as hexylamine, the solutions turned turbid some time afterwards and a dark precipitate of metal sulfides was formed after some hours or days depending on the concentration of the amine. Hexylamine reacts with the metal xanthates and the corresponding organic thiocarbamate compound (R–NH–CS–O–R′) and metal sulfides are formed in an uncontrolled way. However, by replacing hexylamine with the long-chained oleylamine, which is a common capping ligand for metal sulfide nanoparticles,20 oleylamine controls the growth and stabilizes the formed copper indium sulfide, which leads to a defined growth of CuInS2 nanocrystals. The reaction scheme is illustrated in Fig. 1. | ||
| Fig. 1 Schematic illustration of the formation of the copper indium sulfide nanocrystals (stabilized by oleylamine ligands) at room temperature, chemical structures of the precursors and photographs of the solution at the start (directly after adding oleylamine) and at the end of the reaction time. | ||
The copper and indium xanthates form a yellow solution when dissolved together in toluene. After the addition of oleylamine, the solution turns dark red within several seconds. In the UV-Vis spectra in Fig. 2, it can be seen that the visual observation of the dark red color is based on a shift of the absorption onset from 470 to 570 nm and the presence of an additional absorption peak with a maximum at 750 nm. The absorption peak decreases and the dark red color fades again in the next few minutes of the reaction (the absorption peak at 750 nm vanishes completely). This indicates the temporary formation of a metal–amine complex in which the amine as well as the xanthate groups are coordinated to the copper or indium central atom. A similar intermediate was found in the reaction of zinc dithiocarbamates with octylamine by Jung et al.33 In the further course of the synthesis, the light red solution gradually turns red brown and finally into a dark brown solution and the absorption onset shifts from below 600 nm to about 700 nm. After a reaction time of 10 h, this onset shift slows down significantly, as can be also seen in Fig. 2 in the UV-Vis spectra measured in situ during the synthesis.
 | ||
| Fig. 2 Progress of the reaction in the first 35 h followed by UV-Vis spectroscopy. | ||
Typically after 72 h, the reaction is stopped by the addition of methanol, leading to precipitation of the formed nanocrystals. Then, the nanoparticles are purified by repeated dissolution in chloroform and subsequent precipitation into methanol, whereby excess oleylamine is removed and a brown powder is yielded which is well soluble in organic solvents leading to dark brown solutions. Extending the reaction time to some more days does not change the color of the reaction solution any more. Also from NMR spectroscopy measurements, performed at certain times during the synthesis, it can be concluded that all xanthate groups have reacted and thus no reactive species can be formed any more which could drive the reaction further. For example, after two days of reaction, the broad peaks at 230 ppm for the carbon of the dithiocarbonate group (metal–S–C(S)–O–R) and –O–CHR2 at 98 ppm in the 13C NMR spectrum and the peak at 5.04 ppm for the –O–CHR2 in the 1H NMR spectrum have vanished completely (see also Fig. 9 and discussion). It has to be mentioned that, although the pure copper and indium xanthates have different chemical shifts for the thiocarbamate as well as for the –O–CHR2 protons, only one set of broad signals of the xanthate groups can be detected during the reaction.
Structural, optical and thermogravimetric characterization
