
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Guest-induced stereoselective self-assembly of quinoline-containing PdII and PtII metallacycles†
Víctor Blanco*ab,
Dolores Abellab,
Tamara Ramab,
Cristina Alvariñob,
Marcos D. García*b,
Carlos Peinador*b and
José M. Quintelab
aDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Granada, Campus Fuentenueva s/n, 18071, Granada, Spain. E-mail: victorblancos@ugr.es
bDepartamento de Química Fundamental and Centro de Investigacións Científicas Avanzadas (CICA), Facultade de Ciencias, Universidade da Coruña, 15071, A Coruña, Spain. E-mail: carlos.peinador@udc.es; marcos.garcia1@udc.es
First published on 16th August 2016
Abstract
A family of mono- and dinuclear metallacycles has been self-assembled from (en)M(NO3)2 metal centers (M = PdII or PtII) and 4,4′-bipyridinium/2,7-diazapyrenium-based ligands incorporating quinoline subunits in their structures. These receptors recognize aromatic substrates, up to the dimensions of naphthalene, by means of hydrophobic forces, π–π stacking and C–H⋯π interactions. In the case of the M2L2 receptors, the quinoline moieties in the short sides of these molecular rectangles, cause the existence of two atropisomers with different cavity characteristics. The formation of inclusion complexes, derived from the dinuclear hosts and appropriate aromatics, induced stereoselectivity on the self-assembly, which was found to be modulated mainly by the size of the guests and their ability to optimize the number of potential C–H⋯π interactions with the two isomeric receptors. In addition, the π-acceptor and hydrophobic character of the host also contributes to this stereoselectivity. The proposed mechanism for the interconversion of the syn/anti isomers, studied by DFT methods, involves the rotation of the quinoline rings without dissociation of the self-assembled metallacycles.
Introduction
Chemists have long been fascinated by the way nature achieves selectivity through molecular recognition.1–5 In the most prominent example, enzymes reach astonishing levels of substrate discrimination by producing conformational changes in their structures as hosts following Koshland's induced-fit model.6,7 Even though supramolecular chemistry has tried to emulate the efficiency of enzymes as highly selective receptors, the prevalent approximation has relied on the use of the more simplistic Fischer's lock-and-key model;8 with the success of the molecular recognition event relying on the complementarity between the rigid binding partners in terms of size and shape.9,10 Therefore, there is a great need for the development of conformationally-flexible synthetic hosts,11 molecular receptors able to assemble and modify their structures on the basis of the characteristics of their binding partners.12–22Having this into account, coordination-driven self-assembly23–27 has been established as a powerful tool for the rational and modular construction of innumerable 2D/3D metallacyclic receptors.28–34 Among the numerous advantages of these hosts compared with their organic counterparts,23–34 it should be emphasized that the structural features of their inner cavities can be controlled by a cautious choice of ligands. Furthermore, metallic ions facilitate not only their solubility in aqueous or organic media, but also give access to different geometries and allow the introduction of properties on the hosts associated with the metal employed.
In our particular case, part of our research is focused on the synthesis of new square and rectangular-shaped PdII/PtII metallacycles.35–41 These hosts are synthesized by coordination-driven self-assembly of N,N′-dialkyl or N-monoalkyl-4,4′-bipyridinium/2,7-diazapyrenium salts as bidentate ligands around (en)ML2 centers (M = PdII/PtII, L = NO3, OTf),42 an approach that sums up to the above-mentioned advantages of metallacycles the strong π-deficient character of the resulting receptors, implementing π–π and C–H⋯π interactions on the binding processes.43
In the present work,44 we planned the introduction of quinoline moieties in the short sides of our dinuclear molecular rectangles. These heterocycles can rotate around a CH2–N axis, giving rise to the existence of atropisomeric metallacyclic receptors with different cavity characteristics. It will be shown that these conformationally-flexible hosts can modify their shapes on the stereoselective binding with different guests, maximizing in that manner the intermolecular interactions in a way reminiscent to the induced-fit found in enzymes.
Results and discussion
Synthesis of the quinoline-containing ligands L1–L4
Ligands L1, L2,44 L3 and L4 were obtained as their hexafluorophosphate salts by N-alkylation of one of the free nitrogen atoms of the corresponding precursors 1·2PF6,45 3 or 4 with 4-chloromethylquinoline (2a) or 4-chloromethyl-2-methylquinoline (2b), followed by counterion exchange with NH4PF6 (Scheme 1). Anion metathesis of L1·3PF6 and L2–L4·PF6, by subsequent treatment with Amberlite® CG-400 ion-exchange resin and AgNO3, afforded the corresponding water-soluble nitrate salts of the ligands, which were characterized by NMR spectroscopy and mass spectrometry. The identity of L4 was further confirmed by X-ray diffraction of appropriate single crystals obtained for its chloride salt (see the ESI†). | ||
| Scheme 1 Synthesis of the quinoline-containing ligands L1–L4. | ||
 | ||
| Scheme 2 Template-assisted self-assembly of metallacycles M1a,b and M2a,b15 (en = ethylenediamine). | ||
Addition of 1 equiv. of the PdII complex 5a to a 10 mM solution of ligand L1·3NO3 in D2O, led to a mixture of several species as shown by the presence of multiple sets of signals in the 1H NMR spectrum. This behavior is similar to that shown by the simpler analogous ligand L5 (bearing a pyridine ring instead of the quinoline unit, Scheme 2),46 but, unlike this previously reported system, a concentration decrease to 0.5 mM did not resulted in the full reorganization into a single species (Fig. S1, ESI†), suggesting that the presence of the larger quinoline moiety hinders the self-assembly process.
Nevertheless, this limitation could be surpassed by the addition of 3 equiv. of sodium p-phenylenediacetate (PDA) to the initial equimolar mixture of 5a and L1·3NO3. Here, the electron-rich aromatic compound acts as a template for the self-assembly of the metallacycle, producing quantitatively the inclusion complex PDA⊂M1a·5NO3 (Scheme 2), as observed by 1H NMR spectroscopy (i.e. the signals of the quinoline hydrogens Hn, Ho and Hs show a pronounced downfield shift, ΔδHs = 1.80 ppm, ΔδHn = 0.91 ppm and ΔδHo = 0.75 ppm, typical for the coordination of this heterocycle to PdII centers) (Fig. S2, ESI†).
