
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of iron oxide coupled and doped titania nanocomposites: tuning of physicochemical and photocatalytic properties†
Ayyakannu Sundaram
Ganeshraja
ab,
Kanniah
Rajkumar
b,
Kaixin
Zhu
ac,
Xuning
Li
ac,
Subramani
Thirumurugan
b,
Wei
Xu
 de,
Jing
Zhang
d,
Minghui
Yang
f,
Krishnamoorthy
Anbalagan
*b and
Junhu
Wang
*a
de,
Jing
Zhang
d,
Minghui
Yang
f,
Krishnamoorthy
Anbalagan
*b and
Junhu
Wang
*a
aMössbauer Effect Data Center, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: wangjh@dicp.ac.cn; Tel: +86 411 84379159
bDepartment of Chemistry, Pondicherry University, Pondicherry 605014, India. E-mail: kanuniv@gmail.com; Tel: +91 413 2654509
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
eRome International Center for Materials Science, Superstripes, RICMASS, via dei Sabelli 119A, I-00185 Roma, Italy
fDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 26th July 2016
Abstract
A facile hydrothermal method was firstly employed to synthesize iron oxide coupled and doped titania nanocomposites using an aqueous solution of titanium nitrate. The present nanocomposites exhibit altered compositional, optical, electrical, magnetic and photocatalytic properties with respect to varying dosage of iron in the titania matrix. The architecture of characteristic iron oxide such as Fe2O3 coupled with titania was confirmed by 57Fe Mössbauer spectroscopy and X-ray absorption fine structure spectroscopic measurements. The enhanced photocatalytic activity was demonstrated by comparing with that of pure hematite, anatase TiO2, rutile TiO2 and P25 in the degradation of methylene blue under visible light (λ > 480 nm) irradiation in an aqueous suspension. The strategy presented here gives a promising route towards the development of a metal oxide coupled and doped semiconductor material for applied photocatalysis and related applications.
1. Introduction
Metal oxide coupled semiconductor photocatalysts have attracted much attention because of their applications in environmental remediation, in which they are used in the photocatalytic degradation of pollutants in air and water.1,2 Waste water from various industries such as textiles, printing, food processing and cosmetic units containing huge amounts of toxic organic chemicals become harmful to human health. Many methods such as adsorption, flocculation, photocatalysed degradation and chemical oxidation have been used to remove the persistent harmful chemicals released into aquatic environments.3,4 Nowadays, TiO2, is used as an important functional material and finds numerous applications as photocatalyst, in electronic devices, in conversion devices as well as promising electrode material due to its attractive advantages of high abundance, low cost, better safety, and environmental friendliness.5 However, usage of TiO2 photocatalysts in the waste-water treatment is still limited because of major bottleneck and methodology drawbacks associated with this. Titanium dioxide has a high band gap energy (EG = 3.2 eV) and this can be triggered by UV or near UV radiation (λ = 380 nm), which accounts about 3–4% in the solar spectrum.6 Thus, the possibility of utilizing solar light as an energy source in TiO2 photocatalysis is limited. However, it is very difficult to recycle nanosized powdered materials in wastewater treatment which may cause secondary pollution.Recently, a number of attempts have been made to extend the spectral response of TiO2 into the visible light region but also reduce the recombination of photogenerated electron–hole pairs, thus enhancing its photocatalytic activity.7,8 Among these attempts, considerable effort has been devoted to the study of iron doped TiO2 particles in order to obtain better efficiency and a possible structure–property relationships.9 Litter and Navio10 investigated a mechanistic aspect on the effect of Fe3+ in titania during photocatalysed reaction. In fact, FexTi1−xO2 has been proved as a highly efficient visible light tunable photocatalyst and the catalyst illustrated more stability during the decomposition of dyes or other water pollutants.11 At the same time, numerous efforts have also been carried out on the development of new materials with intrinsic photocatalytic activity under visible light response such as nitrogen codoped Fe–TiO2,12 CaIn2O4/Fe–TiO2,13 prussian blue/TiO2,14 Fe–Sn–TiO2 (ref. 15 and 16) etc.
Among them, magnetic iron oxide nanoparticles have attracted considerable research interests because of its unique properties, including decent magnetic, electric, catalytic properties, biocompatibility and low toxicity.17 Recently, several methods have been developed to prepare various composite materials containing iron oxides combined with TiO2,17–19 which usually exhibit improved properties. Liu et al.20 developed an efficient visible-light photocatalyst based on matching the energy levels of surface-grafted and bulk-doped iron ions with similar energy states. This was identified from 57Fe Mössbauer and X-ray absorption fine structure (XAFS) techniques. These techniques are precious for identification of chemical states of surface and bulk doped iron with TiO2. Our group has developed iron containing titania nanocomposites by soft chemical solution process, the results have been different for using different precursors.15,16 Mössbauer results have shown doublet patterns for highly dispersed and homogenous iron doped titania and sextet patterns for iron cluster formation.21 However, it is essential to need to develop an efficient method for controlling well the chemical states of dopant ions in titania matrix and on the surface. Moreover, the efficiencies of these photocatalysts are still far from satisfactory due to a high recombination rate of the photogenerated electron–hole pair. In our previous studies, ferromagnetic photocatalysts such as Sn-doped TiO2,22 Fe codoped Sn–TiO2 and CuO coupled Sn–TiO2 (ref. 23) were successfully developed and used in the photocatalytic degradation of methyl orange and phenol derivatives. According to the energy level theory on semiconductors, presence of iron oxides can enhance effectively in the photocatalytic activity of TiO2.24 Although there are a few reports on the preparation of Fe2O3/TiO2 nanocomposites,24,25 there is none on the synthesis of weak superparamagnetic iron oxide coupled and doped titania nanocomposites by facile hydrothermal method using the same precursor and reagents with a view to compare their photocatalytic, optical and magnetic properties. Probable implications of variations in the molar ratio of iron content into TiO2 and the magnetic characteristics in relation to the photocatalytic activity of the composite particles have not yet been investigated in detail.26
In this investigation, XAFS and 57Fe Mössbauer spectroscopic measurements were applied to investigate two different form of iron such as iron oxide coupled and doped in titania. The important issues, such as (i) enhancement in the photocatalytic activities of titania, (ii) effects of structural, optical, electrical and magnetic properties of iron oxide coupled and doped anatase–rutile mixed phases, and (iii) photodegradation of a model organic dye pollutant such as methylene blue (MB) in an aqueous suspension system was systematically investigated. Moreover, the photocatalytic activity of the prepared nanocomposites after repeated utilization cycles was also tested and discussed. The results reported here will be a critical and necessary input for the efficient development of photocatalytic degradation process in water remediation.