The crystallinity, primary crystallite size and crystal structure of the synthesized nanoparticles after purification by repeated precipitation in methanol are investigated by powder X-ray diffraction. The diffraction pattern depicted in Fig. 3A reveals four broad peaks with maxima at 19.8, 28.0, 46.6 and 54.9° 2θ. The first peak at 20° 2θ stems from the capping agent oleylamine39 and all the other peaks can be ascribed to chalcopyrite CuInS2 and match well with the reference pattern for tetragonal CuInS2 (PDF 01-75-0208). The distinct diffraction peaks, which are also comparable to others reported for CuInS2 nanoparticles prepared at higher temperatures,31,39–42 indicate a good crystallization of the nanoparticles even at room temperature. From the broadening of the peaks and using Scherrer equation, a primary crystallite size of 2–3 nm is estimated.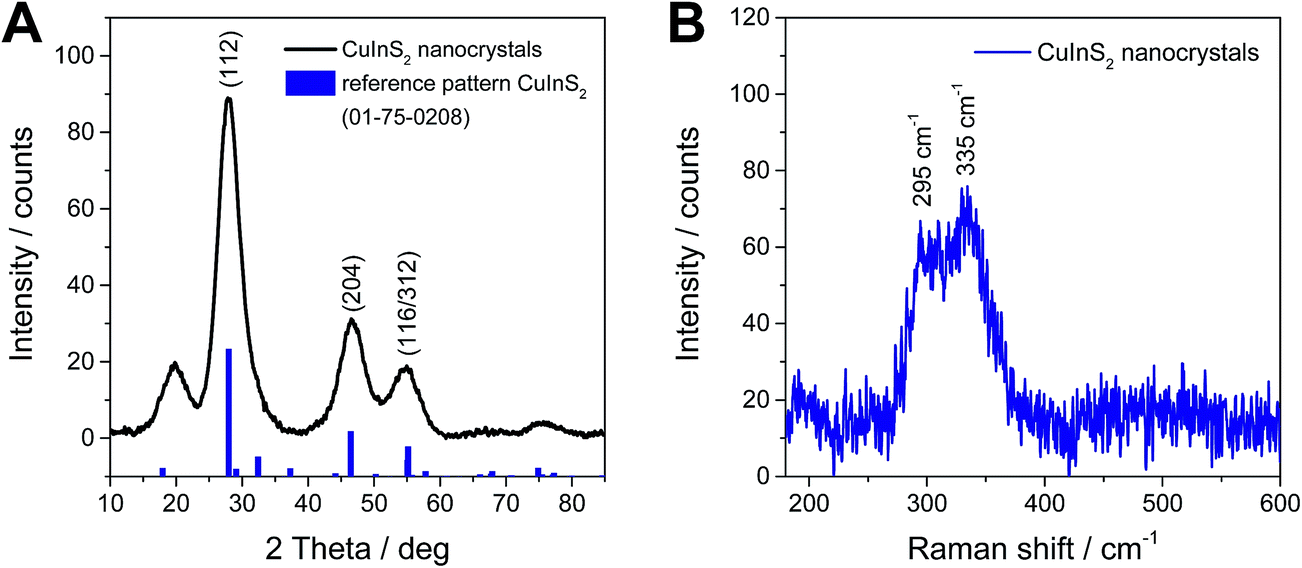 | ||
| Fig. 3 X-ray diffraction pattern (A) and Raman spectrum (B) of CuInS2 nanocrystals synthesized at room temperature. | ||
Also the Raman spectrum proves the presence of chalcopyrite CuInS2 nanocrystals. The spectrum in Fig. 3B, shows two broad peaks, which are partly overlapping, with maxima at 295 cm−1 and 335 cm−1. The first peak at 295 cm−1 stems from the A mode and the peak at 335 cm−1 can be assigned to the E1LO/B12LO modes of chalcopyrite CuInS2.43–45
The shape and size of the nanocrystals were investigated by transmission electron microscopy (TEM) shown in Fig. 4. The formed nanocrystals display an average diameter of approx. 3 nm. The nanocrystals exhibit a globular shape and a comparably narrow particle size distribution. The diameters of several nanocrystals, determined from the TEM images, are given in Fig. S1 in the ESI.† The low contrast of the nanocrystals to the carbon supporting film is due to the small size of the nanoparticles. In the HR-TEM image, see inset in Fig. 4, lattice fringes can be observed. The selected area electron diffraction (SAED) pattern, presented in the inset of Fig. 4, shows diffraction rings, typical for nanocrystalline samples without preferential order of the nanocrystals. The positions of the main diffraction rings in the pattern (112, r = 3.08 nm−1; 204/220, r = 5.09 nm−1; 116/312, r = 6.02 nm−1) are also in good agreement with reference data for chalcopyrite CuInS2 (PDF 01-75-0208). The corresponding radial intensity profile is shown in the ESI (Fig. S2†). TEM-EDX measurements reveal an almost stoichiometric Cu:In ratio in the nanocrystals of 55:45 at%. ICP-OES measurements confirm this result giving a Cu:In ratio of 53:47 at%. Additionally, they disclose that the prepared nanocrystals are sulfur-rich (atomic ratio of Cu:In:S = 1:0.90:2.61).
 | ||
| Fig. 4 TEM-micrograph, SAED pattern and HR-TEM image of the synthesized nanocrystals. | ||
The particle size of about 3 nm as obtained from TEM measurements is additionally confirmed by SAXS measurements. In Fig. 5A, the corresponding SAXS curve is presented and by an analysis of the data via the Guinier plot, a radius of gyration (RG) of 1.06 nm can be extracted from the SAXS data. This corresponds to a diameter of the nanocrystals of 2.74 nm and matches well with the nanoparticle sizes observed by TEM.
 | ||
| Fig. 5 (A) SAXS data and (B) UV-Vis absorption and photoluminescence emission spectra (λex = 560 nm). | ||
The UV-Vis absorption data, presented in Fig. 5B, reveal an absorption onset at around 800 nm and a steep increase of the absorption towards lower wavelengths. In addition, a weak excitonic peak present as a shoulder around 620 nm can be noticed in the spectrum. The prepared CuInS2 nanoparticles also show a characteristic PL emission peak with a maximum at 815 nm (1.52 eV).9,46,47
These structural and optical characterizations show that the properties of these nanocrystals prepared at room temperature are comparable to CuInS2 nanocrystals prepared by colloidal synthesis routes at temperatures around 200 °C.8,9 The size of the synthesized nanocrystals is quite small, however, they clearly exhibit a chalcopyrite crystal structure and the particle size distribution is quite narrow, even though they are prepared at room temperature without a special hot injection protocol. Also, the absorption and emission properties are comparable to CuInS2 nanocrystals prepared via conventional syntheses.