The self-assembly of the corresponding PtII metallacycle M1b showed similar characteristics than those observed for the PdII system. Thus, when an equimolar solution of ligand L1·3NO3 and (en)Pt(NO3)2 (5b) (in the 10–0.8 mM concentration range) was subjected to the conditions required to labilize the more inert Pt–N(Py) bond and achieve thermodynamic control, i.e. heating at 100 °C for 7 days, a mixture of different species was obtained (Fig. S3, ESI†). Conversely, when a 6 mM equimolar mixture of L1·3NO3 and 5b was heated in H2O at 100 °C for 7 days in the presence of 1.7 equiv. of PDA, the self-assembly process was more efficient, showing a higher extent of reorganization into a single species. Moreover, the inert character of the Pt–N(Py) bond at room temperature, that produces concentration-independent PtII species, allowed the isolation of the void metallacycle M1b as its water-insoluble hexafluorophosphate salt, after addition of an excess NH4PF6 to the initial aqueous solution of PDA⊂M1b·5NO3.
As shown in Fig. 1, NMR spectroscopy in CD3NO2 confirmed the presence of M1b·5PF6 as the major species, with signals displaying chemical shifts, compared with those of the free ligand L1·3PF6, compatible with the PtII metallacycle. Along with the expected downfield shift experienced by the signals of the quinoline hydrogens Hn–p and Hr–s upon coordination (for instance, ΔδHs = 1.44 ppm, ΔδHn = 0.59 ppm and ΔδHo = 0.56 ppm), the signals of the bipyridine units (Ha–d and Hi–l) and the phenylene spacer (Hf,g), are consistently shifted to lower frequencies as a result of the shielding experienced by their temporary location inside of the inner metallacycle cavity through rotation of the pyridine rings of the bipyridine units. The structure of M1b·5PF6 was further confirmed by mass spectrometry, with the FAB-MS spectrum showing peaks at m/z = 1393.2, 1248.3, 1102.3 and 957.3,47 corresponding to the loss of one to four counterions, consecutively (Fig. S4, ESI†).
 | ||
| Fig. 1 Partial 1H NMR (CD3NO2, 500 MHz) spectra of: (a) ligand L1·3PF6; (b) metallacycle M1b·5PF6; (c) inclusion complex HQ⊂M1b·5PF6. The lettering corresponds to that shown on Scheme 1. | ||
Isolation of the void mononuclear metallacycle M1b·5PF6 allowed the study of its behavior as host. Beforehand, and with the aim of gain some insight into the structure of the receptor, this was characterized by means of DFT calculations at the B3LYP level (Table 1). Here, the optimized structure of M1b displays the typical rectangular shape of this type of systems,43 with an inner cavity defined by the bipyridinium, phenylene and quinoline moieties arranged orthogonally to the mean plane of the metallacycle. A comparison of the different distances of the cavity of M1b with the simpler pyridine-containing analogue M2b,46 showed that the presence of the quinoline ring has little influence on the size of the host (Table 1). The distance between the bipyridine units within M1b is 3.85–3.98 Å, varying only <0.1 Å respect to the reference species M2b, and quite close to the ideal value for the inclusion of aromatic rings. The length of the long side of the rectangle remains essentially unchanged in both metallacycles (≈6.8 Å). Therefore, and on the basis of the DFT calculations, the quinoline moiety should have no influence on the requirements of size and shape of the potential guests for the metallacyclic host M1b.
|
||||
|---|---|---|---|---|
| Heterocyclic ring on the short side | ab | bb | cc | dd |
| a The dimensions correspond to the centroid–centroid or atom–atom distances after subtracting two times the van der Waals radius of C.b Distances between the centroids of the corresponding pyridine rings.c Distances between the centroids of the pyridine ring in the quinoline moiety and the phenylene ring on the short side of the rectangle.d Distance between methylene groups.e From ref. 46. | ||||
| Quinoline (M1b) | 3.98 | 3.85 | 6.80 | 7.83 |
| Pyridinee (M2b) | 4.02 | 3.87 | 6.76 | 7.80 |

With this information in our hands, we proceeded to study in CD3NO2 the inclusion of a selected series of aromatics (hydroquinone (HQ), 1,4-bis[2-(2-hydroxyethoxy)ethoxy]benzene (BHEB) and 1,5-bis[2-(2-hydroxyethoxy)ethoxy]naphthalene (1,5NPH)) within M1b. Firstly, addition of 3 equiv. of HQ or BHEB resulted in the formation of the corresponding inclusion complexes as can be deduced from the inspection of the NMR spectra (Fig. 1c and S5, ESI†). The signals of the phenylene and quinoline rings show a downfield shift, accounting for the existence of C–H⋯π interactions in solution, between the hydrogen atoms of the guest and the aromatic rings on the short sides of the host. Conversely, hydrogens in β-position of the pyridine rings, located in the central part of the cavity, shift upfield due to the shielding produced by the aromatic ring of the guest. On the other hand, addition of 1 equiv. of 1,5NPH also resulted in changes in the 1H NMR spectrum which, however, could not be analyzed as a result of it is complexity, probably due to the lower symmetry of the aggregate, as a result of the insertion of the guest and the equilibrium established between the two insertion modes of the naphthalene moiety.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the syn and anti isomers.
:1 mixture of the syn and anti isomers.
 | ||
| Scheme 3 Template-assisted self-assembly of metallacycles anti/syn-M3a. | ||
In that case, although the PtII system M3b was efficiently prepared from an equimolar 0.5 mM solution of ligand L2·NO3 and 5b, a template was again needed to obtain the PdII analogue M3a·6NO3 (Scheme 3). When the template-assisted self-assembly was studied with a small group of selected aromatic guests, we observed by NMR spectroscopy that 1,5NPH promoted the completely stereoselective self-assembly of the corresponding inclusion complex. X-ray crystallography showed that the anti-M3a isomer was the sole atropisomer present in 1,5NPH⊂M3a·6NO3, with the observed stereoselectivity being attributed to an increased number of potential C–H⋯π interactions between the guest and the anti-receptor compared to its syn isomer.44
In the present work, we further investigated this stereoselective self-assembly process, trying to discern the influence of the nature and the substitution pattern of the guest in the stereoselectivity. Both phenylene and naphthalene electron-rich derivatives were chosen as substrates, as they show the adequate size to fit into the host.