2. Experimental
2.1 Chemicals
Anhydrous ferric chloride (99%), titanium isopropoxide (99%) and barium sulfate (spectral grade) were purchased from Sigma Aldrich. Ammonium hydroxide, hydrogen peroxide, ferric nitrate, oxalic acid and nitric acid were purchased from Himedia and SD Fine Chemicals (India). Fe2O3 (particle size = 30 nm, 99.5%), P25, rutile TiO2, and MB were purchased from Aladdin chemicals (China). All other chemicals are at least analytical grade with no further treatment. All the solvents were distilled and purified before use.2.2 Instrumentation
X-ray diffraction (XRD) patterns were recorded in 2θ ranging 10–80° with step size 0.02° using CuKα radiation from X-Pert PANalytical X-ray diffractometer. Raman spectra were recorded on Jobin Yvon Horibra LABRAMR1100 micro-Raman spectrophotometer. X-ray photoelectron spectra (XPS) were measured on ESCALAB 250Xi X-ray photoelectron spectrometer with monochromatic source Al Kα and the binding energy (B.E.) was calibrated using contaminated carbon as an internal standard (C 1s B.E. 284.6 eV). Surface morphology and elemental mapping were examined by scanning electron microscope-energy dispersive X-ray detector (SEM-EDX, Hitachi, S-3400N microscope). Brunauer–Emmett–Teller (BET) surface area measurement was made by nitrogen adsorption at −196 °C using a Micromeritics ASAP 2010 apparatus. HRTEM images were taken on JEOL JEM-2000EX microscope. Ti and Fe contents in the nanocomposites were determined by inductively coupled plasma atomic emission spectrometry (ICP-AES) on an IRIS Intrepid II XSP instrument (Thermo Electron Corporation).57Fe Mössbauer spectra were separately collected on a Topologic 500A system at room temperature. Rh(57Co) source was moved in a constant acceleration mode. Doppler velocity and isomer shift were calibrated by metallic α-Fe foil. The spectra were fitted with the appropriate superpositions of Lorentzian lines using the MossWinn 3.0i computer program. In this way, the 57Fe Mössbauer spectral parameters could be determined, including the isomer shift (IS), the electric quadrupole splitting (QS), the full width at half maximum, and the relative resonance areas of the different components of the absorption patterns.
Fe K-edge XAFS spectra were collected at the Beamline 4W1B of the Beijing Synchrotron Radiation Facility (BSRF) with stored electron energy of 2.5 GeV and accumulated currents dropping from 250 to 150 mA. The incident beam is monochromatized using Si(111) double crystal monochromator. The Fe K-edge XAFS spectra of sample and standards were recorded in fluorescence and transmission mode, respectively. The argon-filled gas ion chambers and Lytle detector were adopted. The Fe K-edge spectra were processed following the conventional procedure using the IFEFFIT package.27 After subtracting the atomic background, the spectra were normalized to unit edge jump. The k3 weighted EXAFS signals χ(k) were subjected to the Fourier transform within the k range of [2, 12.17]. Structural fitting was conducted using Fe:8a site of Fe3O4 model structure for first shell fitting. To simulate the doping of Fe in TiO2, we carried out theoretical calculations using the full multiple scattering (FMS) theory within muffin-tin approximation.28 The self-consistent field (SCF) potential and Hedin–Lundqvist exchange correlation potential were employed. To achieve good convergence, the cluster radius for SCF and FMS is 5 and 10 Å, respectively. The FEFF code allows us to extract the angular momentum projected density of states for the atomic cluster while calculating the XANES spectra. Owing to the finite cluster size, the density of states is broadened with respect to the ground states calculations based on density of functional theory (DFT). Nevertheless, it is sufficient for interpreting the variations of electronic structure upon the doping.
UV-vis diffuse reflectance spectra (UV-vis DRS) were obtained for dry-pressed disk samples using a Shimadzu UV 2450 double-beam spectrophotometer equipped with an integrating sphere (ISR-2200) assembly using BaSO4 as a reference sample. Steady state fluorescence emission spectra were recorded on Spex FluoroLog-3 spectrofluorometer (Jobin-Yvon Inc.) using 450 W xenon lamp equipped with a Hamamatsu R928 photomultiplier tube. Time-resolved fluorescence decay measurements were carried out using nano-LED (λexc = 295 nm) source for excitation with repetition rate 10 kHz. Life times were determined by fitting the data to exponential decay models using software packages of the commercially available DAS6 v6.2-Horiba Jobin Yvon. The goodness of fit was assessed by minimizing the reduced chi-squared function (χ2).
Jeol X-Band, JES X 310 EPR spectrometer is the state of the art Electron Paramagnetic Resonance (EPR) used for the measurement of species that contain iron ions in the nanocomposites with 9.4 GHz operating frequency at room temperature. Magnetization measurements were carried out using vibrating sample magnetometer (VSM) in powder form on Lakeshore-7404 at room temperature. The temperature dependent dc magnetization was measured using EverCool 7 Tesla SQUID Magnetometer.
Mott–Schottky analysis was carried out in 0.5 M Na2SO4 (pH 6.4) at a frequency of 1 kHz on CHI660E, using a Pt foil and an SCE as counter electrode and reference electrode, respectively. The working electrode was prepared on ITO via electrophoresis deposition from the suspension of 20 mg of ground powder in 25 mL of 0.2 g−1L I2/acetone solution, and a negative bias of 20 V was applied vs. the counter electrode (an FTO electrode) for 5 minutes via a DC power supplier (Itech IT6874A), then the electrode was washed with acetone carefully and dried at 60 °C before the test.
2.3 Preparation of iron oxide coupled and doped titania nanocomposites
Samples were prepared by a facile hydrothermal method22,23 with some further modifications. Firstly, about 5 mol reddish brown titanium nitrate aqueous solution was prepared.23 In order to prepare various Fe loaded titania, 0.5, 1.0, 2.0, 3.0 and 4.0 M ferric nitrate (100 mL) aqueous solutions were introduced separately into the titanium nitrate aqueous solutions under magnetic stirring. Then 100 mL of oxalic acid solution (0.6 M) was added drop wise within 2 h under magnetic stirring to ensure complete precipitation. The resultant suspension was transferred into Teflon lined stainless steel autoclave, sealed and heated upto 110 °C for 4 h with a pressure maintained at 18 psi. Finally, the precipitates were filtered, washed with distilled water followed by ethanol, and dried in air at 90 °C for 12 h. The dried precipitates were calcined at 400 °C for 4 h in a muffle furnace to get iron oxide coupled and doped titania nanocomposites. These nanocomposites are denoted as S1, S2, S3, S4 and S5, prepared using 0.5, 1.0, 2.0, 3.0 and 4.0 M ferric nitrate (100 mL) in titanium nitrate solutions, respectively. Experimental results reported for pure anatase TiO2 sample in our previous paper,22 are referred for comparison.2.4 Photocatalytic activity
The photocatalytic activity of the samples was evaluated by photodegradation of methylene blue, MB (10−5 M) using the light source of PE. ILC CERMAX xenon illumination system model LX 175/300 attached with CX-04E power supply (10 A) and with a 480 nm long-pass filter. The distance between the lamp and the solution was about 8.0 cm. Photochemical experiments were performed in a dark atmosphere in a box. In a typical photocatalysis experiment, 50 mg of catalyst was dispersed in 80 mL of aqueous solution of MB in a 100 mL glass beaker at neutral pH and at room temperature. Then the suspension was kept in darkness and magnetically stirred for 45 min to reach the adsorption–desorption equilibrium on solid catalyst. Prior to irradiation, the solution was continuously stirred with a magnetic stirrer for a thorough mixing. About 3 mL of aliquots was periodically taken out from the suspension at regular interval of 60 min and centrifuged (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 2 min) to separate the solution. The supernatants were measured by GBC Cintra UV-vis spectrophotometer to record the changes of absorption values at λ = 665 nm in relation to the initially taken aqueous solution of MB. The photocatalytic degradation efficiency (PDE) was calculated using the formula PDE = (A0 − At/A0) × 100%, where A0 is the absorbance of MB at 0 time and At is the absorbance of MB solution measured at various irradiation time intervals. In a typical recycle experiment, the S5 catalyst was isolated from aqueous solution of MB under visible light irradiation at 360 min through centrifuge, washed thoroughly with deionized water, further with ethanol and dried at room temperature.
000 rpm, 2 min) to separate the solution. The supernatants were measured by GBC Cintra UV-vis spectrophotometer to record the changes of absorption values at λ = 665 nm in relation to the initially taken aqueous solution of MB. The photocatalytic degradation efficiency (PDE) was calculated using the formula PDE = (A0 − At/A0) × 100%, where A0 is the absorbance of MB at 0 time and At is the absorbance of MB solution measured at various irradiation time intervals. In a typical recycle experiment, the S5 catalyst was isolated from aqueous solution of MB under visible light irradiation at 360 min through centrifuge, washed thoroughly with deionized water, further with ethanol and dried at room temperature.