From the IR spectra in Fig. 6A, it can be seen that no metal xanthates remain in the final nanocrystal samples as in this spectrum none of the characteristic IR-bands of the copper or indium xanthates, e.g. the bands at 1238 and 1203 cm−1 (asymmetric C–O–C stretching vibrations) and the bands at 1047 and 1017 cm−1 (C–S stretching vibrations), can be identified. In the nanocrystal sample, only the IR-bands typical for oleylamine, which are e.g. the stretching vibration of NH (R–NH2) at 3233 cm−1, the asymmetric and symmetric C–H stretching modes at 2919 and 2850 cm−1 and the C–H deformation vibration at 1443 cm−1, are found.20 The vibration modes of oleylamine are slightly shifted towards lower wavenumbers compared to that of the free amine ligand15 indicating the coordination of oleylamine to the nanocrystal surface through the lone pair of the nitrogen atom.
 | ||
| Fig. 6 (A) IR spectra of copper and indium xanthate and the oleylamine capped CuInS2 nanocrystals before and after heat treatment at 500 °C and (B) thermogravimetric analysis of the nanocrystal sample. | ||
To determine the amount of oleylamine stabilizing the nanocrystals, which is present in the sample, thermogravimetric analysis was performed, which shows a weight loss of 49.1% during a heating run up to 500 °C. The weight loss starts at a temperature of approx. 200 °C and proceeds up to 450 °C. A weight loss in this temperature range is typical for the loss of oleylamine in nanoparticle samples.39,48 Moreover, also a comparison of the IR spectra of the nanoparticle sample before and after heat treatment at 500 °C confirms the absence of the characteristic peaks of oleylamine, which are present before heat treatment.
Investigation of the nanocrystal growth
In a further step, we investigated the kinetics of the nanocrystal growth via time resolved SAXS measurements to follow the evolution of the particle diameter during the reaction. In Fig. 7A, the SAXS data of a nanoparticle synthesis, which was performed in a vial with magnetic stirring and samples for the SAXS measurements being taken in certain intervals, are shown. Additionally, we performed the synthesis also directly in the measuring capillary to acquire SAXS data more frequently (see Fig. S3, ESI†). The evolution of the particle diameter of the nanocrystals is derived from the Guinier plot analysis and is shown in Fig. 7B and C. It is noted here that for the analysis of the evolution of the particle diameter the lowest q-values were excluded, because of an upturn in this q-range (emerging in later stages of the reaction), which is discussed below. The results of the Guinier analysis reveal a fast growth of the nanocrystals at the beginning of the reaction. After the first hour, the nanocrystal growth is slowing down once the crystals have a size of 2.2 nm and after one day, a particle diameter of 2.6 nm is reached. In the further course of the reaction, the nanocrystal size stays constant and no further growth is observed. This exemplifies that even though the reaction is performed at room temperature, the nanocrystals are formed comparably fast. A comparison of Fig. 7B and C reveals that by performing the synthesis with stirring, the nanocrystals become slightly bigger than those formed directly in the measuring capillary without stirring, while the qualitative evolution of the nanocrystal size is similar. | ||
| Fig. 7 SAXS data of the reaction solution at different times during the formation of the CuInS2 nanocrystals in a vial with stirring (A) and the corresponding evolution of the nanocrystal size extracted from the SAXS data (B: reaction performed in a vial with stirring, C: reaction performed directly in the SAXS measuring capillary). | ||
As already mentioned above, the SAXS data show a slight upturn at low q (q < 0.5 nm−1), beginning after 5 h of reaction time (see SAXS data in Fig. 7A). This upturn is characteristic for the formation of bigger particles, such as aggregates or agglomerates. A Guinier plot analysis including also this upturn at very low q-values leads for the first two hours of the reaction to similar particle diameters as already described above (Fig. 7B). However, at later times the scattering intensity at q = 0 I(0) and the radii R, determined from the radius of gyration under the assumption of spherical shape, are increasing considerably as shown in Fig. 8A giving a hint to the tendency of a slight agglomeration of the nanocrystals in the reaction solution.