Not only compounds with the hydroxyl groups in different positions were tested, but also derivatives with diethyleneglycol linkages, which have been shown to contribute to the overall stability of the complexes by means of C–H⋯O hydrogen bonds.35–43
The results obtained are summarized in Table 2, showing several general trends. Firstly, single phenylene compounds did not induce any stereoselectivity (entries 1–5, 18), and only certain naphthalene derivatives (entries 7, 9–11, 13) were found to modify the expected 50:50 syn/anti ratio. Whether the electron donating group is a hydroxyl or a diethyleneglycol moiety has a certain impact on the stereoselectivity, but does not seem to play a key role. Hence, only slight improvements in the isomer ratio are observed when the latter group is present compared with the hydroxy-substituted compounds (entries 9, 11), probably due to its larger size. However, 2,7NPH does not follow this trend and the nature of the electron-donating group showed strong impact on the stereoselectivity (entry 13), as it induces full reorganization to a single isomer, while 2,7DHN, its hydroxy analogue, maintains the initial 50:50 ratio (Table 2, entry 12).
| Entry |
|
Guest | Isomers ratio | |
|---|---|---|---|---|
| a The ratio was determined by integration of 1H NMR signals corresponding to the different isomers.b From ref. 44.c No analysis was possible due to the broad signals observed in the 1H NMR spectrum.d The self-assembly was not complete as other minor species were observed in the 1H NMR spectrum. | ||||
| 1 |  |
HQ | R = H | 50:50b |
| 2 | BHEB | R = (CH2)2O(CH2)2OH | 50:50b |
|
| 3 |  |
RS | R = H | 50:50b |
| 4 | 1,3RS | R = (CH2)2O(CH2)2OH | 50:50 |
|
| 5 |  |
THB | 50:50b |
|
| 6 |  |
1,5DHN | R = H | —c |
| 7 | 1,5NPH | R = (CH2)2O(CH2)2OH | 100:0b |
|
| 8 |  |
1,3DHN | R = H | 50:50 |
| 9 | 1,3NPH | R = (CH2)2O(CH2)2OH | 66:33b |
|
| 10 |  |
2,3DHN | R = H | 60:40d |
| 11 | 2,3NPH | R = (CH2)2O(CH2)2OH | 70:30 |
|
| 12 |  |
2,7DHN | R = H | 50:50d |
| 13 | 2,7NPH | R = (CH2)2O(CH2)2OH | 100:0d |
|
| 14 |  |
1,6DHN | R = H | 50:50d |
| 15 | 1,6NPH | R = (CH2)2O(CH2)2OH | —c | |
| 16 |  |
2,6DHN | R = H | 50:50d |
| 17 | 2,6NPH | R = (CH2)2O(CH2)2OH | —c | |
| 18 |  |
PDA | 50:50 |
|
| 19 |  |
Naphthalene | 50:50d |
|

Whilst 1,5NPH was found to favor the anti-stereoisomer, as previously shown, 1,3NPH and 2,3NPH should favor syn-M3a if we assume that they adopt a similar insertion mode. Here, the steric hindrance between the polyether chains in position 2 or 3 of the guest and one of the quinoline rings in the anti-isomer would be, potentially, the factor determining the preference for the syn-isomer. As anticipated, inspection of the 1H NMR spectra showed that both 1,3NPH and 2,3NPH did in fact induce a change in the syn/anti ratio, although in a less efficient manner that the effect exerted by 1,5NHP, as only 66:33 and 70:30 ratios were achieved, respectively (Table 2, entries 9, 11).44 This behavior is consistent with the number of host–guest C–H⋯π interactions established being a key factor on the observed stereoselectivity, as 1,3NPH and 2,3NPH can only give rise to three C–H⋯π interactions with the quinoline units in the supposedly preferred syn-M3a·6NO3 isomer, instead of the four established in 1,5NPH⊂anti-M3a·6NO3. The higher ratio observed for 2,3NPH compared to the 60:40 ratio induced by 2,3DHN supports the hypothesis of the steric clash between the polyether chains and the quinoline rings of the host being a factor in the stereoselectivity. However, the similarity between the atropisomers precluded the assignment of the two sets of signals observed by NMR spectroscopy and, unfortunately, no good quality single crystals could be obtained for the inclusion complexes 1,3NPH/2,3NPH⊂M3a·6NO3 to unambiguously identify the major metallacycle species as the syn isomer. Nevertheless, the data in Table 2 supports the conclusions of our previous study, allowing us to further corroborate that the nature and the substitution pattern of the aromatic guest plays a key role in the observed stereoselectivity of self-assembly processes.44
For a series of reasons, the proposed mechanism for the syn–anti interconversion was pointed out to be the rotation of the quinoline ring around the M–N(quinoline)–CH2 axis, crucially without dissociation of the M–N(quinoline) bond (Fig. 2). The experimental Gibbs free energy barrier for this process in the corresponding PtII metallacycle M3b·6PF6 was found to be ΔG‡exp = 75.3 kJ mol−1, as measured by the coalescence method,48 and resulted in good agreement with the theoretical value of ΔG‡calc = 69.9 kJ mol−1 determined by DFT calculations.44 To further support the proposed interconversion mechanism, the energy barrier was also determined for the corresponding PdII metallacycle in the inclusion complex HQ⊂M3a·6NO3. In this case, the experimental Gibbs free energy barrier was measured applying the coalescence method to the signals between 8.60 and 8.80 ppm (Fig. S6, ESI†), obtaining a value of ΔG‡exp = 71.2 kJ mol−1, very close to that observed for the PtII metallacycle. DFT calculations at the M06 level in vacuo, using the Synchronous Transit-guided Quasi-Newton method,49,50 showed that both the syn and anti atropisomers are connected through a transition state in which the quinoline unit is rotated by ca. 90° around the M–N(quinoline)–CH2. As in the analogous PtII metallacycle, no dissociation of the Pd–N(quin) is observed. The rotation barrier was calculated as ΔG‡calc = 63.6 kJ mol−1. The good agreement between the experimental values of the energy barriers for M3a and M3b reinforces the validity of our proposed mechanism, as these energies seem to be essentially independent of the nature of the square-planar metal center, despite PdII– and PtII–pyridine complexes showing a quite different degree of kinetic lability. In this direction, when 1,5NPH was added at room temperature to a 1:1 mixture of syn/anti-M3b·6PF6 in CD3NO2, a sole atropisomer was detected in solution, with only one set of signals for the quinoline moieties observed in the 1H NMR spectrum recorded at 253 K (Fig. S7, ESI†). This spectrum is compatible with the inclusion complex 1,5NPH⊂11b·6PF6, with some signals of the naphthalene moiety strongly shielded (δ = 2.5 and 3.5 ppm) due to its inclusion inside the cavity, and all the resonances of the bipyridinium units being non-equivalent as a result of the loss of symmetry produced by the slow exchange between the two possible insertion modes for the naphthalene guest. As the Pt–N(Py) bond is kinetically inert at room temperature, the reorganization of the system towards a single stereoisomer further supports the proposed mechanism for the syn/anti interconversion of the atropisomers.
