
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAb initio and first principles theoretical investigations of triplet–triplet fluorescence in trimethylenemethane biradicals†
Yasunori Matsui‡
ab,
Kosuke Usui‡c,
Hiroshi Ikedaab and
Stephan Irle*cd
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka Prefecture University, Sakai, Osaka 599-8531, Japan
bThe Research Institute for Molecular Electronic Devices (RIMED), Osaka Prefecture University, Sakai, Osaka 599-8531, Japan
cDepartment of Chemistry, Graduate School of Science, Nagoya University, Nagoya, 464-8601, Japan. E-mail: sirle@chem.nagoya-u.ac.jp
dInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Nagoya 464-8601, Japan
First published on 11th August 2016
Abstract
Theoretical studies on triplet–triplet (T1 → T0) fluorescence of the arylated trimethylenemethane (TMM) biradicals, 32˙˙, were carried out using post-Hartree–Fock ab initio and various first principles density functional theory methods. Analysis of optimized geometries including bond alternations and spin distributions indicates that the triplet ground (32˙˙) and excited (32˙˙*) states of these biradicals have aromatic and quinoidal characteristics, respectively. Inspection of their calculated electronic structures shows that, in comparison to 32˙˙, one of the spins in 32˙˙* is more delocalized onto arene-rings linked to the TMM framework.
The trimethylenemethane1 (TMM, Fig. 1) biradical is a representative non-Kekulé molecule that has a triplet ground state (T0) owing to its D3h symmetry and pair of degenerate singly-occupied molecular orbitals (SOMOs). As a result of this characteristic, many theoretical and experimental studies have been carried out probing the curious electronic structures and magnetic properties of TMM biradicals.2 The fact that these biradicals emit triplet–triplet (T1 → T0) fluorescence has received great attention.3,4