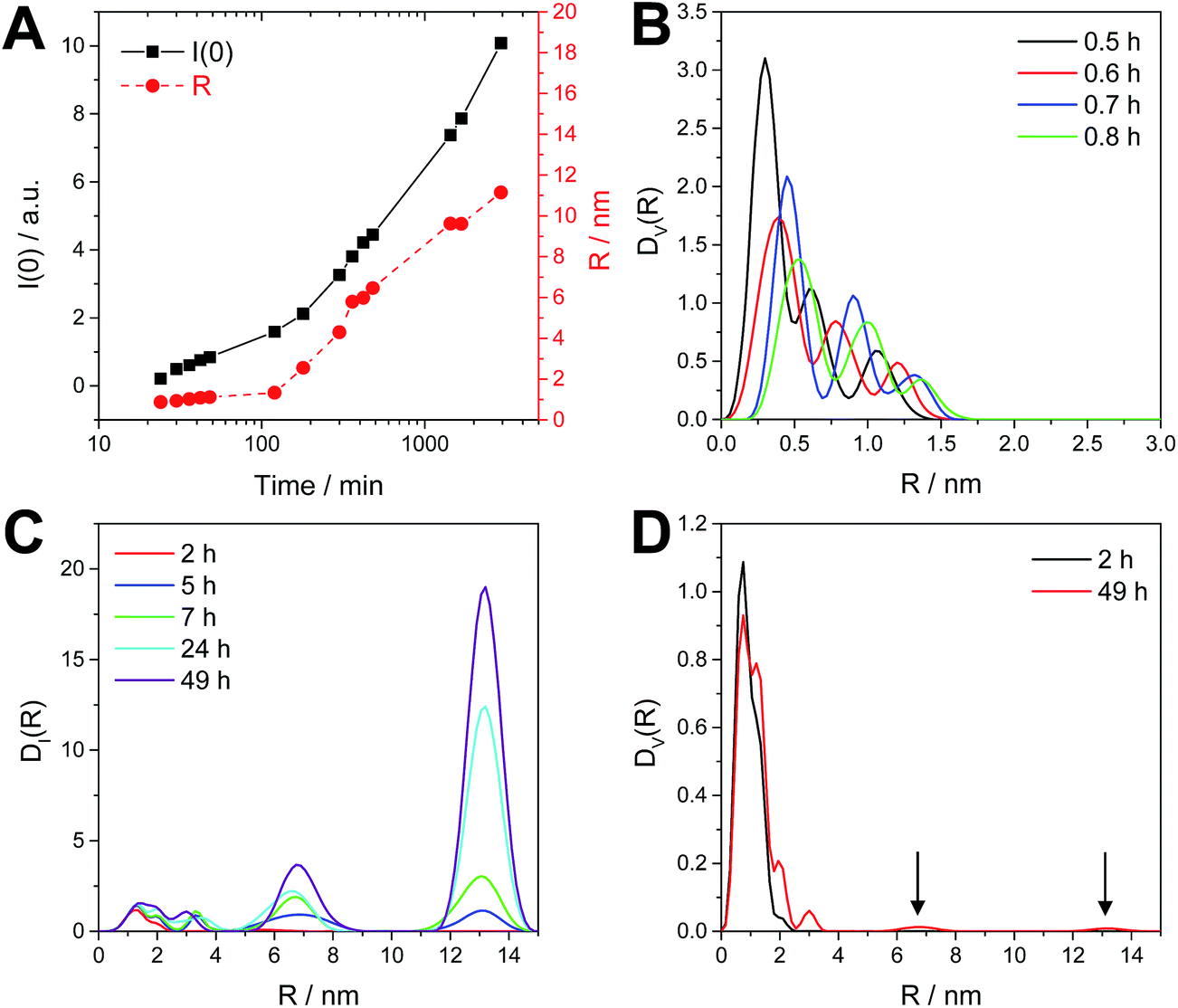 | ||
| Fig. 8 Guinier plot results of I(0) and radius (R) as a function of time (A) and the size distributions DV(R) and DI(R) (B–D) calculated for the reaction performed in a vial with stirring. | ||
To further elucidate this observation, a polydispersity analysis was performed resulting in size distributions weighted by volume (DV(R)) and intensity (DI(R)) (Fig. 8B and C). The size distribution DV(R) shows slowly growing populations at small hydrodynamic radii (0.3 to 1.5 nm) during the first hour of reaction (Fig. 8B). The size distribution DI(R) reveals also the presence of two bigger populations, centered at 6.7 and 13.2 nm at reaction times of more than 2 h (Fig. 8C). However, these larger particles or agglomerates found in the polydispersity analysis represent only a negligible amount of mass or volume as DV(R) after 2 h as well after 49 h of reaction time show only very little contributions at higher radii (see arrows in Fig. 8D).