 | ||
| Fig. 2 Proposed mechanism for the interconversion of the quinoline-containing dinuclear metallacycles exemplified for syn/anti-M3a. | ||
 | ||
| Scheme 4 Crystallization-mediated self-assembly of inclusion complexes guest⊂anti-M4·6NO3. | ||
Nevertheless, X-ray diffraction-quality single crystals of the inclusion complex HQ⊂anti-M4·6NO3 could be grown from a solution of ligand L3·NO3, (en)Pd(NO3)2 (5a) and HQ in NaNO3(aq) (3 M) at 4 °C (Scheme 4). The obtained crystal structure shows the inclusion of a hydroquinone molecule inside the cavity of the host, with only the anti-M4 stereoisomer present in the structure (Fig. 3). The distances between HQ and the bipyridinium units are 3.39 and 3.42 Å, adequate to maximize the π–π interactions. Other structural features of the inclusion complex in the solid state are in good agreement to those reported to analogous systems,43 with the quinoline units of the host almost orthogonal (85°) to the mean plane of the metallacycle. As a result, the CH3 groups are, as expected, in the proximity of the Pd centers (C–Pd distance: 3.12 Å), creating a steric clash that hinders the self-assembly process in solution, as evidenced also by the Pd–N(quinoline) distance being of 2.07 Å, slightly longer than the average values for the Pd–N bonds in this kind of metallacycles.
 | ||
| Fig. 3 Capped-stick representation of the crystal structure of inclusion complex HQ⊂anti-M4·6NO3. Solvent molecules, counterions and hydrogen atoms are omitted for clarity. Color scheme: C, grey; O, red; N, blue; Pd, yellow. Only one of the two disordered positions of the guest is shown. | ||
Interestingly, the 1H NMR spectrum recorded immediately after dissolving the crystals of HQ⊂anti-M4·6NO3 in D2O was much simpler than that obtained by solution self-assembly (Fig. 4). This spectrum showed the presence of a major species (ca. 85%) as one set of signals for the metallacycle was observed in vast majority, which corresponds to one of the host stereoisomers, likely the anti (Fig. 4b). The shift to higher frequencies of the signals of Hk and the methyl group (ΔδHk = 1.94 ppm and ΔδHg = 0.83 ppm), confirms the coordination of the quinoline unit to the PdII center resulting in the Hk and the methyl group being located above the coordination plane of the metal center. In addition, the shift to lower frequencies of the signal of the HQ (Δδ = −1.83 ppm) together with the upfield shift of the signals of the bipyridinium H in β position (ΔδHb = −0.58 ppm and ΔδHc = −0.72 ppm) indicate the inclusion of the guest within the metallacycle cavity. A comparison of the chemical shift of the aromatic hydrogens of the hydroquinone with previously reported systems, shows that the inclusion process should be fast on the NMR timescale and the signal observed corresponds to an average between the complexed and uncomplexed HQ.
 | ||
| Fig. 4 Partial 1H NMR (D2O, 500 MHz) spectra of: (a) ligand L3·NO3; (b) HQ⊂anti-M4·6NO3, recorded straightaway after dissolution of crystals; (c) solution (b) after 7 d. The lettering corresponds to that shown on Scheme 1. * Signal corresponding to 1,4-benzoquinone, obtained from a partial oxidation of HQ. | ||
Therefore, NMR spectroscopy probed the existence of the metallacycle M4 in aqueous media, suggesting that the cause for the solution self-assembly to fail was related to kinetic reasons rather than the thermodynamic stability of the system. Interestingly, and as expected for the results obtained for metallacycle M3a (vide supra), the 1H NMR spectrum acquired from the same sample after one week at room temperature, showed a mixture of the inclusion complexes derived from the syn and anti atropisomers in a 1:1 relationship, indicating that the expected equilibrium ratio between both isomers had been reached (Fig. 4c). This result is in good agreement with the expected higher rotational barrier of the quinoline moieties around the CH2–C and Pt–N bonds, as a consequence of the steric hindrance owed to the methyl group that slows down the interconversion process between the atropisomers.
To further study this isomerization barrier, VT NMR experiments were carried out with a solution of HQ⊂anti-M4·6NO3 in D2O (Fig. S9, ESI†). When the temperature was raised to 333.5 K, a 1:1 ratio between the syn and anti stereoisomers was readily observed, showing that at this temperature the interconversion process is faster enough to allow the process to reach equilibrium within minutes. However, slow exchange in the NMR timescale is observed, even when the temperature was raised to 358.5 K, and as it can be deducted from the 1H NMR spectrum which displays two well-defined sets of signals for the host with no evidences of coalescence.
This behavior exhibited for M4 is clearly in contrast with the results obtained for the non-substituted quinoline-based system HQ⊂syn/anti-M3a·6NO3, for which coalescence was observed at 333.5 K and fast exchange in the NMR timescale between the atropisomers at 351.5 K, a result in good agreement with a higher barrier for the methylquinoline rotation. Because the experimental value of the interconversion barrier could not be calculated experimentally as coalescence was not reached, we estimated its value as ΔG‡calc = 118.8 kJ mol−1 by means of DFT calculations at the M06 level in vacuo, using the Synchronous Transit-guided Quasi-Newton method. The comparison of the calculated transition state for the syn/anti interconversion in M4 and M3a clearly explains the higher value for the energy barrier observed in the former (Fig. S11, ESI†).
As expected, both transition states show an intermediate arrangement between the syn and anti conformations, with one of the quinoline moieties located approximately orthogonal to its orientation in both isomers (see Fig. S11, ESI†). Crucially, both systems show different degrees of distortion of the metallacyclic structure. Therefore, whilst the shape and size of the metallacycle are essentially maintained for the transition state geometry of M3a, although displaying an enlargement of Pd–N(quinoline) bond length (ca. 0.2 Å) and with the (en)Pd and the quinoline groups slightly out of the equatorial plane of the structure. In contrast, the structure of the transition state of M4 shows a more pronounced alteration. In this case, the (en)Pd group is also out of the equatorial plane of the metallacycle, yet the plane defined by the Pd and the N atoms of the ethylenediamine moiety and the mean plane of the metallacycle, defined by the remaining three corners of the molecular rhomboid, form an angle of ca. 27° in sharp contrast to the ca. 4° angle observed in M3a. Moreover, the distance between the centroids of the pyridine rings closest to the rotational center is increased by 0.55 Å in M4, while this distance remains essentially unchanged in M3a (0.03 Å increase). The origin of this pronounced distortion for M4, and therefore its increased rotational barrier when compared with M3a, can be undoubtedly attributed to the methyl group in the substituted quinoline pointing towards one of the rings of the bipyridine unit and giving rise to a significant steric clash.