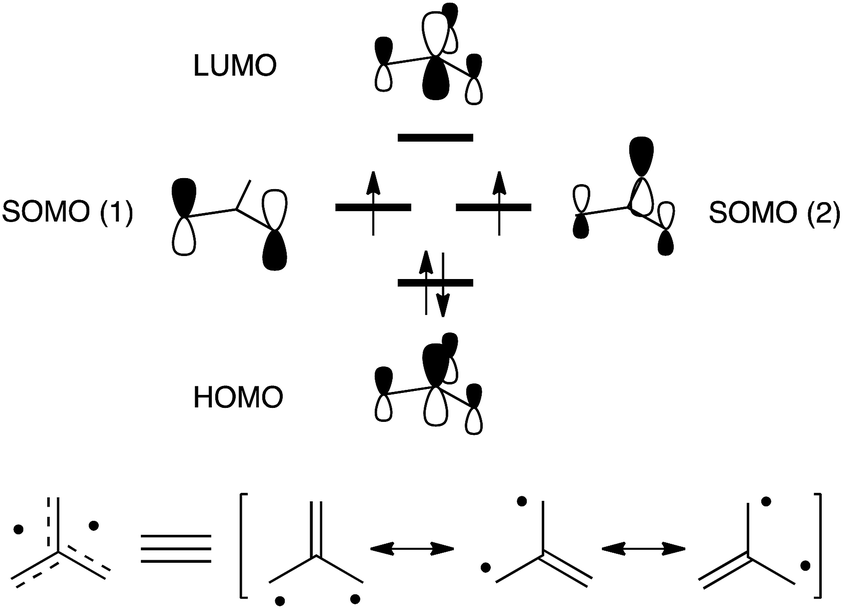 | ||
| Fig. 1 (Top) Frontier molecular orbitals of the parent TMM biradical and (bottom) its corresponding Lewis resonance structures. | ||
In previous studies, Matsui, Ikeda, and coworkers described the observation of T1 → T0 fluorescence from the excited states of the 1-methyl-1-phenyl-, 1,1-diphenyl-, and 1-(2-naphthyl)-1-phenyl-substituted TMM biradicals, 32a–c˙˙* (Scheme 1). For this purpose, thermoluminescence (TL)5 and two-color two-laser flash photolysis measurements were conducted using the corresponding aryl-substituted methylenecyclopropanes 1a–c.6 Moreover, T1 → T0 fluorescence of these biradicals was utilized as the basis for the design of a new organic light-emitting diode, termed “organic radical light-emitting diode”.5a
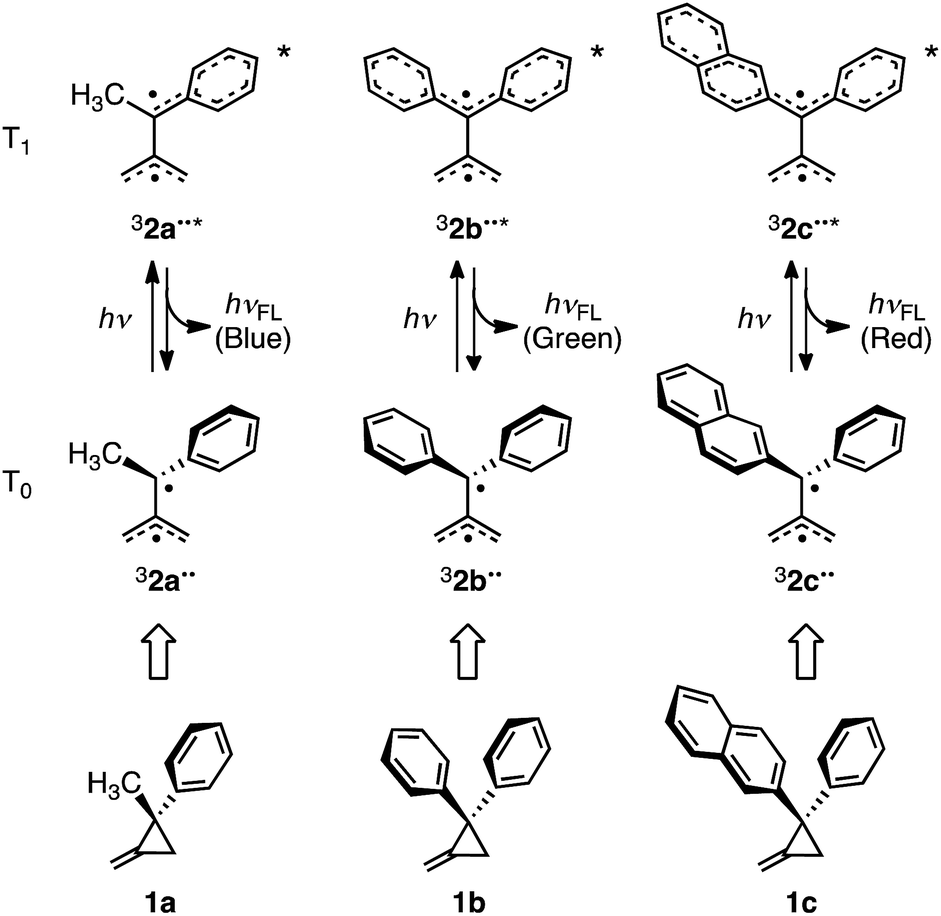 | ||
| Scheme 1 Chemical structures of 1a–c and electronic structures of 32a–c˙˙ and 32a–c˙˙* associated with T0–T1 and T1–T0 transitions accompanying fluorescence. | ||
The electronic configurations of the excited T1 states of the TMM biradicals are complicated by their multi-configuration character. Additionally, electronic transition wavelengths and T0 geometries of compound 32b, computed using time-dependent (TD) density functional theory (DFT)6 with the B3LYP/cc-pVDZ method, are considerably different from the absorption and emission wavelengths observed experimentally.7 Thus, more accurate and rigorous ab initio quantum chemical studies, and a detailed analysis of geometry relaxation occurring in the T1 state are required to have a greater understanding of the electronic structures of the excited states of TMM biradicals.