Investigation of the reaction mechanism
The thermal reaction of metal xanthates to metal sulfides proceeds via the Chugaev reaction, which was proven also by the identification of reaction products, e.g. of the volatile alkene and COS via mass spectrometry.17 However, Pradhan et al. showed that in the presence of alkyl amines, milder reaction temperatures can be used leading to metal sulfide nanoparticles but it was still suggested that the reaction pathway proceeds via the Chugaev elimination reaction.32 However, as already discussed above, by the addition of small amounts of hexylamine the corresponding N-hexyl-thiocarbamate product could be identified indicating that another reaction pathway is possible. Nevertheless, the reaction conditions in this particular set up (only approx. 10 mol% hexylamine in relation to the metal xanthates and temperatures of 140 °C and above were used) led us to the conclusion that the main course of the reaction is still proceeding according to the Chugaev reaction.31However, using larger amounts of alkyl amine – here more than one equivalent in relation to xanthate groups is used – the question arises if the proposed Chugaev reaction (cf. Pradhan et al. in ref. 32) is the driving force for the formation of metal sulfides especially if low temperatures are used. We assume that in this route, the formation of the nanocrystals proceeds via the following steps: first, when the oleylamine is added to the metal xanthate solution, it coordinates to the metal center in the copper xanthate or indium xanthate, respectively. The dark reddish color in the initial step of the synthesis stems most likely from the coordination to the copper ions as, when we added oleylamine to individual copper and indium xanthate solutions in toluene, only the copper xanthate solution turned red while the indium xanthate solution remained colorless (see discussion of the UV-Vis spectra above). Then, the amino group acts as a nucleophile and attacks the (thio)carbonyl center of the xanthate group, forming a tetrahedral intermediate. In the next phase of the reaction, the corresponding metal hydrosulfide and the corresponding organic thiocarbamate product is formed. This thiocarbamate product (O-(2,2-dimethylpentan-3-yl)-(E)-octadec-10-en-1-ylcarbamothioate) was isolated from the reaction mixture. To investigate the reaction mechanism in more detail, 1H and 13C NMR spectroscopy was carried out to identify possible other reaction products and especially in order to clarify if the Chugaev reaction also takes place under these conditions. If this is the case, it should be possible to identify the corresponding elimination product i.e. 4,4-dimethylpent-2-ene. Thus, the reaction was carried out in CDCl3 as solvent using the same concentrations of the metal xanthates and oleylamine as above and NMR spectra were measured during the synthesis. The recorded spectra are shown in Fig. S9 and S10 in the ESI.† As already mentioned above, only a set of broad singlets of the metal xanthate groups is visible and they are continuously decreasing with increasing reaction time. However, due to the many different and overlapping signals of oleylamine with the emerging signals, an additional model experiment using only indium xanthate and hexylamine has been carried out. Although hexylamine cannot stabilize the nanoparticles in solution, it is expected to react in the same way as oleylamine. Fig. 9 shows the 1H NMR spectra of this experiment shortly after mixing as well as at the end of the reaction in comparison with pure hexylamine and indium xanthate. The 13C NMR spectra as well as 2D NMR spectra can be found in the ESI (Fig. S5–S7†). The most characteristic chemical shift of the indium xanthate, the triplet of the O–CH-group at 4.75 ppm, has shifted downfield after the addition of hexylamine and gives only a broad signal at lower field at 5.04 ppm. The signal of the adjacent CH2-groups is slightly shifted upfield from 1.76 ppm to 1.72 ppm. This, together with the shift of the NH-proton of oleylamine from 1.07 ppm to ∼4 ppm, also indicates a change of the coordination sphere at the metal center due to the binding of the amine. However, this chemical shift is strongly dependent on the concentration of the amine as well as on the reaction time, the peak can be found below 4 ppm at shorter reaction times and higher than 4 ppm towards the end of the reaction. Furthermore, the N–CH2-signal is shifted from 2.68 ppm to 2.83 ppm upon start and finally to 2.99 ppm at the end of the reaction. In addition, new peaks attributable to the thiocarbamate (O-(2,2-dimethylpentan-3-yl)-hexyl-carbamothioate) arise at 5.36 ppm (O–CH), 3.30 ppm and 3.58 ppm (both NH–CH2) as well as broad small signals appear at 6.8 ppm and 6.3 ppm (NH) in the spectrum. In the 13C NMR spectrum a series of new peaks, always appearing in pairs, e.g. at 191.7 ppm and 190.9 ppm (O–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–NH), 90.7 ppm and 88.1 ppm (O–CH–), 45.3 and 43.1 ppm (NH–CH2–) is observed. Due to the limited rotation of the C(S)–NH-bond, two isomers are formed giving also different NMR signals. With increased reaction time, further signals emerge, e.g. at 3.45 ppm and 3.10 ppm. The latter chemical shift is characteristic for the free alcohol 2,2-dimethyl-3-pentanol. The other signal is typical for the corresponding N-hexyl-dithiocarbamate. Consequently, also the characteristic shifts at 205 ppm (NH–(CS)–S) and 51.1 ppm (CH2–NH–) are found in the corresponding carbon NMR spectrum and 2D NMR spectra (see ESI, Fig. S5–S7 for more details†). Thus, it is obvious that the synthesis proceeds also via a reaction mechanism different to the Chugaev elimination. At the same time as these new products are formed, the peaks from the xanthates and the hexylamine decrease.
S)–NH), 90.7 ppm and 88.1 ppm (O–CH–), 45.3 and 43.1 ppm (NH–CH2–) is observed. Due to the limited rotation of the C(S)–NH-bond, two isomers are formed giving also different NMR signals. With increased reaction time, further signals emerge, e.g. at 3.45 ppm and 3.10 ppm. The latter chemical shift is characteristic for the free alcohol 2,2-dimethyl-3-pentanol. The other signal is typical for the corresponding N-hexyl-dithiocarbamate. Consequently, also the characteristic shifts at 205 ppm (NH–(CS)–S) and 51.1 ppm (CH2–NH–) are found in the corresponding carbon NMR spectrum and 2D NMR spectra (see ESI, Fig. S5–S7 for more details†). Thus, it is obvious that the synthesis proceeds also via a reaction mechanism different to the Chugaev elimination. At the same time as these new products are formed, the peaks from the xanthates and the hexylamine decrease.