Following the above-mentioned crystallization-mediated self-assembly, single crystals of the inclusion complexes RS- and 1,5NPH⊂M4·6NO3 were also obtained. The solid state structures show the formation of the expected inclusion complexes, with the corresponding aromatic guest inside the cavity of the metallacycle in its anti atropisomeric form. The dimensions of the void cavity and, other structural features of the host in both crystals, are very similar to those observed in HQ⊂anti-M4·6NO3. In particular, it should be noted that host–guest C–H⋯π interactions play a key role again on the observed atroposelectivity. For the RS substrate within RS⊂anti-M4·6NO3, these interactions are found between the hydrogen in position C4 and the benzene ring of the closest quinoline unit. In the case of 1,5NPH⊂anti-M4·6NO3, the solid state structure displays four C–H⋯π interactions between the hydrogen atoms in positions C4 and C8 and the pyridine ring of the quinoline units, and the hydrogen atoms in positions C3 and C7 and the benzene ring of the quinoline units. Additionally, C–H⋯O hydrogen bonds are established between the methylene hydrogens and those in α position of the pyridinium ring and the oxygen atoms of the polyether chains of 1,5NPH (Fig. 5).
 | ||
| Fig. 5 Solid state structure of inclusion complexes RS⊂anti-M4·6NO3 (top), and 1,5NPH⊂anti-M4·6NO3 (bottom) showing the C–H⋯π (a–c) and C–H⋯O (d–f) interactions. [H⋯π] or [H⋯O] and [C⋯π] or [C⋯O] distances and [C–H⋯π/O] angles: (a) 3.09 Å, 3.97 Å, 154°; (b) 2.69 Å, 3.61 Å, 163°; (c) 2.80 Å, 3.58 Å, 140°; (d) 2.36 Å, 3.20 Å, 148°; (e) 2.65 Å, 3.43 Å, 140°; (f) 2.43 Å, 3.34 Å, 153°. Solvent molecules, counterions and remaining hydrogen atoms are omitted for clarity. Color scheme: C, grey; O, red; N, blue; H, white; Pd, yellow. | ||
The crystals of RS⊂anti-M4·6NO3 were dissolved in D2O and the 1H NMR spectrum recorded soon thereafter. The spectrum is similar to that obtained for HQ⊂anti-M4·6NO3, with a single atropisomer as a major species (ca. 90%) and chemical shifts for the metallacycle signals similar to those observed in the inclusion complex with HQ (ΔδHk = 1.95 ppm, ΔδHg = 0.86 ppm, ΔδHb = −0.56 ppm and ΔδHc = −0.71 ppm). As expected, the signals of the substrate RS are shifted upfield (ΔδH2 = −1.85 ppm, ΔδH4 = −1.68 ppm, and ΔδH5 = −2.09 ppm, supporting the presence of the inclusion complex (Fig. S10, ESI†).
Unlike previous systems, in which no complete self-assembly was observed in the absence of a template, addition of 1 equiv. of 5a to a 10 mM solution of L4·NO3 in D2O induced complete self-assembly of metallacycle M5·6NO3 as a mixture of the chiral syn and meso anti atropisomers (Scheme 5). Hence, the 1H NMR spectrum shows two sets of signals in a 1:1 ratio, with very similar chemical shifts, as expected, for the syn and anti atropisomers. Assignment of the signals by means of 2D NMR spectroscopy, showed that the quinoline and diazapyrenium protons Hf–k and Ha are shifted to higher frequencies, indicating the coordination of the bidentate ligand to the PdII centers. In contrast, the remaining signals of the diazapyrenium moieties are shifted to lower frequencies, due to the influence of the hydrophobic inner cavity formed. Additionally, the methylene protons become non-equivalent as a result of the rotation of the quinoline units around the CH2–Pd axis being slow on the NMR timescale. This trend is similar to that observed in the other systems studied herein and further confirms the self-assembly of the metallacyclic host syn/anti-M5·6NO3. A plausible reason for the more efficient self-assembly process observed in this case, can be attributed to the more favored solvation of the dimeric metallacycle compared to other linear and higher cyclic oligomers, a fact clearly linked to the increased hydrophobic character of the diazapyrene acceptor unit compared to 4,4-bipyridine.
 | ||
| Scheme 5 Self-assembly of metallacycles anti/syn-M5·6NO3. | ||
Subsequently, we studied the self-assembly of an inclusion complex between HQ and metallacycle M5·6NO3. Addition of 1 equiv. of the guest to the previously self-assembled species promoted the formation of the corresponding inclusion complex, as evidenced by the inspection of the 1H NMR spectra (Fig. 6). In this occasion, the signals for the quinoline unit are once more shifted downfield, suggesting the establishment of C–H⋯π interactions with the HQ molecule within the inner cavity. Moreover, the diazapyrenium signals are broad, probably due to a coalescence situation for the inclusion process on the NMR timescale. This behavior represents a clear difference respect to the bipyridine-based metallacycles M1–M4, as in those cases the inclusion process was fast on the NMR timescale, a fact related to the more intense π–π and hydrophobic interactions that can be established between diazapyrenium units of the host and the aromatic guest. More interestingly, upon formation of HQ⊂M5·6NO3, a 2:1 ratio between the two atropisomers was obtained, instead of the 1:1 ratio observed with metallacycles M3·6NO3 and M4·6NO3 in the equilibrium. This result shows how the stereoselectivity of the self-assembly also depends on the strength of the π and hydrophobic interactions between host and guest in addition to the C–H⋯π interactions. The interconversion between the stereoisomers is slow on the NMR timescale at room temperature as in previous systems, with VT NMR spectroscopy giving a coalescence temperature of 358 K (Fig. S12, ESI†). However, the interconversion barrier could not be measured in this case, as the coalescence method can only be applied when the initial species exist in a 1:1 ratio.
 | ||
| Fig. 6 Partial 1H NMR (D2O, 500 MHz) spectra of: (a) ligand L4·NO3; (b) metallacycle syn/anti-M5·6NO3; (c) inclusion complex HQ⊂syn/anti-M5·6NO3. The lettering corresponds to that shown on Scheme 1. * Signal corresponding to 1,4-benzoquinone, obtained from a partial oxidation of HQ. | ||
When 2,7DHN was used as guest instead of HQ, X-ray diffraction-quality single crystals were obtained from a solution of the inclusion complex in NaNO3(aq), allowing the study of its structure in the solid state. This crystal shows the presence of the anti-stereoisomer with one molecule of 2,7DHN within its cavity. The metallacycle displays similar structural features as those commented for the other inclusion complexes reported in this work, showing three C–H⋯π interactions between the H atoms in positions C1, C4 and C5 of the guest and the pyridine or benzene rings of the quinoline groups (Fig. 7). These C–H⋯π interactions determine a transversal insertion mode of the guest within the cavity of the receptor, with the O–O vector of 2,7DHN forming an angle of 81° with the mean plane of the metallacycle and the C2 axis of the guest being almost parallel to it (angle ca. 7°), allowing in this manner the establishment of the C–H⋯π interactions as the hydrogen atoms of the guest are directly oriented towards the corresponding aromatic rings of the quinoline moieties. Due to the lower symmetry of the 2,7DHN compared with M5, there are two equivalent transversal insertion modes for the guest within the cavity of the receptor. An inspection of the structure packing revealed as well the presence of three additional molecules of 2,7DHN outside the cavity of the metallacycle. Whilst one of these seems to be filling empty space in the lattice but not interacting significantly with the metallacycle, a second one, however, is located between the diazapyrenium units of two different metallacycles, at 3.39 Å from them, evidencing the presence of π–π interactions outside the host cavity. The third one is located between the quinoline moieties of different metallacycles, although the stacking between the units shows a more distorted geometry.