In the investigation described below, we carried out a theoretical study of the triplet–triplet fluorescence mechanism to accurately predict structural, electronic and T1 → T0 emission properties of triplet aryl-substituted trimethylenemethane biradicals 32˙˙*. Ab initio post-Hartree–Fock methods and three different (TD-)DFT functionals were used for this purpose. When treating open-shell systems, DFT methods often suffer from spin contamination, which usually is more severe when excited rather than ground states are computed. To validate the (TD-)DFT results, high-level ab initio benchmark calculations where carried out on 32˙˙ and the calculated spin densities and excitation energies obtained by using both types of methods were compared. In this effort, the triplet excited states of three model aryl-substituted TMM biradicals were theoretically studied because they each emit one of the three primary colors (blue, green, and red) (Scheme 1).5a
All (TD-)DFT geometry optimizations were performed using the spin-unrestricted UB3LYP, UM06-2X, and CAM-UB3LYP functionals8 in conjunction with the 6-31G(d) basis set.9 (TD-)DFT single point energies were obtained using the same methods, however, the larger 6-311+G(d)9c basis set was employed. For the smallest, methyl-phenyl substituted biradical, 32a˙˙, complete ab initio active space self-consistent field (CASSCF)10 single point energy calculations with 10 π-orbitals and 10 electrons were performed, followed by a multireference second-order Møller–Plesset perturbation (MRMP2) treatment.11 All DFT calculations were conducted using Gaussian 09,12 while the GAMESS-US13 and MOLPRO14 program packages were employed for the post-Hartree–Fock calculations. Below, we will refer to T1 and T0 states of 2˙˙ simply as 32˙˙* and 32˙˙, respectively.
In the first phase of this study, molecular geometry optimizations were carried out on 32a–c˙˙ and 32a–c˙˙* using various DFT functionals.15 The geometries of triplet ground (T0) and excited (T1) states of these biradicals, obtained by using UB3LYP/6-31G(d), are displayed in Fig. 2, and selected bond lengths are listed in Table 1. The lengths of the C5–C6, C6–C7 and C7–C8 bonds (see Fig. 2 for atom numbering) for 32a˙˙ are in the range of 1.39–1.42 Å, which is typical for benzene-rings having a small degree of bond alternation. The results of applying a simulated harmonic oscillator model of the aromaticity (HOMA)16 of the phenyl group in 32a˙˙ (0.927) suggests that its aromaticity is relatively high (i.e., roughly equal to that of benzene). In contrast, the respective C5–C6, C6–C7 and C7–C8 bond lengths in 32a˙˙* are 1.47, 1.37, and 1.44 Å, suggesting that the degree of bond alternation is higher than in the ground state and, as a result, that the aromatic character of the phenyl group is reduced upon excitation to the T1 state. The greatly lower HOMA value (0.187) calculated for the phenyl group of 32a˙˙* also indicates that its aromaticity is strongly reduced.