 | ||
| Fig. 9 1H NMR spectra of the pure educts indium xanthate and hexylamine, and spectra of the reaction shortly after mixing and at the end of reaction (corresponding 13C NMR spectra can be found in Fig. S5 in the ESI†). | ||
In addition, H2S was detected by the characteristic smell of the solution after the reaction and by the classical H2S detection test using lead acetate paper showing the typical black color of PbS if placed above the reaction solution in the headspace of the reaction vial. With these findings, we are now able to propose the following reaction pathway shown in Fig. 10. After coordination of the amine to the metal center of the methyl xanthates, the amine reacts with the xanthate group either to the corresponding thiocarbamate product (product C) and metal hydrosulfides, which subsequently eliminate H2S and form the CuInS2-nanoparticle core, or in a side reaction via the elimination to the alcohol (E) and the dithiocarbamate (D). The first reaction to the thiocarbamate (C) is the faster reaction as this product appears quickly after the addition of the amine, whereas products E and D are formed slowly during the reaction. As can be seen from the NMR spectra in Fig. 9, the chemical shifts for D at 3.45 ppm and the characteristic doublet of doublets for E at 3.10 ppm are hardly visible at the beginning of the reaction but getting more intense towards the end of the reaction. It has to be noted that the distribution of the products also depends on the amine concentration, i.e. a higher concentration of the amine increases the formation of the thiocarbamate C. This indicates that the formation of D proceeds via the coordinated amine. If the dithiocarbamate D remains coordinated to the metal center or the nanoparticle surface is not clear from the undertaken experiments but both might be plausible. However, in all cases it was not possible to detect any traces of the corresponding Chugaev reaction product, the corresponding olefin. This indicates that the reaction does not proceed via the Chugaev elimination, but via the nucleophilic reaction of the amine directly at the thiocarbonyl-carbon. The same NMR experiments were carried out with oleylamine. Although the peaks of the products and educts largely overlap, the characteristic peaks of the thiocarbamate, the dithiocarbamate and the alcohol can be clearly identified (see ESI, Fig. S9 and S10 for spectra and detailed interpretation†).
 | ||
| Fig. 10 Reaction scheme illustrating the conversion of metal xanthates into metal sulfide nanoparticles at room temperature in the presence of alkyl amines. | ||
Synthesis of CdS nanocrystals via the presented room temperature approach
To demonstrate the versatility of the developed method, we also prepared CdS nanocrystals with the same synthesis route. In this case, cadmium-O-2,2-dimethylpentan-3-yl-dithiocarbonate was used as precursor. Here, the solution turned from pale yellow to yellow within 3 days, and after precipitation and purification of the nanoparticle sample, a well soluble intense yellow powder was yielded. Fig. 11A shows the X-ray diffraction pattern of this sample and reference patterns for hexagonal CdS (PDF 41-1049) and cubic CdS (PDF 01-080-0019). According to this, CdS is formed in a mixed cubic and hexagonal crystal structure, which can originate from a mixed cubic and hexagonal stacking as also already observed in other studies on CdS nanocrystals.49 The TEM image in Fig. 11B shows that the nanocrystals are also spherical and slightly larger (3–4 nm) compared to the CuInS2 ones prepared via this synthesis route. The optical properties of these CdS nanoparticles are depicted in Fig. 11C. The absorption has an onset at 480 nm corresponding to an optical band gap of 2.6 eV. The PL spectrum recorded using an excitation wavelength of 440 nm reveals an emission peak with a maximum at 640 nm. | ||
| Fig. 11 X-ray diffraction pattern (A), TEM image (B) and UV-Vis and PL spectra (C) of the prepared CdS nanocrystals. | ||
First experiments using other metal xanthates (e.g. lead or bismuth) indicate that either very fast reaction kinetics can also lead to insoluble metal sulfide precipitates or limited crystallization to amorphous metal sulfide particles. In the first case, changes in the reaction conditions might slow down the reaction, in the second case a thermal treatment might be necessary in any case to yield nanoparticles with high crystallinity.
Conclusion
The reaction of copper and indium xanthates with oleylamine already proceeds at ambient temperatures yielding CuInS2 nanocrystals, which have an average size of about 3 nm and a chalcopyrite crystal structure. The nanoparticles are well soluble and exhibit optical properties characteristic for CuInS2 nanoparticles. Thus, it is not necessary to heat up the metal xanthates to induce a thermal decomposition as usually carried out in literature (for CuInS2 nanocrystals cf.ref. 27–30). Whereas the thermal pathway follows the Chugaev elimination mechanism, the presented room temperature synthesis proceeds via a direct reaction of the amine with the metal xanthate as identified by NMR investigations. First, oleylamine coordinates to the metal center followed by a substitution reaction on the thiocarbonyl group of the xanthate giving the N-oleyl-thiocarbamate derivative and a metal hydrosulfide. Condensation of the metal hydrosulfide groups with concurrent elimination of H2S leads to copper indium sulfide nanocrystals. Furthermore, the formation of N-oleyldithiocarbamate and 2,2-dimethylpentan-3-ol takes place as side reaction. In all experiments carried out within this study, we could not identify any products, e.g. the corresponding elimination product, 2,3-dimethylpent-3-ene, which would support a Chugaev mechanism.In principle, this approach can also be transferred to other oleylamine capped metal sulfide nanocrystals, as we have shown for the synthesis of CdS as an example. Also in this case, luminescent nanocrystals with a diameter of 3–4 nm have been obtained.