3. Results and discussion
3.1 Structural characterization
X-ray diffractograms and Raman spectra of the prepared samples are displayed in Fig. 1. These imply that the composites are basically composed of anatase and rutile forms of titania,29 whereas small impurity of iron exists as hematite (Fe2O3) phase. It was observed that the intense anatase phase is combined with impurity phase of rutile in S1–S3 samples, while rutile phase decreases as increasing iron content in the case of S4 and S5 samples as shown in Fig. 1a. A perusal of Raman spectra reveals the presence of residual amounts of the Fe2O3 phase of iron, such impurity phase is proposed to be responsible for the broadening and poorly resolved features of the X-ray diffractogram detected from 30 to 60°. Average particle size of samples was obtained from XRD peaks broadening using Scherrer equation30 and calculated to be approximately 6–17 nm (Table 1). Moreover formation of Fe2O3 crystalline phase was confirmed from Raman spectral analysis as shown in Fig. 1b (insert), the characteristic Raman peak positions are little different from that of pure Fe2O3 nanoparticles.31 This indicates that iron exists as oxide as a separate crystalline oxide phase, however, is coupled with titania. Raman spectral analysis strongly supports the results of XRD for the samples under study. Although, the undoped TiO2 was prepared by a similar experimental method22 the respective XRD pattern and Raman spectra differ and are shown in Fig. S1a and b.† It could be concluded that pure anatase phase was observed for undoped TiO2 nanoparticles. Moreover, we observed little higher peak shifting of prepared S1–S5 samples with undoped TiO2 in the both the XRD and Raman spectra. These results led to the conclusion that iron ions present in titanium lattice site, moreover, some of the Fe2O3 crystals are coupled on the surface of TiO2. | ||
| Fig. 1 XRD patterns (a) and Raman spectra (b) (insert figure short range Raman spectra) of S1–S5 samples (where A = anatase, R = rutile and # = hematite (Fe2O3)). | ||
| Sample | E G (eV) | D (nm) | Lifetime | BET | |||||
|---|---|---|---|---|---|---|---|---|---|
| τ 1, ns | τ 2, ns | τ 3, ns | χ 2 | Surface area (m2 g−1) | Langmuir surface area (m2 g−1) | Pore size (nm) | |||
| S1 | 3.04 | 17.25 | 1.99 | 32.92 | 0.005 | 1.88 | 74.87 | 517.55 | 7.70 |
| S2 | 2.84 | 13.74 | 1.65 | 31.49 | 0.008 | 1.78 | 97.08 | 695.16 | 7.79 |
| S3 | 2.26 | 10.33 | 1.38 | 29.06 | 0.009 | 1.78 | 95.51 | 632.84 | 7.29 |
| S4 | 1.98 | 7.51 | 1.75 | 32.82 | 0.155 | 1.88 | 105.43 | 675.70 | 6.54 |
| S5 | 1.95 | 5.65 | 2.42 | 33.19 | 0.103 | 2.00 | 97.06 | 601.64 | 6.08 |
57Fe Mössbauer, EPR and XAFS spectroscopic measurements were used in determining the chemical state of iron in the prepared nanocomposites. Liu et al.20 reported a detailed account on the states of surface-grafted and bulk-doped iron with TiO2 in more detail on account of 57Fe Mössbauer spectra. Both bulk doped and surface grafted iron with TiO2 exhibited quadrupole splitting (QS) and showed isomer shifts (IS) of around 0.38 mm s−1, indicating that both the bulk-doped and surface-grafted Fe3+ ions are in paramagnetic state with a quadrupole doublet as an octahedral structure. As shown in Fig. 2a, the spectral peaks of S1–S5 samples could be fitted by the simple addition of spectra for iron oxide coupled and doped TiO2 in equal proportion. It is noteworthy that the QS for the surface-coupled and bulk-doped iron oxide are different. According to the Mössbauer parameters listed in Table 2, the doublets were assigned to Fe3+ states and showed IS = 0.35–0.38 mm s−1 and QS = 0.50–0.61 mm s−1 for iron oxide doped TiO2, whereas IS = 0.33–0.36 mm s−1 and QS = 0.94–1.08 mm s−1 for iron oxide surface coupled TiO2, indicates surface iron oxide having a larger structural degree of freedom as compared to that of doped iron oxide. It may be due to the formation of higher symmetric state, and oxygen vacancies or structural defects.20 The peak area of bulk doped and coupled iron sites are 18.4–45.7% and 54.3–81.6% for S1, S3, S4 and S5 samples, respectively. It could be concluded that bulk doped iron sites are smaller than surface coupled iron sites, however contrast behavior was observed for S2 sample. Our group recently reported paramagnetic doublet for Fe doped and Fe–Sn codoped TiO2 by a soft-chemical solution process.21,32 Alternatively, the value of isomer shift for the superparamagnetic doublet is around 0.34 mm s−1 with larger QS values of the sample with particle diameter of less than 10 nm, which is larger than those of the amorphous samples.33 This could be concluded that the nanoparticles begin to show superparamagnetic behavior in the case of hematite. The superparamagnetic behavior34 of these nanoparticles leads to a complete lack of magnetic hyperfine splitting of the spectrum and represents in quadrupole doublet with IS of ∼0.34 mm s−1 and a QS of ∼0.99 mm s−1.35 These results indicate that bulk doped Fe3+ ions are substitutionally introduced into TiO2 crystal at Ti4+ sites and superparamagnetic Fe2O3 nanocrystals are in coupled state with titania surface matrix.
 | ||
| Fig. 2 57Fe Mössbauer (a) and EPR (b) spectra of S1–S5 samples at room temperature. | ||
| Sample | EPR | 57Fe Mössbauer | VSM | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g | ΔHpp (G) | g | Site 1 | Peak area (%) | Site 2 | Peak area (%) | M s (emu g−1) × 10−3 | |||||
| IS (mm s−1) | QS (mm s−1) | LW (mm s−1) | IS (mm s−1) | QS (mm s−1) | LW (mm s−1) | |||||||
| S1 | 2.00 | 255 | 4.47 | 0.36 | 0.51 | 0.30 | 32.8 | 0.36 | 0.97 | 0.50 | 67.2 | 100.52 |
| S2 | 2.00 | 920 | 4.43 | 0.38 | 0.61 | 0.39 | 54.4 | 0.34 | 1.08 | 0.48 | 45.6 | 90.18 |
| S3 | 2.03 | 1718 | 4.33 | 0.37 | 0.50 | 0.28 | 18.4 | 0.33 | 0.94 | 0.58 | 81.6 | 147.04 |
| S4 | 2.01 | 1225 | 4.36 | 0.37 | 0.56 | 0.38 | 39.7 | 0.34 | 1.00 | 0.53 | 60.3 | 187.64 |
| S5 | 2.04 | 1616 | 4.36 | 0.35 | 0.61 | 0.41 | 45.7 | 0.33 | 1.03 | 0.57 | 54.3 | 181.35 |
Fig. 2b exhibits the EPR spectra of S1–S5 samples recorded at room temperature. There appears broad signals with respect to gyromagnetic factor (g) at 2.00–2.04 and 4.33–4.47 (Table 2), whereas no such signals were observed for amorphous Fe2O3, α-Fe2O3, TiO2, and a physically mixed oxide (10 wt% Fe2O3/TiO2).36 The resonance lines at 2.00 (ΔHpp ∼60–98 G), 2.00–2.16 (ΔHpp ∼1000–800 G) and 4.30 are usually attributed to the isolated octahedral Fe3+ in anatase, Fe3+ in iron oxides-type clusters and isolated rhombic Fe3+ ions segregated on the TiO2 surfaces, respectively.20,37 It suggests that iron oxide such as Fe2O3 particles are present on the TiO2 surfaces and furthermore isolated rhombic Fe3+ ions doped in the TiO2 bulk in S1–S5 samples. As Fe content increased, both the intensity and line width increased. This is because the increase in iron oxide loading would increase the magnetic interaction between Fe3+ ions.