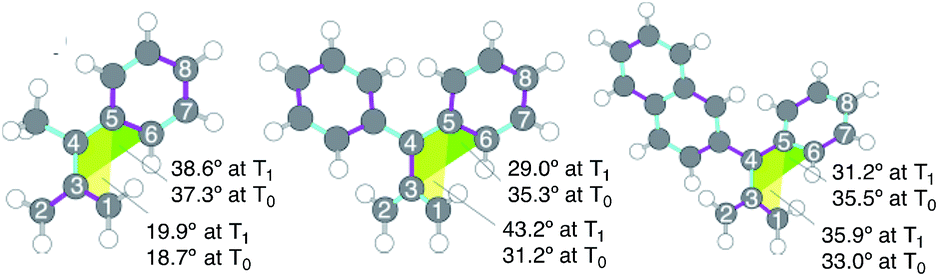 | ||
| Fig. 2 Optimized structures and atom labeling of (left) 32a˙˙*, and (center) 32b˙˙*, and (right) 32c˙˙*. Pink and blue bonds indicate elongated and shortened bonds of 32˙˙* relative to 32˙˙. | ||
| Biradicals | Bond lengths/Å | HOMAa | ||||
|---|---|---|---|---|---|---|
| C3–C4 | C4–C5 | C5–C6 | C6–C7 | C7–C8 | ||
| a Aromaticity indices of the phenyl group. | ||||||
| 32a˙˙ | 1.45 | 1.46 | 1.42 | 1.39 | 1.40 | 0.927 |
| 32a˙˙* | 1.44 | 1.44 | 1.47 | 1.37 | 1.44 | 0.187 |
| 32b˙˙ | 1.47 | 1.47 | 1.42 | 1.39 | 1.40 | 0.929 |
| 32b˙˙* | 1.49 | 1.43 | 1.45 | 1.37 | 1.42 | 0.583 |
| 32c˙˙ | 1.47 | 1.47 | 1.42 | 1.39 | 1.40 | 0.931 |
| 32c˙˙* | 1.47 | 1.45 | 1.39 | 1.42 | 1.40 | 0.896 |
The changes in the C–C bond lengths taking place in proceeding from the diphenyl-substituted TMM biradical, 32b˙˙, to 32b˙˙* are similar but somewhat less pronounced in comparison to those seen for the 32a˙˙/32a˙˙* system (Table 1). The calculated HOMA value changes are in accord with the reduction of aromaticity taking place in the phenyl group of 32a–b˙˙ upon electronic excitation, with the largest change occurring in the monophenyl-substituted TMM biradical 32a˙˙. On the other hand, the calculated bond lengths and HOMA values for 32c˙˙/32c˙˙* deviate from this trend. In this case, the degree of aromaticity of the single phenyl group is less affected by excitation, presumably because excitation is localized mainly in the larger π-conjugated naphthyl group. The calculated dihedral angles composed of C1–C3–C4–C5 (yellow, Fig. 2) and C3–C4–C5–C6 (green) for the 32˙˙/32˙˙* systems also follow these trends.
An analysis of their electronic structures provides further information about the nature of the excited TMM biradicals 32˙˙*. Spin distributions of 32a–c˙˙ and 32a–c˙˙* at Franck–Condon geometries were determined at the (TD-)UB3LYP/6-31G* level of theory (Fig. 3a). The results show that the spin densities of both 32a˙˙ and 32b˙˙ are almost completely localized at C4 (benzylic), C1 (allylic), and C2 (allylic). For 32a–c˙˙, the spin densities at C1 and C2 are almost identical to those of the corresponding ground state, 32a–c˙˙. In contrast, the spin density at C4 of 32a˙˙* is located close to the ipso-position (C5) of the phenyl group. The change