Acknowledgements
The authors would like to thank Christopher Fradler for NMR measurements, Josefine Hobisch for thermogravimetric analyses and Helmar Wiltsche for performing ICP-OES measurements. Financial support from the Austrian Science Fund (FWF) is gratefully acknowledged by TR (grant number: J3515-N20).References
- Y. Min, G. D. Moon, C.-E. Kim, J.-H. Lee, H. Yang, A. Soon and U. Jeong, J. Mater. Chem. C, 2014, 2, 6222–6248 RSC.
- J. Jiea, W. Zhang, I. Bello, C.-S. Lee and S.-T. Lee, Nano Today, 2010, 5, 313–336 CrossRef.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033–3087 RSC.
- D. Aldakov, A. Lefrançois and P. Reiss, J. Mater. Chem. C, 2013, 1, 3756–3776 RSC.
- M. V. Kolvalenko, L. Manna, A. Cabot, Z. Hens, D. V. Talapin, C. R. Kagan, V. I. Klimov, A. L. Rogach, P. Reiss, D. J. Milliron, P. Guyot-Sionnest, G. Konstantatos, W. J. Parak, T. Hyeon, B. A. Korgel, C. B. Murray and W. Heiss, ACS Nano, 2015, 9, 1012–1057 CrossRef PubMed.
- S. R. Thomas, C.-W. Chen, M. Date, Y.-C. Wang, H.-W. Tsai, Z. M. Wang and Y.-L. Chueh, RSC Adv., 2016, 6, 60643–60656 RSC.
- B. Tell, J. L. Shay and H. M. Kasper, Phys. Rev. B: Solid State, 1971, 4, 2463-2471 Search PubMed.
- A. D. P. Leach and J. E. Macdonald, J. Phys. Chem. Lett., 2016, 7, 572–583 CrossRef CAS PubMed.
- J. Kolny-Olesiak and H. Weller, ACS Appl. Mater. Interfaces, 2013, 5, 12221–12237 CAS.
- C. Zhao, Z. Bai, X. Liu, Y. Zhang, B. Zou and H. Zhong, ACS Appl. Mater. Interfaces, 2015, 7, 17623–17629 CAS.
- S. Khanchandani, S. Kumar and A. K. Ganguli, ACS Sustainable Chem. Eng., 2016, 4, 1487–1499 CrossRef CAS.
- Z. Bai, W. Ji, D. Han, L. Chen, B. Chen, H. Shen, B. Zhou and H. Zhong, Chem. Mater., 2016, 28, 1085–1091 CrossRef CAS.
- Z. Pan, I. Mora-Seró, Q. Shen, H. Zhang, Y. Li, K. Zhao, J. Wang, X. Zhong and J. Bisquert, J. Am. Chem. Soc., 2014, 136, 9203–9210 CrossRef CAS PubMed.
- N. Guijarro, E. Guillén, T. Lana-Villarreal and R. Gómez, Phys. Chem. Chem. Phys., 2014, 16, 9115–9122 RSC.
- D. H. Jara, S. J. Yoon, K. G. Stamplecoskie and P. V. Kamat, Chem. Mater., 2014, 26, 7221–7228 CrossRef CAS.
- J. E. Halpert, F. S. F. Morgenstern, B. Ehrler, Y. Vaynzof, D. Credgington and N. C. Greenham, ACS Nano, 2015, 9, 5857–5867 CrossRef CAS PubMed.
- T. Rath, M. Edler, W. Haas, A. Fischereder, S. Moscher, A. Schenk, R. Trattnig, M. Sezen, G. Mauthner, A. Pein, D. Meischler, K. Bartl, R. Saf, N. Bansal, S. A. Haque, F. Hofer, E. J. W. List and G. Trimmel, Adv. Energy Mater., 2011, 1, 1046–1050 CrossRef CAS.
- M. Arar, M. Gruber, M. Edler, W. Haas, F. Hofer, N. Bansal, L. X. Reynolds, S. A. Haque, K. Zojer, G. Trimmel and T. Rath, Nanotechnology, 2013, 24, 484005 CrossRef PubMed.
- C. Krause, D. Scheunemann, J. Parisi and H. Borchert, J. Appl. Phys., 2015, 118, 205501 CrossRef.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- T. Rath and G. Trimmel, Hybrid Mater., 2014, 1, 15–36 Search PubMed.
- X. Tang, W. Cheng, E. S. G. Choo and J. Xue, Chem. Commun., 2011, 47, 5217–5219 RSC.
- C. Czekelius, M. Hilgendorff, L. Spanhel, I. Bedja, M. Lerch, G. Müller, U. Bloeck, D.-S. Su and M. Giersig, Adv. Mater., 1999, 11, 643–646 CrossRef CAS.
