
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nioplexes encapsulated in supramolecular hybrid biohydrogels as versatile delivery platforms for nucleic acids†
Santiago
Grijalvo
abc,
Gustavo
Puras
cd,
Jon
Zárate
cd,
Ramon
Pons
b,
Jose Luis
Pedraz
cd,
Ramon
Eritja
bc and
David Díaz
Díaz
*ab
aInstitute of Organic Chemistry, University of Regensburg, Universitätstrasse. 31, D-93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Fax: +49 941 943 4121; Tel: +49 941 943 4373
bInstitute of Advanced Chemistry of Catalonia (IQAC-CSIC), Spain
cBiomedical Research Networking Center in Bioengineering, Biomaterials and Nanomedicine (CIBER BBN), Spain
dNanoBioCel group, University of the Basque Country (EHU-UPV), Spain
First published on 11th April 2016
Abstract
Supramolecular hydrogels based on N-protected phenylalanine (Fmoc–Phe–OH) were used to encapsulate non-ionic surfactant vesicles (niosomes). The niosomes consisted of an amphiphilic lipid mixed with polysorbate-80 and electrostatically complexed with a fluorescently labelled oligodeoxynucleotide (FITC–ODN) as a model nucleic acid derivative. The diffusion properties of the supramolecular hydrogel were conveniently tuned by adding a small amount of κ-carrageenan (≤1% w/v) as a crosslinking agent. Interestingly, neither cationic niosomes nor the biopolymer additive significantly affected the hydrogelation properties of the amino acid-based low molecular weight (LMW) gelator. In vitro drug release experiments from Fmoc–Phe–OH hydrogels containing cationic niosomes were successfully carried out in the absence and in the presence of κ-carrageenan. The niosomal ODN liberation in solution was fitted using Higuchi, Korsmeyer–Peppas and Weibull drug release models, showing the prevalence of diffusion mechanisms in each case. Moreover, the time release was easily prolonged by increasing the concentration of κ-carrageenan. Preliminary transfection studies indicate the suitability of these supramolecular hybrid hydrogels to embed niosomal formulations and, consequently, for being used as tunable delivery vehicles for nucleic acids.
1. Introduction
Since Wichterle and Lim reported for the first time the synthesis of crosslinked poly(hydroxyethylmethacrylate) (pHEMA) for biological use,1 synthetic and natural polymer-based hydrogels have gained substantial importance over the years in a good number of biomedical, pharmaceutical and biotechnological applications such as tissue engineering, drug delivery systems or cell culture scaffolds.2–5 The research in this area has contributed to the development of novel smart polymer gel networks having specific responses to external stimuli such as pH, light, temperature or chemical additives,6–8 which could facilitate the controlled liberation of embedded therapeutic drugs.9In contrast to polymer or chemical gels10 that are based on covalent bonds, physical or supramolecular gels11 are typically made of low molecular weight (LMW) compounds self-assembled through non-covalent interactions (e.g., hydrogen-bonding, π–π stacking, van der Waals, dipole–dipole, charge-transfer and coordination interactions). In general, such non-covalent nature of supramolecular gels provides reversible gel-to-sol phase transitions as response to external stimuli. The solid-like appearance and rheological properties of the gels result from the immobilization of the liquid (major component) into the interstices of a solid matrix (minor component) mainly through capillary forces.12 The formation of the 3D-network with numerous junction zones results from the entanglement of 1D-suprapolymeric strands of gelator molecules usually of nm diameters and mm lengths.13 The synthetic versatility of many LMW gelators together with their biocompatible properties have allowed their use in a number of biomedical applications including cell adhesion, cell growth, tissue engineering and controlled drug delivery.14–17 Among the plethora of LMW compounds that are able to form supramolecular hydrogel networks, aminoacid derivatives and peptide conjugates18–23 constitute one of the most studied groups. Within this context, the use of phenylalanine (Phe) derivatives as hydrogelator was reported for the first time in 2006.24,25 In 2011, Roy and Banerjee26 reported the ability of N-terminally Fmoc protected Phe, Fmoc–Phe–OH, to form stable and transparent hydrogels at concentrations as low as 0.1% w/v.