taking place in spin densities at each atom in the 32b˙˙ → 32b˙˙* transition is similar to those occurring in the 32a˙˙ → 32a˙˙* transition. Interestingly, the spin density changes caused by the 32c˙˙ → 32c˙˙* transition occur most prominently in the 2-naphthyl group. The alterations in electronic structure are consistent with the geometrical changes that take place in all of the 32˙˙ → 32˙˙* transitions (see the discussion in the previous section).
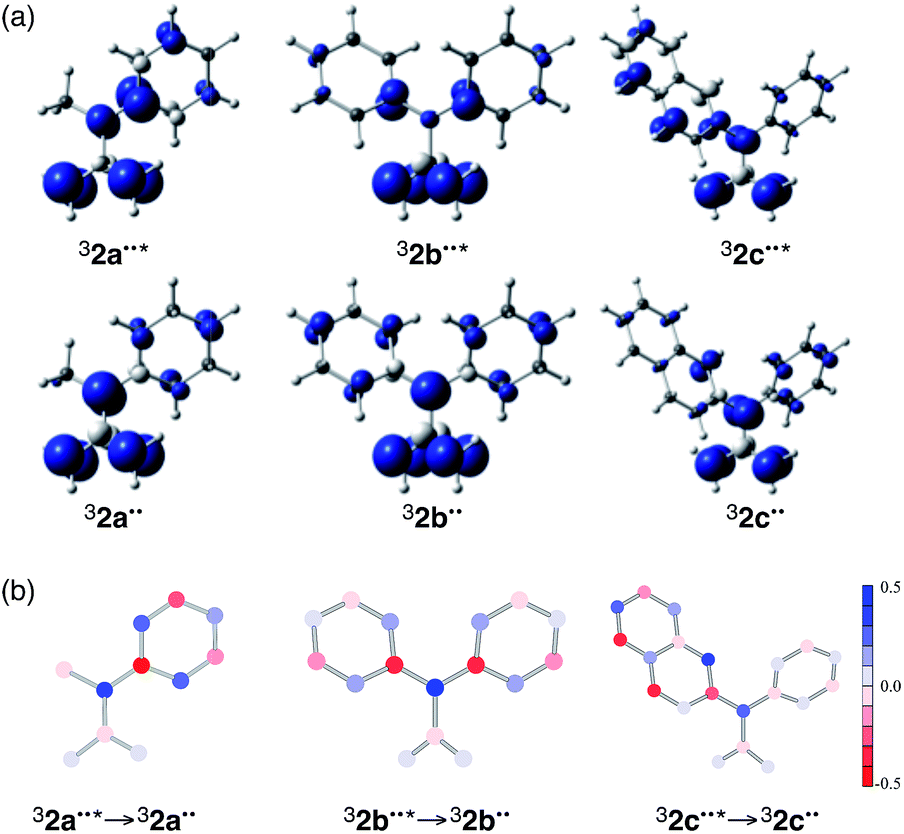 | ||
| Fig. 3 (a) Spin distributions for 32a–c˙˙ and 32a–c˙˙* at Franck–Condon geometries. Blue and gray represent positive and negative areas of spin densities, respectively. (b) T1–T0 difference charges occurring in the 32˙˙* → 32˙˙ transitions calculated from natural orbitals for α- and β-spin. Red and blue represent the increase and decrease of α-spin density, respectively, upon excitation. | ||
To express the spin distribution changes that take place in 32˙˙* → 32˙˙ transitions in a quantitative manner, we express the difference in the spin density of the ith atom, 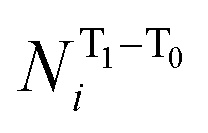 by the equation given in eqn (1),
by the equation given in eqn (1),
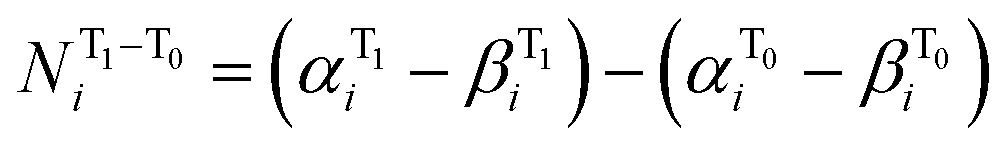 | (1) |
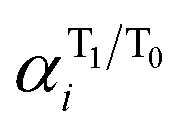 and
and  are the respective natural spin densities estimated from α and β natural orbitals for 32˙˙* and 32˙˙ (see Tables S7–S9†). Inspection of the data listed in Fig. 3a shows that the spin distributions at C1, C2 and C3 (the allyl moiety) do not significantly change during the 32˙˙* → 32˙˙ transition. Moreover, the sum of their spin densities is approximately 1.1 each for 32a–c˙˙ and 32a–c˙˙* (see Table S10†). Thus, it appears that one unpaired electron is delocalized over the allyl moieties of both 32a–c˙˙ and 32a–c˙˙*. The other unpaired electron in the T1 excited state (32˙˙*) is delocalized over the remainder of the molecule, whereas it is localized on C4 in 32a–c˙˙. The electronic structure change occurring upon excitation is also made apparent by inspection of the flow of spin density from C4 to C5 (Fig. 3b). The excitation-promoted, spin density reorganization decreases on proceeding from 32a˙˙ to 32c˙˙. In addition, introduction of benzene or naphthalene rings leads to an enhancement in electron delocalization in the T0 state (Tables S7–S9†). The analysis presented above indicates that electronic structures of 32a–c˙˙ and 32a–c˙˙* can be schematically depicted in the manner shown in Scheme 1. As a result, the distribution of the second unpaired electron is controlled by introduction of aromatic groups on the TMM biradical backbone (e.g., 32c˙˙).