- F. M. Courtel, R. W. Paynter, B. Marsan and M. Morin, Chem. Mater., 2009, 21, 3752–3762 CrossRef CAS.
- J. J. Nairn, P. J. Shapiro, B. Twamley, T. Pounds, R. von Wandruszka, T. R. Fletcher, M. Williams, C. Wang and M. G. Norton, Nano Lett., 2006, 6, 1218–1223 CrossRef CAS PubMed.
- T. Rath, C. Padeste, M. Vockenhuber, C. Fradler, M. Edler, A. Reichmann, I. Letofsky-Papst, F. Hofer, Y. Ekinci and T. Griesser, J. Mater. Chem. A, 2013, 1, 11135–11140 CAS.
- D. P. Dutta and G. Sharma, Mater. Lett., 2006, 60, 2395–2398 CrossRef CAS.
- S. H. Lu, T. F. Chen, A. J. Wang, D. Zheng, Y. L. Li and Y. S. Wang, Mater. Sci. Eng., B, 2016, 203, 19–26 CrossRef CAS.
- A. Kharkwal, K. Jain, S. B. Tyagi, A. K. Singh, S. N. Sharma and M. Kharkwal, Colloid Polym. Sci., 2014, 292, 2913–2926 CAS.
- A. Kharkwal, S. N. Sharma, K. Jain, L. Arora, P. Chawla, A. K. Singh and S. Chand, Colloid Polym. Sci., 2013, 291, 2607–2617 CAS.
- C. Fradler, T. Rath, S. Dunst, I. Letofsky-Papst, R. Saf, B. Kunert, F. Hofer, R. Resel and G. Trimmel, Sol. Energy Mater. Sol. Cells, 2014, 124, 117–125 CrossRef CAS.
- N. Pradhan, B. Katz and S. Efrima, J. Phys. Chem. B, 2003, 107, 13843–13854 CrossRef CAS.
- Y. K. Jung, J. I. Kim and J.-K. Lee, J. Am. Chem. Soc., 2010, 132, 178–184 CrossRef CAS PubMed.
- D. S. Koktysh, J. R. McBride and S. J. Rosenthal, Nanoscale Res. Lett., 2007, 2, 144–148 CrossRef CAS.
- A. J. MacLachlan, T. Rath, U. B. Cappel, S. A. Dowland, H. Amenitsch, A.-C. Knall, C. Buchmaier, G. Trimmel, J. Nelson and S. A. Haque, Adv. Funct. Mater., 2015, 25, 409–420 CrossRef CAS PubMed.
- D. Orthaber, A. Bergmann and O. Glatter, J. Appl. Crystallogr., 2000, 33, 218–225 CrossRef CAS.
- O. Glatter, J. Appl. Crystallogr., 1980, 13, 7–11 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- A. Pein, M. Baghbanzadeh, T. Rath, W. Haas, E. Maier, H. Amenitsch, F. Hofer, C. O. Kappe and G. Trimmel, Inorg. Chem., 2011, 50, 193–200 CrossRef CAS PubMed.
- H. Zhong, S. S. Lo, T. Mirkovic, Y. Li, Y. Ding, Y. Li and G. D. Scholes, ACS Nano, 2010, 4, 5253–5262 CrossRef CAS PubMed.
- W. Yue, S. Han, R. Peng, W. Shen, H. Geng, F. Wu, S. Tao and M. Wang, J. Mater. Chem., 2010, 20, 7570–7578 RSC.
- J. Hua, H. Cheng, X. Yuan, Y. Zhang, M. Liu, X. Meng, H. Li and J. Zhao, RSC Adv., 2015, 5, 30981–30988 RSC.
- Y.-K. Kim, S.-H. Ahn, K. Chung, Y.-S. Cho and C.-J. Choi, J. Mater. Chem., 2012, 22, 1516–1520 RSC.
- M. Nanu, J. Schoonman and A. Goosens, Thin Solid Films, 2004, 451–452, 193–197 CrossRef CAS.
- F. M. Courtel, A. Hammami, R. Imbeault, G. Hersant, R. W. Paynter, B. Marsan and M. Morin, Chem. Mater., 2010, 22, 3752–3761 CrossRef CAS.
- D. So and G. Konstantatos, Chem. Mater., 2015, 27, 8424–8432 CrossRef CAS.
- D. Deng, Y. Chen, J. Cao, J. Tian, Z. Qian, S. Achilefu and Y. Gu, Chem. Mater., 2012, 24, 3029–3037 CrossRef CAS.
- W. Huang, Q. Li, Y. Chen, Y. Xia, H. Huang, C. Dun, Y. Li and D. L. Carroll, Sol. Energy Mater. Sol. Cells, 2014, 127, 188–192 CrossRef CAS.
- T. Di Luccio, A. M. Laera, L. Tapfer, S. Kempter, R. Kraus and B. Nickel, J. Phys. Chem. B, 2006, 110, 12603–12609 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM, SAXS and NMR data. See DOI: 10.1039/c6ra22813e |
| This journal is © The Royal Society of Chemistry 2016 |