As shown in Fig. S2,† the XAFS spectra were used to compare in energy, k and R-space. Firstly, there are subtle variations in the intensity of the spectra in energy space. The iron K-edge XANES probes the transition of 1s electrons to the 4p empty states. All spectra show similar spectral features including the pre-edge peak A′ and main peak A to peak C, followed by a broad feature D and E. The pre-edge feature A′ originates from the transition of 1s electrons to the Fe centred d states hybridized with ligand p states. The subsequent features are contributed by the electronic structure as well as the geometric structure due to multiple scattering events of the photoexcited electrons. The intensity of feature B gradually smeared out from S1 to S5, suggesting that the structural disordering is increased in samples with high iron content.
In Fig. 3, we compared the XANES spectra with the reference iron compounds as well as the theoretically obtained spectra of Fe doped TiO2. Judging from the spectral features, it is concluded that the spectra of all species are quite similar to the spectra of Fe2O3 and/or Fe3O4, due to the excellent agreement of intensity and position of peak features. On the other hand, the intensity ratio between feature A and feature B of samples (Fig. 3) is not in agreement with that of Fe2O3 suggesting the existence of another form of Fe besides iron oxide, for instance, some of the Ti4+ sites in titania are replaced by iron.
 | ||
| Fig. 3 Comparison of XANES spectra with reference iron oxides and two theoretical spectra of iron replacing Fe in anatase or rutile TiO2 at Fe K-edge for all species. | ||
By resorting to the EXAFS fitting, one can extract the coordinate structure of iron atoms. As shown in Fig. S3,† the first shell in R-space EXAFS can be readily fitted using the Fe(8a site, Fe3+) coordinates of Fe3O4 but with a phase shift for χ(k) and the bond length difference in χ(R). The iron is coordinated by four oxygen atoms equally separated. To compare the results consistently, the EXAFS fitting is performed using k3 weight with k and R range of [2, 12] Å−1 and [1, 2.34] Å, respectively. We summarized the structural parameters including the number of coordinates, Fe–O bond distances, and the mean square relative displacement (MSRD) of Fe–O bond as presented in Table 3. The coordinate number for first shell oxygen is 4.3–5.3 varying with the concentrations and the Fe–O bond length varies from 1.96–1.97 Å. The bond distance of samples measured is in nearer value to that of the bond length of Fe2O3 but coordinate number is lower than that of the Fe2O3, which are 6 for first shell coordinates. Hence the iron atoms in the sample may have oxygen vacancies, structural defects and small size of the hematite makes large surface area with significantly reduced coordinate number. The coordinate number of iron in samples are in between 4 and 6; therefore we can, by combining XANES and EXAFS analysis, conclude that there are two types of iron in the compounds, one is the distorted Fe2O3 and the other is iron replacing Ti sites in titania. The surface elementary composition and chemical states of iron in titania nanocomposites were characterized by XPS as shown in Fig. S4.†
| Sample | N | R Fe–O (Å) | MSRDFe–O (Å2) | R-Factor | k weight | k range | R range |
|---|---|---|---|---|---|---|---|
| S1 | 5.3 | 1.969 ± 0.007 | 0.008 ± 0.0005 | 0.009 | 3 | [2, 12.2] | [1, 2.34] |
| S2 | 4.8 | 1.968 ± 0.006 | 0.008 ± 0.0005 | 0.008 | 3 | [2, 12.0] | [1, 2.34] |
| S3 | 4.6 | 1.964 ± 0.005 | 0.008 ± 0.0004 | 0.006 | 3 | [2, 12.1] | [1, 2.34] |
| S4 | 4.4 | 1.961 ± 0.005 | 0.008 ± 0.0004 | 0.005 | 3 | [2, 12.2] | [1, 2.34] |
| S5 | 4.3 | 1.959 ± 0.005 | 0.009 ± 0.0004 | 0.006 | 3 | [2, 12.2] | [1, 2.34] |
3.2 Surface morphology analyses
The surface morphology and textural structure of the as-prepared nanocomposites were characterized by SEM, HRTEM and N2 physisorption analysis. The SEM micrographs of samples S1–S5 are in close resemblance in morphology as shown in Fig. S5.† There were large aggregates of tiny particles and the aggregates did not possess any definite shape. Hence the growth of TiO2 even in the presence of Fe did not form different morphology. This can be attributed to the fact that the nanoparticles themselves are composed of many smaller particles, while composite becomes more agglomerated. However, the size distribution of the powder was not calculated, the size of the particles are varies in the range of 0.24–1.20 μm for S1–S5 samples, it could be indicated that the Fe2O3 coupled and doped with TiO2 to form large agglomerated particles. Although, we could not find separate Fe2O3 particles in SEM images. Additionally, EDX microanalysis yielded the elementary composition of the material (Fig. S6–S9†). The chemical compositions of the samples were further ensured from ICP elemental analysis. The EDX and ICP values are tabulated in Table S1,† which shows the existence of a considerable amount of iron present in the catalyst along with basic elements titanium and oxygen. EDX results imply that coupled and doped iron oxide primarily exists in the TiO2 surface region. Moreover, line spectra and X-ray mapping analysis were also performed to locate the amount of each element in terms of percentage distribution within the stipulated contour area. It is evident that the growth of particle is restrained by iron oxide dispersion, which is significant importance not only for the design of surface properties and surface area,22 but also for tuning the electronic structure and characteristics.Fig. S10† and 4 show the TEM, STEM, HRTEM images and SAED patterns of the typical S1 and S5 samples, respectively. As shown in Fig. 4a, the prepared samples are nearly spherical in shape and did not have a core–shell structure. Closer observation of Fig. 4b and c reveals that most of the prepared nanocomposites belong to a Janus structure,38 and the single size of the S5 nanocomposites was about 6–17 nm. In Fig. 4d, the HRTEM image of typical S5 nanocomposite shows that Fe2O3 is coupled with anatase and/or rutile TiO2 phase to form a Janus structure. Moreover, the interplanar distance of three parts are 0.25, 0.32 and 0.35 nm, which corresponds well with the (110) plane of Fe2O3, (110) plane of TiO2(R) and the (101) plane of TiO2(A), respectively. The SAED patterns revealed a clear characteristic rings corresponding to high crystallinity and additionally complicated bright spots are clearly observed for S1 sample (Fig. S10e†), indicating the coexistence of anatase–rutile or iron oxide–anatase–rutile crystalline phases. However, due to the small interface between iron oxide and TiO2 nanoparticles in Janus structure, the recombination of light-induced electrons and holes would be decreased, which could enhance the photocatalytic activity.38
 | ||
| Fig. 4 TEM (a) and (b), STEM (c), HRTEM (d) images and SAED pattern (e) of S5 sample. | ||
The textural properties were evaluated by N2 physisorption analysis as shown in Fig. S11.† The BET surface area, Langmuir surface area and pore size of the particles are calculated and presented in Table 1. These parameters are important in the application of this material as a catalyst. The pure TiO2 physisorption isotherm is classified as type IV with a H2 hysteresis loop, characteristic of mesoporous nanoparticles.39 The N2 sorption isotherm of S1–S5 samples (Fig. S11a†) were also showed a type IV isotherm and the pore size distribution curve which indicates the predominance of pores 6.08–7.79 nm in diameter (Fig. S11b†). This property implies the presence of mesoporous structure. The adsorption equilibrium isotherms of all samples are similar in the textural properties. However, when the iron content is increased, the specific surface area shows a downward trend. The structural, chemical and morphological characterizations performed mainly using 57Fe Mössbauer spectroscopy, EPR, XAFS and other conventional techniques have clearly explored that iron oxide (Fe2O3) is coupled and doped with anatase–rutile mixed phase of titania in the all five S1–S5 samples.
