
Fe3O4@Pt/MWCNT/carbon paste electrode for determination of a doxorubicin anticancer drug in a human urine sample
Abstract
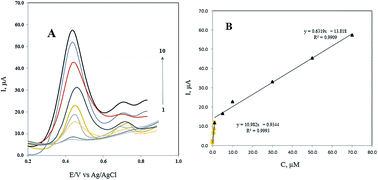
In this study, a Fe3O4@Pt nanoparticle and multi-walled carbon nanotube (MWCNT) modified carbon paste electrode was used as a fast and sensitive tool for the electrochemical determination of doxorubicin (DOX). The electrochemical oxidation of DOX was investigated at Fe3O4@Pt/MWCNT/CPE using the differential pulse voltammetry method. The developed electrode exhibited excellent electrochemical activity towards the electrochemical oxidation of the investigated anticancer drug. Under the optimized experimental conditions, a linear calibration curve in the range of 0.05 to 70.0 μmol L−1 with two different slopes and a detection limit of 1 nmol L−1 was obtained. Finally, the method was successfully employed for the voltammetric determination of DOX in a urine sample at trace levels with good recoveries.
 Please wait while we load your content...
Please wait while we load your content...