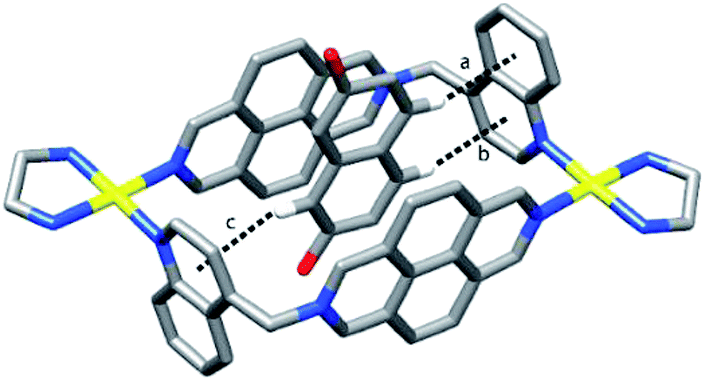 | ||
| Fig. 7 Capped sticks drawing of the solid state structure of the inclusion complex 2,7DHN⊂anti-M5·6NO3 showing the C–H⋯π interactions. [H⋯π] and [C⋯π] distances and [C–H⋯π] angles: (a) 2.77 Å, 3.69 Å, 163°; (b) 3.02 Å, 3.94 Å, 165°; (c) 3.02 Å, 3.94 Å, 165°. Solvent molecules, counterions and remaining hydrogen atoms are omitted for clarity. Only one of the two disordered positions of the guest is shown. | ||
Conclusions
A series of mono- and dinuclear metallacycles were prepared by self-assembly of (en)M(NO3)2 (M = PdII or PtII) and 4,4′-bipyridinium/2,7-diazapyrenium-based ligands incorporating quinoline subunits in their structures. We have shown that the mononuclear metallacycle M1 act as receptor for π-donor phenylene and naphthalene aromatic guests by means of hydrophobic forces, π–π stacking and C–H⋯π interactions. The structure optimized by DFT methods showed that the quinoline unit promoted little changes in the size and shape of host in comparison with metallacycle M2, in which the quinoline unit is replaced by a phenylene moiety. The presence of quinoline moieties in the short sides of the dinuclear rectangular metallacycles M3–5, gives rise to the existence of two different stereoisomers. The inclusion process of appropriate aromatics in solution or crystallization-mediated induced stereoselectivity on the self-assembly. This stereoselectivity was found to be modulated mainly by the size of the guests and their ability to optimize the number of potential C–H⋯π interactions with the two isomeric receptors. In addition, the increased π-acceptor and hydrophobic character of the host also contributes to an enhanced stereoselectivity. The proposed mechanism for the interconversion of the syn/anti isomers, studied by DFT methods, involves the rotation of the quinoline rings on the metallacycles without dissociation of the metal-pyridine bonds. The interconversion energy barrier in M3a and M4 was studied by VT-NMR and DFT methods. A comparison of the values obtained for M3a and M4 also support the proposed interconversion mechanism as the energy barrier increased with the steric hindrance around the quinoline moiety.Experimental
Crystal structure analysis
X-ray diffraction quality single crystals of L4·Cl were grown by slow evaporation of a concentrated solution of the ligand in water. Single crystals of the inclusion complexes HQ⊂anti-M4·6NO3, RS⊂anti-M4·6NO3, 1,5NPH⊂anti-M4·6NO3 and 2,7DHN⊂anti-M5·6NO3 were obtained from an equimolecular solution of ligand L3·NO3 or L4·NO3, (en)Pd(NO3)2 (5a) and the corresponding guest in NaNO3(aq) (3 M) maintained at 4 °C. The X-ray diffraction data were collected on a Bruker Smart 1k or a Bruker X8 APEX(II) diffractometer. The structures were solved by direct methods (SHELXT52 or SIR53,54) and refined with the full-matrix least-squares procedure (SHELX 2014)55 against F2 by using the WinGX56 software. Hydrogen atoms were placed in idealized positions (Ueg(H) = 1.2Ueg(C) or Ueg(H) = 1.5Ueg(C)) and were allowed to ride on their parent atoms. The X-ray diffraction experimental and refinement data are summarized in Table 3. The HQ molecule in the inclusion complex HQ⊂anti-M4·6NO3 is disordered into two positions and a RIGU instruction was used in the refinement. Two of nitrate anions were disordered and modeled accordingly into two half-occupied positions using RIGU and PART instructions where appropriate. The lower symmetry of 2,7DHN compared with M5 in 2,7DHN⊂anti-M5·6NO3 results in two possible insertion modes of the guest within the cavity of the metallacycle. The resulting disorder into two positions of 2,7DHN was modeled by using the PART instruction in combination with the EXYZ and EADP instructions and DFIX, DANG, SIMU, DELU and ISOR restraints and constraints. Disordered 2,7DHN molecules outside the cavity were modeled following the same strategy. Moreover, one of the nitrate counterions in the asymmetric unit is disordered and could not be modelled adequately by assigning specific positions to those atoms. These 8 nitrate anions in the unit cell have been treated as a diffuse contribution to the overall scattering by applying the SQUEEZE57 routine included in PLATON.58 Additionally, some of the nitrate counterions in HQ⊂anti-M4·6NO3, 1,5NPH⊂anti-M4·6NO3 and 2,7DHN⊂anti-M5·6NO3 show disorder that was treated using the SIMU, DELU, FLAT, DFIX, DANG and ISOR restraints and constraints where appropriate. CCDC-1478616 (L4·Cl), CCDC-1478617 (HQ⊂anti-M4·6NO3), CCDC-1478618 (RS⊂anti-M4·6NO3), CCDC-1478619 (1,5NPH⊂anti-M4·6NO3) and CCDC-1478620 (2,7DHN⊂anti-M5·6NO3) contain the supplementary crystallographic data for this paper.†| L4·Cl·2H2O | HQ⊂M4·6NO3·5H2O | RS⊂M4·6NO3·11H2O | 1,5NPH⊂M4·6NO3·12H2O | 2,7DHN⊂M5·6NO3·8H2O | |