are the respective natural spin densities estimated from α and β natural orbitals for 32˙˙* and 32˙˙ (see Tables S7–S9†). Inspection of the data listed in Fig. 3a shows that the spin distributions at C1, C2 and C3 (the allyl moiety) do not significantly change during the 32˙˙* → 32˙˙ transition. Moreover, the sum of their spin densities is approximately 1.1 each for 32a–c˙˙ and 32a–c˙˙* (see Table S10†). Thus, it appears that one unpaired electron is delocalized over the allyl moieties of both 32a–c˙˙ and 32a–c˙˙*. The other unpaired electron in the T1 excited state (32˙˙*) is delocalized over the remainder of the molecule, whereas it is localized on C4 in 32a–c˙˙. The electronic structure change occurring upon excitation is also made apparent by inspection of the flow of spin density from C4 to C5 (Fig. 3b). The excitation-promoted, spin density reorganization decreases on proceeding from 32a˙˙ to 32c˙˙. In addition, introduction of benzene or naphthalene rings leads to an enhancement in electron delocalization in the T0 state (Tables S7–S9†). The analysis presented above indicates that electronic structures of 32a–c˙˙ and 32a–c˙˙* can be schematically depicted in the manner shown in Scheme 1. As a result, the distribution of the second unpaired electron is controlled by introduction of aromatic groups on the TMM biradical backbone (e.g., 32c˙˙).
Previously, Ikeda and co-workers experimentally determined the wavelength maxima for T1 → T0 fluorescence of 32˙˙* by using TL methods.5a The TL maxima of 32a˙˙*, 32b˙˙* and 32c˙˙* in methylcyclohexane matrices at ca. 130 K are 451, 501, and 602 nm, respectively. In the present work, we calculated the fluorescence wavelengths of 32a–c˙˙* using various TD-DFT functionals (Table 2). Interestingly, the fluorescence wavelengths obtained by using the UB3LYP method match well with the experimentally determined values.5 Although use of the UM06-2X method also gives maxima that are close to the experimental values, it slightly overestimates the excitation energies. Utilization of the long-range corrected CAM-UB3LYP functional does not lead to improved results, suggesting that charge-transfer interactions are not important in 32˙˙*. The use of UHF in this functional for treatment of large electronic separations suffers from a more serious spin contamination (〈S2〉 ∼2.54–4.01), especially in the case of 32c˙˙.
| Biradical | Expa | TD-DFTb | ||
|---|---|---|---|---|
| TL | UB3LYP | UM06-2X | CAM-UB3LYP | |
| λTL/nm | λEm/nm (〈S2〉, f) | λEm/nm (〈S2〉, f) | λEm/nm (〈S2〉, f) | |
| a From ref. 5a.b The 6-311+G(d) and 6-31G(d) basis sets were employed for single point calculations and optimizations, respectively. | ||||
| 32a˙˙ | 451 | 443 (2.43, 0.003) | 438 (2.28, 0.005) | 440 (3.10, 0.001) |
| 32b˙˙ | 501 | 479 (2.24, 0.010) | 464 (2.23, 0.018) | 443 (2.54, 0.007) |
| 32c˙˙ | 602 | 613 (2.63, 0.002) | 554 (2.39, 0.004) | 903 (4.01, 0.000) |
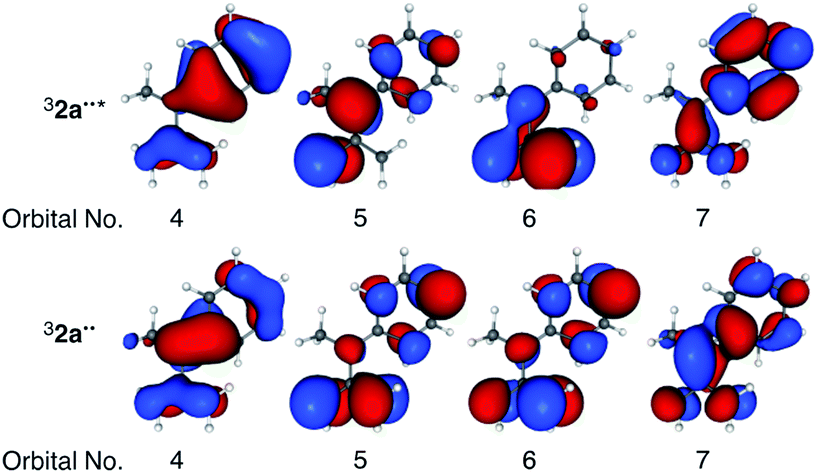