3.3 Optical characterization
The optical properties of S1–S5 nanocomposites were measured in absorbance mode. Absorption characteristics of TiO2 are greatly modified because of coupling and doping of iron oxide with TiO2. Fig. 5a shows the DRS spectra of pure anatase TiO2 and S1–S5 samples. The spectrum of Fe2O3 showed high energy absorbance peaks at 550 and 650 nm (Fig. S12†). The DRS spectra of S1–S5 samples illustrate that the materials absorb more effectively in the 400–800 nm range. Three absorption bands are observed in the UV region at 216, 250 and 320 nm as shown in Fig. 5a, which are characteristic of TiO2 nanoparticles.40 However, the strong absorption of iron oxides causes significant changes in the absorbance spectrum of composite samples. The band range from 410 to 420 nm is probably due to excitation of 3d electrons from Fe3+ levels to the TiO2 CB (a charge transfer transition), whereas the broad band at ∼500 nm likely originates from 2T2g → 2A2g, 2T1g, d–d transitions of Fe3+ or from charge transfer between two Fe3+ ions as a form of iron oxide.41 Moreover, the shift in absorption edge maximum is due to change in the band gap of titania, suggesting a significant quantity of Fe3+ ions doped into a few layers of titania tetrahedral as well as octahedral coordination sites because of the overlap of 3d orbital of Fe3+ with semiconductor lattice and in addition cocatalyst of iron oxide coupled below the conduction band of TiO2.42 Absorption edge of S1–S5 samples illustrates a red shift with significantly a strong absorption in comparison with that of pure TiO2 in the visible region. As the iron content increases in titania composite system, the absorption edge shifts towards longer wavelength side with simultaneous reduction in band gap energy from 3.04 to 1.95 eV. This indicates the existence of empty states of the iron oxide conduction band, most probably from t2g Fe(B), are positioned above the Fermi level and below the conduction band of TiO2.42 It implies that iron oxide coupled and doped titania nanocomposites could absorb radiant energy in a wider range of wavelength, most probably in the visible region, are excellent and suitable materials than pure TiO2, which absorbs only UV light. | ||
| Fig. 5 UV-vis DRS (a) and photoluminescence emission spectra (b) of anatase TiO2 and S1–S5 samples at room temperature. | ||
More precisely, optical absorption properties of photocatalysts are a chief driver on the efficiency and enhancement of activity of photocatalysts. In addition, photoluminescence (PL) can be another indicator; the signals and their intensity are closely related to the activities of photocatalysts.43 Specifically, PL can be an effective tool to study the lattice defects in the metal oxides coupled and doped TiO2 samples. The PL spectra of TiO2 based materials are attributed to three kind of physical origins: self trapped excitons, oxygen vacancies, and surface states (defects).44 Photoemission spectra (λexc = 320 nm) of S1–S5 samples are depicted in Fig. 5b. The increase of ferric nitrate content in the preparation process of S1–S5 samples may play a major role and modifies the intensity of photoemission. However, peak position is not sensitive to changes in nanocomposites, but the emission intensity is decreased considerably due to the inclusion of iron oxide in titania. The S1–S5 samples show alarming and variable emission intensity between 350 and 550 nm (3.55–2.26 eV) depending on the conditions of preparations. It is observed from emission spectra that the time duration curves of S1–S5 samples are similar to that of TiO2 (Fig. S13a†). The emission band at 328 nm in the UV region is associated with the intrinsic emission of TiO2 lattice. The peak at 418 nm in the blue-violet region originates from charge recombination in the defect states of the surface. These surface states arise due to the interaction of iron oxide and TiO2 matrix. This peak shows a slight blue shift in violet band attributable to the charge transfer due to the interaction of iron oxide and TiO2. The bands at 466 nm in the blue area are due to different intrinsic defects in the TiO2 lattice such as oxygen vacancies, cation (Ti) vacancies, and interstitial defects. A green emission observed at 490 nm is viewed because of donor–acceptor recombination or charge transitions from the conduction band to oxygen vacancies.
The excitation states of S1–S5 samples were further characterized by lifetime measurements as shown in Fig. S14† and respective parameters are presented in Table 1. The lifetime decays were fitted in tri-exponential kinetics, in which the prominent lifetime τ1 is due to recombination of photoinduced charge carriers radiantly. In an earlier study, we reported the lifetime of pure anatase TiO2 (τ1 = 1.48 ns, τ2 = 10.07 ns and τ3 = 0.22 ns).22 In response to the results, the τ1 values for the currently prepared nanocomposites are longer than pure anatase TiO2, it means the charge carriers could be relaxed in deep-trap levels related to oxygen vacancies of the nanostructure. Consequently, the charge carriers recombine in iron oxide coupled and doped TiO2 illustrates radiant process with lifetime (τ2), which is fairly larger than that of radiant process life time of pure anatase TiO2. The fast component τ3 may be attributable due to near band edge relaxation of TiO2. In general, it was observed that emission lifetimes are altered and exist longer for the nanocomposites under study. It is a good indication on the lower recombination rate of the photoproduced electron–hole pairs, subsequently enhances the efficiency in photocatalytic performance.45 Therefore, photocatalytic activity of TiO2 nanoparticles may be altered and enhanced by combining titania lattice with Fe3+ ions and implantation of iron oxide, this can influence a “charge separation effect”, of the photogenerated excitons.46
3.4 Magnetic and electrochemical characterizations
Magnetic aspects of nanocomposites were investigated by room temperature VSM and temperature dependent magnetization measurements. Room temperature magnetization curves of the as-prepared S1–S5 nanocomposites are presented in Fig. 6a. The curve of the samples makes very weak saturation magnetization (Ms) (Table 1) even at a magnetic field of 15 kG, and no hysteresis is found. It can be understood that the as-prepared nanocomposites perform weak paramagnetic with superparamagnetic behavior as noticed from both magnetic and Mössbauer studies. Literature reports indicated that the superparamagnetic TiO2–Fe2O3 composites showed rapid aggregation under the influence of a magnetic field, which can be useful for separation of these particles after remediation and in applications.47 In case the system is focused for magnetic recovery and regeneration, then the Ms value of the iron oxides is anticipated to be no less than 1 emu g−1, so as to separate using external magnetic field.8 In the present case, the Ms value (90.2–187.6 × 10−3 emu g−1) of nanocomposites are very weak with absence of coercivity (Hc) and remanence (Mr), which makes it very difficult to re-collect the composite particles through an external magnetic field. This weak superparamagnetic behavior is also an indication that iron oxide is not separately present in nanocomposites, but is in coupled state with TiO2 surface matrix. However, magnetic hystereses loop of undoped TiO2 as shown in Fig. S13b,† implies that very weak ferromagnetic behavior may be found due to the formation of oxygen vacancies.22 | ||
| Fig. 6 Magnetization curve of S1–S5 samples at room temperature (a), temperature dependence of ZFC/FC magnetization plots measured between 2 and 300 K (b), where the critical phase transition temperature (Tc) is indicated on the ZFC/FC data set, and variation in inverse magnetization with temperature (c) for the S5 sample. | ||
Temperature-dependent zero field cooling (ZFC) and field cooling (FC) magnetization data for typical S5 sample is shown in Fig. 6b at 500 Oe. In proper scanning between 2 and 300 K, magnetization decreased appreciably upto Tc nearer to 23 K then it has slowly decreased. No significant difference was observed between ZFC and FC data, showing clear evidence of paramagnetism, while magnetization values are less than 0.06 emu g−1 at from the 23 to 300 K temperature range. It could be concluded EPR and 57Fe Mössbauer spectroscopic measurements at room temperature supported the existence of weak superparamagnetism. Further evidence of the weak superparamagnetism and the possible very weak ferromagnetic behavior at below Tc = 23 K can be observed by inspection of magnetic moment vs. temperature plot and the reciprocal susceptibility χ−1 for sample S5 (Fig. 6b and c).