|---|---|---|---|---|---|
| a In common: refinement method, full-matrix least-squares on F2. Wavelength, 0.71073 Å (Mo-Kα). Absorption correction method, multi-scan. | |||||
| Chemical formula | C24H16ClN3O2 | C52H58N16O25Pd2 | C52H63N16O31Pd2 | C64H76N16O36Pd2 | C92H80N16O34Pd2 |
| Mr | 413.85 | 1519.94 | 1620.98 | 1858.20 | 2166.52 |
| Temperature [K] | 293 | 100 | 100 | 100 | 100 |
| Crystal size [mm3] | 0.33 × 0.21 × 0.065 | 0.24 × 0.14 × 0.06 | 0.29 × 0.13 × 0.12 | 0.43 × 0.38 × 0.22 | 0.31 × 0.13 × 0.085 |
| Crystal system | Orthorhombic | Triclinic | Triclinic | Triclinic | Monoclinic |
| Space group | Pca21 | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
C2/c |
| a [Å] | 14.075(2) | 9.7510(15) | 9.7594(2) | 12.4048(12) | 19.126(5) |
| b [Å] | 7.9884(11) | 11.3294(18) | 18.3309(4) | 13.2527(13) | 20.379(5) |
| c [Å] | 17.570(2) | 14.580(2) | 19.7117(4) | 13.3614(13) | 25.002(5) |
| α [°] | 90 | 80.294(3) | 97.4150(10) | 66.448(2) | 90 |
| β [°] | 90 | 89.934(3) | 90.7800(10) | 80.549(2) | 102.801(5) |
| γ [°] | 90 | 83.204(3) | 102.7820(10) | 83.081(2) | 90 |
| V [Å3] | 1975.6(5) | 1576.2(4) | 3407.00(12) | 1982.6(3) | 9503(4) |
| Z | 4 | 1 | 2 | 1 | 4 |
| ρcalcd [Mg m−3] | 1.391 | 1.601 | 1.580 | 1.556 | 1.514 |
| μ [mm−1] | 0.220 | 0.665 | 0.627 | 0.554 | 0.473 |
| F(000) | 856 | 774 | 1654 | 952 | 4432 |
| θ range [°] | 2.32 to 27.02 | 1.42 to 28.46 | 1.04 to 29.87 | 1.68 to 28.37 | 1.48 to 28.31 |
| hkl ranges | −17,17/−10,10/−22,22 | −10,13/−15,15/−19,19 | −13,13/−25,25/−27,27 | −16,16/−17,17/−17,17 | −25,15/−27,27/−33,33 |
| Reflections collected | 22683 |
21927 |
123471 |
26214 |
46176 |
| Independent reflections | 4314 | 7797 | 19223 |
9786 | 11712 |
| Rint | 0.1501 | 0.0712 | 0.0407 | 0.0292 | 0.0590 |
| Completeness [%] | 100.0 | 99.0 | 99.6 | 99.9 | 99.8 |
| Final R indices [I > 2σ(I)] | R1 = 0.0676 | R1 = 0.0697 | R1 = 0.0667 | R1 = 0.0645 | R1 = 0.0840 |
| wR2 = 0.1519 | wR2 = 0.1710 | wR2 = 0.1722 | wR2 = 0.1827 | wR2 = 0.2475 | |
| R indices (all data) | R1 = 0.1968 | R1 = 0.1237 | R1 = 0.0925 | R1 = 0.0800 | R1 = 0.1321 |
| wR2 = 0.2065 | wR2 = 0.2127 | wR2 = 0.1934 | wR2 = 0.1947 | wR2 = 0.2962 | |
| Goodness-of-fit on F2 | 0.975 | 1.047 | 1.039 | 1.051 | 1.067 |
Computational methods
DFT calculations were performed using the Gaussian 09 (Revision B.01)59 program package with the B3LYP three-parameter hybrid density functional (M1b) or the hybrid meta-GGA approximation with the M06 (ref. 60) functional for syn/anti-M3a and M4. In these calculations we used the standard 6-31G(d,p) basis set for C, H, and N atoms, whereas for Pt and Pd the LanL2DZ effective core potential of Wadt and Hay61 (Los Alamos ECP) and its associated basis set were applied. No symmetry constraints have been imposed during the optimizations. The stationary points found on the potential energy surfaces as a result of geometry optimizations of M1b and syn/anti-M3a and M4 were tested to represent energy minima rather than saddle points via frequency analysis. The diastereoisomer interconversion process in M3a and M4 was studied in vacuo by using the synchronous transit-guided quasi-Newton method49,50 implemented in Gaussian 09. TS-M3a and TS-M4 were also tested by frequency analysis to confirm they correspond to transition states.Acknowledgements
This research was supported by the Ministerio de Economía y Competitividad (MINECO FEDER, CTQ201341097-P) and Xunta de Galicia (EM2014/056). V. B. and T. R. thank the Spanish Ministerio de Economía y Competitividad (MINECO) for a ‘Juan de la Cierva’ postdoctoral contract and Ministerio de Educación, Cultura y Deporte (MECD) for a FPU predoctoral fellowship, respectively. The authors are indebted to Centro de Servicios de Informática y Redes de Comunicaciones (CSIRC), Universidad de Granada, for providing the computer facilities. The authors also thank Prof. J. M. Cuerva for his assistance with the DFT calculations.Notes and references
- M. Eigen, Naturwissenschaften, 1971, 58, 465 CrossRef CAS PubMed.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1154 CrossRef.
- M. Groll, L. Dizel, J. Lowe, D. Stock, M. Bochter, H. D. Bartunik and R. Huber, Nature, 1997, 386, 463 CrossRef CAS PubMed.
- P. Ball, The Self-Made Tapestry: Pattern Formation in Nature, Oxford Univ. Press, Oxford, 1999 Search PubMed.
- S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555 RSC.
- D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 98 CrossRef CAS.
- D. E. Koshland, Angew. Chem., Int. Ed. Engl., 1994, 33, 2375 CrossRef.
- The Lock and Key Principle. The State of the Art–100 Years On, ed. J.-P. Behr, Wiley, New York, 1994 Search PubMed.
- Inclusion Phenomena and Molecular Recognition, ed. J. L. Atwood, Plenum, New York, 1990 Search PubMed.