To validate the (TD-)DFT results presented above, the electronic configuration of 32a˙˙* was evaluated by employing post-Hartree–Fock CASSCF and multireference MP2 calculations. First, 10 π-orbitals of 32a˙˙ were obtained using the RHF/6-311+G(d) method for the singlet spin state. Subsequent CASSCF calculations were carried out using a full π-orbital (10e, 10o) active space for the triplet ground state. In Table 3 are listed the corresponding natural orbital occupation numbers (NOON), where 2, 1, and 0 indicates doubly-, singly-, and un-occupied orbitals, respectively. Importantly, a deviation of NOON from integer numbers implies that the wavefunction has multireference character.17 Analysis of the NOON values for 32a˙˙ indicates the existence of four doubly-occupied (NOON > 1.87), two singly-occupied (NOON ∼ 1.00), and four un-occupied (NOON < 0.13) orbitals within the active space. On the other hand, the NOON values of orbitals numbers 4 and 7 in 32a˙˙* deviate largely from integer values, signaling that the wavefunction has multireference character. Except for natural orbitals 4–7, the NOON values and orbital shapes of 32a˙˙* and 32a˙˙ are almost identical. The singly-occupied natural orbital of 32a˙˙ is localized on the TMM moiety, while singly occupied natural orbitals of 32a˙˙* (Table 3) are delocalized on the phenyl group. The fact that this finding is consistent with the natural charge distributions obtained by using (TD-)DFT calculations (Fig. 3) validates the DFT-based results.
 |
|||||||||||
| NOON | Orbital no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 32a˙˙* | 1.92 | 1.90 | 1.88 | 1.47 | 1.02 | 0.97 | 0.55 | 0.12 | 0.09 | 0.07 | |
| 2a˙˙ | 1.96 | 1.92 | 1.90 | 1.87 | 1.00 | 1.00 | 0.13 | 0.10 | 0.08 | 0.04 | |
The CASSCF-determined emission energy of the benchmark model 32a˙˙ is 2.62 eV (473 nm) and that arising from MRMP2 is 2.51 eV (495 nm). The MRMP2 result is in good agreement with that determined experimentally (2.74 eV, 451 nm), and the TD-DFT determined emission energy (2.82 eV, 440 nm) is also close to the experimental value. These findings suggest that a well-balanced use of dynamic and non-dynamic correlations is necessary for quantitatively accurate assessment of the emission wavelengths of 32˙˙*. The somewhat surprising similarity of the MRMP2 and UB3LYP results show that conceptually more simple density functional approaches can be applied to the complex electronic and molecular structures of 32˙˙*. This is most likely a consequence of fortuitous error compensations.
In conclusion, studies aimed at understanding the T1–T0 fluorescence of aryl-substituted TMM biradicals 32˙˙* were carried out using quantum chemical calculations. The results of calculations using high-level ab initio and various DFT functionals reveal that the wavefunctions of 32˙˙* possesses significant multireference character. However, use of DFT approaches with sufficiently large basis sets leads to predictions of reasonable emission wavelengths of 32˙˙*. In addition, the calculated changes of bond lengths and spin distributions demonstrate that significant relaxation of molecular geometries occurs in T1 on going from an aromatic ground state to quinoid structured excited state. The overall results of this work demonstrate that it is possible to use DFT-based methods to estimate accurately the fluorescence properties of organic open-shell species.18 This ability should accelerate the development and application of luminescent radical materials.