The electronic property of the modified S5 sample film on ITO prepared via electrophoresis method was investigated by using Mott–Schottky (M–S) plot as shown in Fig. 7, which was obtained at room temperature under a dark condition at a frequency of 1 kHz in 0.5 M Na2SO4 (pH 6.4). The positive slope of the M–S plot reveals the n-type property of the dominant component of typical S5 sample. The flat band potential can be estimated from M–S plot (Fig. 7) using the following M–S relation:48

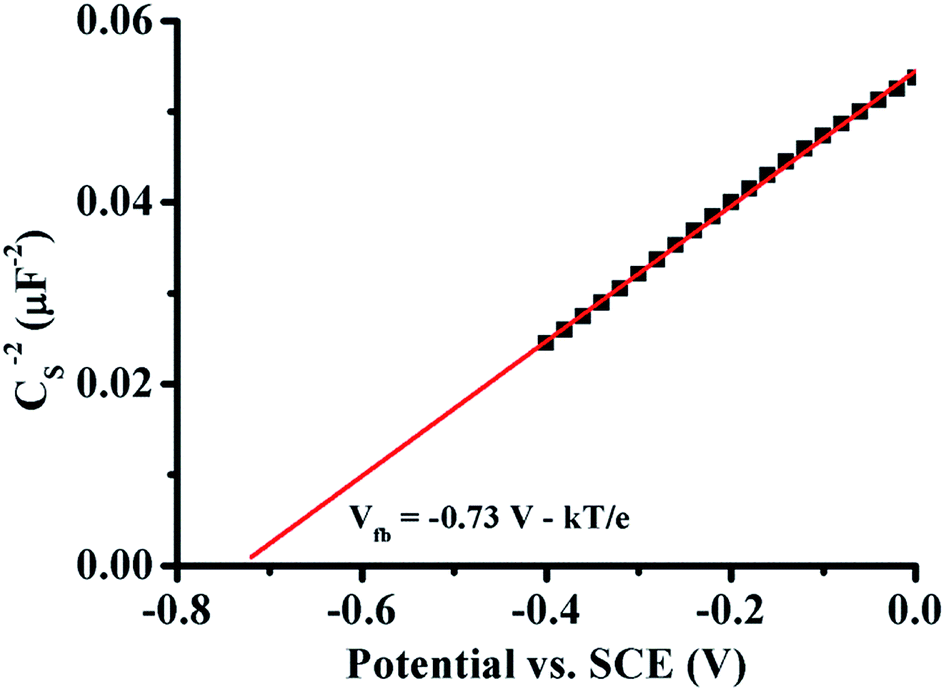 | ||
| Fig. 7 Mott–Schottky plot of S5 sample obtained at frequency of 1 kHz with 0.5 M Na2SO4 solution (pH 6.4), electrode area = 0.5 cm2. | ||
3.5 Photocatalytic activity characterization
To evaluate the photocatalytic activity of S1–S5 nanocomposites, MB solution was used as the model. The temporal UV-vis spectral changes of MB aqueous solution during the photocatalytic degradation reactions are depicted in Fig. S15.† As seen from the figure, when the MB solution was irradiated under the visible light, the main MB absorbance markedly decreased with irradiation time and almost ∼91% disappeared in about 360 min of irradiation. MB solution could not be degraded in the presence of prepared catalyst under the dark conditions (Fig. S16†), signifying that MB degradation in the present study is indeed through a photocatalytic process. Comparison experiments demonstrated that MB photolysis (without any catalyst) was very slow. Further compared to pure Fe2O3, rutile TiO2, anatase TiO2 and P25 nanoparticles, the photocatalytic activity (Fig. 8a) of our samples are efficient, which may be due to the compositional modification.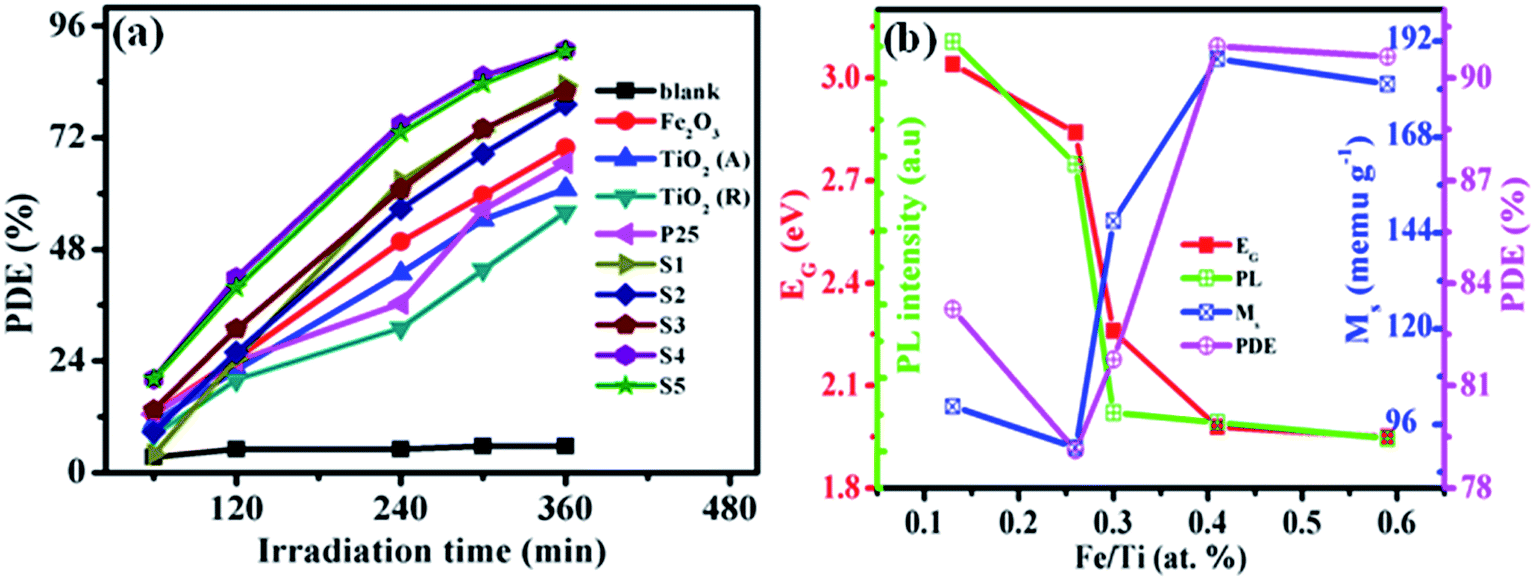 | ||
| Fig. 8 PDE of MB blank solution, with reference catalysts and prepared S1–S5 samples under visible light (λ > 480 nm) irradiation at various time intervals in water (a) and variation of band gap energy (EG), PL intensity of emission band, magnetization (Ms) and PDE values of S1–S5 samples with different Fe/Ti atomic ratio level obtained from ICP results (b). | ||
Correlation analysis was performed between iron content, photocatalytic activity, optical and magnetic behavior of prepared samples. The band gap energy and PL peak intensity values are correlated with Ms and photocatalytic activity results for the as-prepared samples is illustrated in Fig. 8b. Therefore, it can be concluded that implantation of iron oxides subsequent electronic energy levels in the S1–S5 samples are mainly responsible for optical, magnetic and photocatalytic activity of present samples. It has been shown that high levels of iron loaded TiO2 can strongly reduce the band gap energy and PL intensity, resulting in the formation of new trap level below the conduction band of TiO2.18 This will reduce e−–h+ recombination rate and alternatively lead to high level of electron transformation between neighboring metal oxides due to synergetic effect,50 making the catalyst more efficient in photocatalytic activity and strong magnetic behavior in metal oxides coupled semiconductor nanocomposites.22,23 An increase in the value of Ms can be attributed to the increase in the spin–spin transformation at higher iron loading level in prepared nanocomposites. The increase in the photocatalytic activity at higher iron loading level was also confirmed by PL spectroscopic and lifetime profile results. The formation of trapping levels in metal oxide semiconductors are found to be the main cause for enhanced photocatalytic activity of metal oxide nanocomposite structures,51 due to stabilizing the excitation energy level in semiconductor materials. These new and weak superparamagnetic in S1–S5 samples are extremely effective for the MB degradation, and in maintaining relatively high activity. In addition, the amounts of doped (site 1) and coupled (site 2) iron oxides could be correlated with the Ms and photocatalytic activity as shown in Fig. S17.† It is indicated that magnetic and photocatalytic activity increased as increasing doped and coupled iron oxides and the optimum level was obtained for 40% doped (site 1) and 60% coupled (site 2) S4 sample. After increasing both sites decreasing trend was observed. It is concluded that among all samples more efficient photocatalytic and magnetic behavior was observed for S4 sample.