- Host–Guest Complex Chemistry: Synthesis Structure, Applications, ed. F. Vögtle and E. Weber, Springer-Verlag, Berlin, 1985 Search PubMed.
- J. K. M. Sanders, Chem.–Eur. J., 1998, 4, 1378 CrossRef CAS.
- T. W. Bell, Z. Hou, Y. Luo, M. G. B. Drew, E. Chapoteau, B. P. Czech and A. Kumar, Science, 1995, 269, 671 CAS.
- W. Jiang and J. Rebek Jr, J. Am. Chem. Soc., 2012, 134, 17498 CrossRef CAS PubMed.
- S. Le Gac, J. Marrot, O. Reinaud and I. Jabin, Angew. Chem., Int. Ed., 2006, 45, 312 CrossRef PubMed.
- O. Taratula, P. A. Hill, N. S. Khan, P. J. Carroll and I. J. Dmochowski, Nat. Commun., 2010, 1, 148 CrossRef PubMed.
- J. K. Clegg, J. Cremers, A. J. Hogben, B. Breiner, M. M. J. Smulders, J. D. Thoburnad and J. R. Nitschke, Chem. Sci., 2013, 4, 68 RSC.
- T. K. Ronson, A. B. League, L. Gagliardi, C. J. Cramer and J. R. Nitschke, J. Am. Chem. Soc., 2014, 136, 15615 CrossRef CAS PubMed.
- A. Brugnara, L. Fusaro, M. Luhmer, T. Prange, B. Colasson and O. Reinaud, Org. Biomol. Chem., 2014, 12, 2754 CAS.
- A. Specht, P. Bernard, M. Goeldner and L. Peng, Angew. Chem., Int. Ed., 2002, 41, 4706 CrossRef CAS PubMed.
- A. Cooper, M. Nutley, E. J. MacLean, K. Cameron, L. Fielding, J. Mestres and R. Palin, Org. Biomol. Chem., 2005, 3, 1863 CAS.
- T. Mecca, G. M. L. Consoli, C. Geraci, R. L. Spina and F. Cunsolo, Org. Biomol. Chem., 2006, 4, 3763 CAS.
- T. Sawada, H. Hisada and M. Fujita, J. Am. Chem. Soc., 2014, 136, 4449 CrossRef CAS PubMed.
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502 CrossRef CAS.
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417 RSC.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975 CrossRef CAS.
- S. Seidel Russell and P. J. Stang, Acc. Chem. Res., 2002, 35, 952 CrossRef.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554 CrossRef CAS PubMed.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972 CrossRef CAS PubMed.
- E. Zangrando, M. Casanova and E. Alessio, Chem. Rev., 2008, 108, 4979 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed.
- A. Mishra, S. C. Kang and K.-W. Chi, Eur. J. Inorg. Chem., 2013, 5222 CrossRef CAS.
- S. Mukherjee and P. S. Mukherjee, Chem. Commun., 2014, 50, 2239 RSC.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed.
- C. Peinador, E. Pía, V. Blanco, M. D. García and J. M. Quintela, Org. Lett., 2010, 12, 1380 CrossRef CAS PubMed.
- V. Blanco, M. D. García, A. Terenzi, E. Pía, A. Fernández-Mato, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2010, 16, 12373 CrossRef CAS PubMed.
- V. Blanco, M. D. García, C. Peinador and J. M. Quintela, Chem. Sci., 2011, 2, 2407 RSC.
- C. Alvariño, A. Terenzi, V. Blanco, M. D. García, C. Peinador and J. M. Quintela, Dalton Trans., 2012, 41, 11992 RSC.
- C. Alvariño, E. Pía, M. D. García, V. Blanco, A. Fernández, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2013, 19, 15329 CrossRef PubMed.
- T. Rama, E. M. López-Vidal, M. D. García, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2015, 21, 9482 CrossRef CAS PubMed.
- T. Rama, C. Alvariño, O. Domarco, C. Platas-Iglesias, V. Blanco, M. D. García, C. Peinador and J. M. Quintela, Inorg. Chem., 2016, 55, 2290 CrossRef CAS PubMed.
- M. D. García, C. Alvariño, E. M. López-Vidal, T. Rama, C. Peinador and J. M. Quintela, Inorg. Chim. Acta, 2014, 417, 27 CrossRef.
- C. Peinador, V. Blanco, M. D. García and J. M. Quintela, PdII and PtII Metal-Directed Self-Assembly of Supramolecular Structures Based on N-Monoalkyl-4,4′-Bipyridinium Derivatives, in Molecular Self-Assembly: Advances and Applications, ed. A. D. Q. Li, Pan Stanford Publishing, Singapore, 2012, p. 351 Search PubMed.
- A preliminary communication on the stereoselective self-assembly of two atropisomeric PdII metallacycles induced by an aromatic guest has already been published: D. Abella, V. Blanco, E. Pía, M. Chas, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Chem. Commun., 2008, 2879 RSC.
- Z. Zhu, H. Li, Z. Liu, J. Lei, H. Zhang, Y. Y. Botros, C. L. Stern, A. A. Sarjeant, J. F. Stoddart and H. M. Colquhoun, Angew. Chem., Int. Ed., 2012, 51, 7231 CrossRef CAS PubMed.
- M. Chas, D. Abella, V. Blanco, E. Pía, G. Blanco, A. Fernández, C. Platas-Iglesias, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2007, 13, 8572 CrossRef CAS.
- In FAB mass spectrometry the successive loss of anions for bipyridinium systems can be compensated by the incorporation of electrons so the ions formed have always only one positive charge: W. Ong, J. Grindstaff, D. Sobransingh, R. Toba, J. M. Quintela, C. Peinador and A. E. Kaifer, J. Am. Chem. Soc., 2005, 127, 3353 CrossRef CAS PubMed.
- I. O. Sutherland, Annu. Rep. NMR Spectrosc., 1971, 4, 71–235 CrossRef CAS : The coalescence method is based on the equation kc = π(Δν)/(2)1/2 to estimate the rate constant at the coalescence temperature (Tc), where Δν is the initial chemical shift difference (in Hertz) between the coalescing signals in the absence of exchange. The Eyring equation was subsequently employed to calculate ΔG‡c value at Tc.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1994, 33, 449 CrossRef.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49 CrossRef CAS.
- This explanation is also supported by the previously reported results obtained with a ligand bearing an acridine unit, with a fused benzene ring instead of the methyl group as the bulky substituent, which did not undergo self-assembly in solution with (en)Pd(NO3)2 either. See ref. 46.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- SIR92: A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- SIR2014: M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1478616–1478620. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra14909j |
| This journal is © The Royal Society of Chemistry 2016 |