Acknowledgements
The authors acknowledge valuable discussions with Assistant Professor Daisuke Yokogawa in Nagoya University. This research was financially supported by A-STEP (No. AS231Z02594D) and ALCA (No. 1125217700) by Japan Science and Technology Agency (JST). K.U. appreciates support by the IGER program of Nagoya University. H. I. gratefully acknowledges financial support in the form of a Grant-in-Aid for Innovative Areas “π-Space” (No. 21108520 and 23108718 in the Area No. 2007) and “Stimuli-responsive Chemical Species” (No. 24109009 in the Area No. 2408), and the Scientific Research (B) (No. JP20044027 and JP23350023 in the Area No. 2408), and the Challenging Exploratory Research (No. JP21655016 and JP24655037) from JSPS, Japan. S. I. was partially supported by CREST grant from JST.Notes and references
- (a) P. Dowd, Acc. Chem. Res., 1972, 5, 242–248 CrossRef CAS; (b) J. A. Berson, Acc. Chem. Res., 1978, 11, 446–453 CrossRef CAS; (c) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- L. V. Slipchenko and A. I. Krylov, J. Chem. Phys., 2003, 118, 6874–6883 CrossRef CAS.
- (a) J. A. Berson, R. J. Bushby, J. M. McBride and M. Tremelling, J. Am. Chem. Soc., 1971, 93, 1544–1546 CrossRef CAS; (b) M. S. Platz, J. M. McBride, R. D. Little, J. J. Harrison, A. Shaw, S. E. Potter and J. A. Berson, J. Am. Chem. Soc., 1976, 98, 5725–5726 CrossRef CAS; (c) N. J. Turro, M. J. Mirbach, N. Harrit, J. A. Berson and M. S. Platz, J. Am. Chem. Soc., 1978, 100, 7653–7658 CrossRef CAS.
- H. Ikeda, H. Namai, H. Taki and T. Miyashi, J. Org. Chem., 2005, 70, 3806–3813 CrossRef CAS PubMed.
- (a) H. Namai, H. Ikeda, Y. Hoshi, N. Kato, Y. Morishita and K. Mizuno, J. Am. Chem. Soc., 2007, 129, 9032–9036 CrossRef CAS PubMed; (b) H. Ikeda, Y. Matsui, I. Akimoto and K. Kan'no, Aust. J. Chem., 2010, 63, 1342–1347 CrossRef CAS; (c) Y. Matsui, H. Namai, I. Akimoto, K. Kan'no, K. Mizuno and H. Ikeda, Tetrahedron, 2011, 67, 7431–7439 CrossRef CAS.
- (a) P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef; (b) W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef; (c) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS; (d) M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS; (e) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- (a) Y. Matsui, T. Kido, E. Ohta and H. Ikeda, Chem. Commun., 2014, 50, 13963–13966 RSC; (b) Y. Matsui, D. Kawahara, E. Ohta and H. Ikeda, Phys. Chem. Chem. Phys., 2013, 15, 7064–7069 RSC.
- (a) Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (d) T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- (a) P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213–222 CrossRef CAS; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (c) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- B. O. Roos, Adv. Chem. Phys., 1987, 69, 399–446 CrossRef CAS.
- (a) K. Hirao, Chem. Phys. Lett., 1992, 190, 374–380 CrossRef CAS; (b) K. Hirao, Chem. Phys. Lett., 1992, 196, 397–403 CrossRef CAS; (c) K. Hirao, Chem. Phys. Lett., 1993, 201, 59–66 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A Boatz, S. T. Elbert, M. S. Gordon, J. J. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- H. J. Werner, et al., MOLPRO, version 2010, a package of ab initio programs; see http://www.molpro.net.
- For the details, see the ESI.†.
- HOMA is an index of aromaticity obtained by considering bond lengths of phenyl-rings. HOMA values close to 1 and 0 indicate high and low aromaticity, respectively. See: J. Kruszewskia and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- S. Yamanaka and K. Yamaguchi, Bull. Chem. Soc. Jpn., 2004, 77, 1269–1286 CrossRef CAS.
- (a) H. Namai, H. Ikeda, Y. Hoshi and K. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 7396–7398 CrossRef CAS PubMed; (b) Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2015, 54, 3731–3734 CrossRef CAS PubMed; (c) Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef CAS PubMed; (d) Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of all optimized structures, natural spin densities, natural charges, full citations for ref. 12 and 14. See DOI: 10.1039/c6ra16580j |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2016 |