The suggested photocatalytic mechanism of Fe2O3 coupled and doped TiO2 under visible light response is shown in Fig. 9. Ultimately, it indicates that the major process could be the formation, separation, and transfer of electron from trapped Fe doped TiO2 level to a similar energy level in coupled Fe2O3,20 which improves the multi-electron reduction in addition to decomposition of MB. Photoinduced electrons in the doped Fe3+ states under visible-light efficiently are transferred to the surface of the coupled Fe2O3 co-catalysts, as the doped Fe3+ ions in bulk produced energy levels below the conduction band of TiO2, which matches well with the potential of Fe3+/Fe2+ redox couple in the surface coupled Fe2O3 particles. For the photocatalytic process, photoinduced electrons and holes exists over a metal doped TiO2 photocatalysts directly or indirectly react with surface O2 and OH− to form ˙O2− and ˙OH active oxidants, respectively,52 these active species are responsible for the decomposition reactions.53 In this system, the real photocatalytic mechanism of the magnetic catalyst is still not clear and further work need to be carried out.
 | ||
| Fig. 9 The charge transfer in a Fe2O3 coupled and doped TiO2 photocatalyst induced degradation of MB under visible light irradiation in water. | ||
In addition, as practical recyclable catalysts, high catalytic activity in each cycle is necessary. Renewable photocatalytic activity was also investigated, and the results are presented in Fig. S18a.† We can see that the PDE of the typical S5 nanocomposite is very stable. After the degradation experiment was carried out nearly five times, the PDE at different cycles is still higher than 83%. In conclusion, the as-prepared nanocomposites have good photocatalytic activity in degradation of MB in aqueous solution and the efficiency of this catalytic activity can maintain 83% even after being reused five times. Thus, the as-prepared catalyst can be used as a photocatalyst in practical applications. In case of the decrease of the catalytic activity after each recycle may partly result from incomplete separation of the catalyst. Furthermore, the structure of S5 sample was also probed by 57Fe Mössbauer spectroscopy, TEM, HRTEM and STEM-EDS analysis were made on the five times used samples and are presented in Fig. S18b–f.† No significant difference was observed in the structure, surface morphology, chemical states and elemental composition after waste water treated with S5 sample compared with the fresh S5 sample. Hence, prepared nanocomposites are proven to be stable and renewable photocatalysts.
3.6 Theoretical calculation
As shown in Fig. 10, the partially projected density of states (DOS) is compared for the Ti replacement by Fe in rutile and anatase TiO2, respectively. The energy scale in DOS of rutile is 0.3 eV higher than that of anatase while the characteristic electronic structure resembles with each other. It is found that the valence band is dominated by O p states, while the conduction band is mainly due to Ti d states, in consistent with the DFT calculations.20 Upon doping of Fe in place of Ti of TiO2, the Fe 3d states introduce doped energy states within the energy gap in both rutile and anatase TiO2, which is also in consistent with the DFT calculations.20 There are number of hybridized states between Ti d and Fe d states in the vicinity of the Fermi energy level. The partial density of states calculations indicate the drastic variation of the electronic structure upon doping of Fe at Ti site in TiO2. However, previous report demonstrated that surface-grafted and bulk-doped Fe3+ have nearly identical energy levels.20 | ||
| Fig. 10 DOSs for Fe replacing Ti in rutile and anatase TiO2. | ||
4. Conclusions
The visible light active iron oxide coupled and doped titania nanocomposites were successfully synthesized by facile hydrothermal method. Various spectroscopic measurements exposed the presence of structural, optical, magnetic and photocatalytic effects in the system. The surface morphological analysis indicates that the shape and uniformity of the nanomaterials changes with different iron dosage on titania and elemental composition of prepared nanocomposites. The electrochemical study revealed the n-type semiconductor property of prepared sample. The photocatalytic properties of prepared samples were tested, and the results showed that all of the samples have the improved photocatalytic activity due to an efficient separation of photoinduced electron/hole pairs on its surface. The major effects of iron loading are considered to induce: (i) formation of iron oxide coupled and doped anatase–rutile mixed phases, (ii) enhanced light absorption property in visible region and predominantly reduced band gap energy, (iii) weak superparamagnetic character and absence of magnetic separation in applied magnetic field of the iron oxide coupled and doped titania and (iv) photocatalytic activity that mainly showed structural, optical and magnetic dependences. Furthermore, the iron oxide coupled and doped titania has an excellent long-term stability and could perform renewable photocatalytic activity. Results indicate that this material will be a promising visible light active photocatalyst for the treatment of wastewater.Acknowledgements
Prof. J. Wang is grateful to National Natural Science Foundation of China (No. 21476232). Dr ASG acknowledges the CAS President's International Fellowship Initiative (PIFI) programme for a postdoctoral researcher funded by the Chinese Academy of Sciences (No. 2016PT023) and International Young Scientists Research Fund project funded by the NSFC (No. 2161101071). Prof. KA is thankful to CSIR, New Delhi (No. 01 (2570)/12/EMR-II/3.4.2012) for financial support through a major research project. The authors acknowledge the technical support of the 1W2B station of Beijing Synchrotron Radiation Facility. Prof. KA thanks Dr Alok Banerjee for SQUID instrumental facility at UGC-DAE-CSR, Indore, India.References
- B. M. Pirzada, O. Mehraj, N. A. Mir, M. Z. Khan and S. Sabir, New J. Chem., 2015, 39, 7153–7163 RSC.
- F. Zheng, H. Lu, M. Guo, M. Zhang and Q. Zhen, J. Mater. Chem. C, 2015, 3, 7612–7620 RSC.
- Y. Chen, N. Li, Y. Zhang and L. Zhang, J. Colloid Interface Sci., 2014, 422, 9–15 CrossRef CAS PubMed.
- K. Zhu, J. Wang, Y. Wang, C. Jin and A. S. Ganeshraja, Catal. Sci. Technol., 2016, 6, 2296–2304 CAS.
- J. S. Chen, H. Liu, S. Z. Qiao and X. W. Lou, J. Mater. Chem., 2011, 21, 5687–5692 RSC.
- Y. Yang, S. Zhang, L. Wan and D. Xu, Appl. Surf. Sci., 2012, 258, 3469–3474 CrossRef.
- Z. Li, H. Wang, L. Zi, J. Zhang and Y. Zhang, Ceram. Int., 2015, 41, 10634–10643 CrossRef CAS.
- W. Wu, C. Jiang and V. A. L. Roy, Nanoscale, 2015, 7, 38–58 CAS.
- J. Sun, Y. Yang, J. I. Khan, E. Alarousu, Z. Guo, X. Zhang, Q. Zhang and O. F. Mohammed, ACS Appl. Mater. Interfaces, 2014, 6, 10022–10027 CAS.
- M. I. Litter and J. A. Navio, J. Photochem. Photobiol., A, 1996, 98, 171–181 CrossRef CAS.
- R. Dholam, N. Patel, M. Adami and A. Miotello, Int. J. Hydrogen Energy, 2009, 34, 5337–5346 CrossRef CAS.
- H. Sun, G. Zhou, S. Liu, H. M. Ang, M. O. Tadé and S. Wang, Chem. Eng. J., 2013, 231, 18–25 CrossRef CAS.
- W. Yan, Y. Zhang, W. Xie, S. Sun, J. Ding, J. Bao and C. Gao, J. Phys. Chem. C, 2014, 118, 6077–6083 CAS.
- X. Li, J. Wang, A. I. Rykov, V. K. Sharma, H. Wei, C. Jin, X. Liu, M. Li, S. Yu, C. Sun and D. D. Dionysiou, Catal. Sci. Technol., 2015, 5, 504–514 CAS.
- Y. Wang, Y. Zhang, F. Yu, C. Jin, X. Liu, J. Ma, Y. Wang, Y. Huang and J. Wang, Catal. Today, 2015, 258, 112–119 CrossRef CAS.
- F. Yu, Y. Wang, H. Tang, Y. Zhang, C. Jin, X. Liu, M. Li and J. Wang, Catal. Commun., 2014, 51, 46–52 CrossRef CAS.
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397–415 CrossRef CAS PubMed.
- X. Zhang, Y. Zhu, X. Yang, Y. Zhou, Y. Yao and C. Li, Nanoscale, 2014, 6, 5971–5979 RSC.
- S. Shen, X. Guo, L. Wu, M. Wang, X. Wang, F. Kong, H. Shen, M. Xie, Y. Ge and Y. Jin, J. Mater. Chem. B, 2014, 2, 5775–5784 RSC.
- M. Liu, X. Qiu, M. Miyauchi and K. Hashimoto, J. Am. Chem. Soc., 2013, 135, 10064–10072 CrossRef CAS PubMed.
- Y. Wang, K. Nomura, X. Liu, A. I. Rykov, C. Jin, T. Liu and J. Wang, Eur. J. Inorg. Chem., 2016, 2131–2135 CrossRef CAS.
- A. S. Ganeshraja, S. Thirumurugan, K. Rajkumar, K. Zhu, Y. Wang, K. Anbalagan and J. Wang, RSC Adv., 2015, 6, 409–421 RSC.
- A. S. Ganeshraja, A. S. Clara, K. Rajkumar, Y. Wang, Y. Wang, J. Wang and K. Anbalagan, Appl. Surf. Sci., 2015, 353, 553–563 CrossRef CAS.
- A. K. Patra, A. Dutta and A. Bhaumik, ACS Appl. Mater. Interfaces, 2012, 4, 5022–5028 CAS.
- B. Palanisamy, C. M. Babu, B. Sundaravel, S. Anandan and V. Murugesan, J. Hazard. Mater., 2013, 252–253, 233–242 CrossRef CAS PubMed.
- Y. X. Li, M. Zhang, M. Guo and X. D. Wang, Rare Met., 2009, 28, 423–427 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513 RSC.
- S. Shen, X. Wang, T. Chen, Z. Feng and C. Li, J. Phys. Chem. C, 2014, 118, 12661–12668 CAS.
- K. Wantala, P. Khemthong, J. Wittayakun and N. Grisdanurak, Korean J. Chem. Eng., 2011, 28, 2178–2183 CrossRef CAS.
- J. I. Peña-Flores, A. F. Palomec-Garfias, C. Márquez-Beltrán, E. Sánchez-Mora, E. Gómez-Barojas and F. Pérez-Rodríguez, Nanoscale Res. Lett., 2014, 9, 499 CrossRef PubMed.
- J. Wang and Y. Sakai, Hyperfine Interact., 2012, 218, 9–14 CrossRef.
- T. K. Kundu, M. Mukherjee, D. Chakravorty and T. P. Sinha, J. Mater. Sci., 1998, 33, 1759–1763 CrossRef CAS.
- A. F. Lehlooh and S. H. Mahmood, J. Magn. Magn. Mater., 1995, 151, 163–166 CrossRef CAS.
- M. Grigorova, H. J. Blythe, V. Blaskov, V. Rusanov, V. Petkov, V. Masheva, D. Nihtianova, L. M. Martinez, J. S. Munoz and M. Mikhov, J. Magn. Magn. Mater., 1998, 183, 163–172 CrossRef CAS.
- Q. Sun, W. Leng, Z. Li and Y. Xu, J. Hazard. Mater., 2012, 229–230, 224–232 CrossRef CAS PubMed.
- C. Fàbrega, T. Andreu, A. Cabot and J. R. Morante, J. Photochem. Photobiol., A, 2010, 211, 170–175 CrossRef.
- L. Zeng, W. Ren, L. Xiang, J. Zheng, B. Chen and A. Wu, Nanoscale, 2013, 5, 2107–2113 RSC.
- K. S. W. Sing, Pure Appl. Chem., 1982, 54, 2201–2218 CrossRef.
- M. Stefan, O. Pana, C. Leostean, C. Bele, D. Silipas, M. Senila and E. Gautron, J. Appl. Phys., 2014, 116, 114312 CrossRef.
- W. Wu, X. Xiao, S. Zhang, F. Ren and C. Jiang, Nanoscale Res. Lett., 2011, 6, 533 CrossRef PubMed.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- A. Banisharif, S. H. Elahi, A. A. Firooz, A. A. Khodadadi and Y. Mortazavi, Int. J. Biomed. Nanosci. Nanotechnol., 2013, 9, 193–202 Search PubMed.
- K. Anbalagan, J. Phys. Chem. C, 2011, 115, 3821–3832 CAS.
- B. Liu and L. Peng, J. Alloys Compd., 2013, 571, 145–152 CrossRef CAS.
- T. Hirakawa and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 3928–3934 CrossRef CAS PubMed.
- T. Xin, M. Ma, H. Zhang, J. Gu, S. Wang, M. Liu and Q. Zhang, Appl. Surf. Sci., 2014, 288, 51–59 CrossRef CAS.
- D. Chauhan, V. Satsangi, S. Dass and R. Shrivastav, Bull. Mater. Sci., 2006, 29, 709–716 CAS.
- N. Yusoff, S. Vijay Kumar, A. Pandikumar, N. M. Huang, A. R. Marlinda and M. N. Anamt, Ceram. Int., 2015, 41, 5117–5128 CrossRef CAS.
- J. Xu, Z. Gao, K. Han, Y. Liu and Y. Song, ACS Appl. Mater. Interfaces, 2014, 6, 15122–15131 CAS.
- Y. Zhang, X. Yu, Y. Jia, Z. Jin, J. Liu and X. Huang, Eur. J. Inorg. Chem., 2011, 5096–5104 CrossRef CAS.
- S. Banerjee, D. D. Dionysiou and S. C. Pillai, Appl. Catal., B, 2015, 176–177, 396–428 CrossRef CAS.
- F. E. Oropeza, B. Mei, I. Sinev, A. E. Becerikli, M. Muhler and J. Strunk, Appl. Catal., B, 2013, 140–141, 51–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra13212j |
| This journal is © The Royal Society of Chemistry 2016 |