On the other hand, the development of more efficient and safe strategies for the administration of a drug is a key step for the success in modern therapeutic processes. Besides the classical therapeutic approaches to find a drug, gene therapy and gene silencing strategies have emerged as promising treatment options for a number of diseases. However, the search of an optimal administration is a key to getting an efficient therapeutic action. Thus, colloidal drug delivery systems including nanoparticles, liposomes and hydrogels, among others, have experienced significant growth in recent years based on the necessity of optimizing the transportation of poorly-water soluble drugs as well as improving the bioavailability and pharmacokinetics of numerous nucleic acid therapeutics.27 Within this context, niosomes (non-ionic surfactant vesicles) have become promising candidates for drug delivery applications28,29 and an alternative to the use of liposomes, which are considered the keystone of particle-based carrier systems. This growing interest of niosomes is mainly due to their good physical stabilities, cost-effectiveness, easy large-scale synthesis and their structural similarities to liposomes. Moreover, they are able to efficiently entrap hydrophilic and lipophilic drugs either in aqueous compartments or in bilayer domains, respectively. This efficiency has been recently reported by some of us with the use of nioplexes, elaborated with cationic niosomes, for both in vitro and in vivo transfection experiments.30,31
Although liposomes, niosomes and other conventional lipid particles have mainly prepared to obtain more efficient non-druggable therapeutic formulations, the stability of such particles may be lessened by the action of macrophages and their rapid renal clearance. Several approaches have been reported to overcome this drawback, including variation of particle size, surface charge, lipid composition or the addition of different ligands and/or receptors.32,33 Alternatively, the encapsulation of such colloidal particles either in synthetic34 or natural polymers-based hydrogels35 has also generated promising and fascinating hybrid materials that has helped to improve the efficacy of such liposomal formulations.36,37 In fact, this kind of entrapment has generated efficient colloidal particle protections and thereby has reduced both undesirable burst release effects and improved drug delivery when therapeutic molecules have been administered38,39 More recently, novel hybrid materials like injectable hydrogels containing liposomes with self-healing or shear-thinning properties have also proven to be another promising approach employed for therapeutic purposes.40 In particular, hydrogel synthesis from polymer–nanoparticle interactions41 or responsive hydrogel materials for intratumoral delivery have been designed for suitable in vitro and in vivo drug delivery processes and for some cancer treatments42 by increasing the efficacy and reducing toxicity, respectively. Recently, multidomain peptides (MDPs), which have the ability to self-assemble and generate nanofiber hydrogels, have also been used to entrap liposomes containing several growth factors.43,44
In this work we describe the synthesis of lipid-based nioplexes bearing an oligodeoxynucleotide as a model biomolecule. These nioplexes were subsequently encapsulated within supramolecular hydrogels based on self-assembled Fmoc–Phe–OH molecules.23 Some hydrogels were also physically crosslinked by the addition of κ-carrageenan. In vitro release kinetics of the niosomal formulation from these hydrogels was investigated and adjusted to several mathematical models. Finally, the suitability of hybrid materials in the transfection of nucleic acids (e.g. antisense oligonucleotide and plasmid DNA) in cell culture was also evaluated. The overall concept of this work is illustrated in Fig. 1. As far as we are aware, the use of LMW hydrogels to encapsulate nioplexes derived from cationic niosomes for the controlled delivery of therapeutic molecules has not yet been reported.
 | ||
| Fig. 1 Strategy used for the preparation of Fmoc–Phe–OH supramolecular hydrogels containing cationic nioplexes and FITC–ODN as a model nucleic acid. (A) Preparation of cationic niosomes containing FITC–ODN. (B) Illustration of the encapsulation and in vitro release studies of niosomal FITC–ODN from Fmoc–Phe–OH–based hydrogels (hydrogel-1) and Fmoc–Phe–OH hydrogels physically crosslinked with κ-carrageenan (hydrogels-(2–4)). | ||
2. Results and discussion
2.1 Synthesis and characterization of hydrogels containing nioplexes
The synthesis of the amphiphilic lipid-1 (ESI, Fig. S1†) and the subsequent preparation of the cationic niosomal formulation were successfully accomplished following the experimental procedures described in our previous studies.30,31 Briefly, the cationic nioplexes used in this studied were made upon mixing lipid-1 with a non-ionic surfactant agent (polysorbate-80) and a fluorescently labelled oligodeoxynucleotide (FITC–ODN) as model antisense inhibitor (Fig. 1). The so obtained nioplexes were subsequently mixed with a suitable amount of the LMW gelator (Fmoc–Phe–OH) in phosphate buffered saline (PBS, pH 7.4) solution and gently heated in a sealed tube.26 After cooling the mixture down to room temperature, a transparent hydrogel was obtained (hydrogel-1; ESI, Fig. S2A†). Additionally, we prepared similar cationic nioplexes loaded-hydrogels but physically crosslinked with κ-carrageenan with the aim of tuning the diffusional properties of the matrix under physiological conditions. Thus, nioplexes-containing Fmoc–Phe–OH–based hydrogels-(2–4) crosslinked with increasing concentration of κ-carrageenan were prepared in PBS buffer (i.e., hydrogel-2, hydrogel-3 and hydrogel-4 containing 0.5, 0.8 and 1% w/v of κ-carrageenan, respectively) (ESI, Fig. S2B†). All hydrogels (1–4) were able to encapsulate nioplexes with high efficiency (EE > 90%) and the gelation ability of Fmoc–Phe–OH was not significantly influenced by the presence of both κ-carrageenan and niosomes (ESI, Table S1†).The mechanical properties of representative composite hydrogels (i.e., hydrogel-1 and hydrogel-4) were investigated by oscillatory rheological measurements and compared to control gels prepared in the absence of niosomes (i.e., native hydrogel-5 and hydrogel-6, which was only tuned with κ-carrageenan at 1% w/v) (Fig. 2). The storage modulus (G′) and the loss modulus (G′′) were measured as a function of frequency (DFS, dynamic frequency sweep), strain (DSS, dynamic strain sweep) and time (DTS, dynamic time sweep). The viscoelastic nature of all samples was evidenced by G′ values ca. one order of magnitude greater than G′′ and a low dependence of G′ with the applied frequency. The addition of cationic niosomes to non-crosslinked hydrogels (i.e., hydrogel-1vs. hydrogel-5) did not change the mechanical properties of the network, which displayed similar dissipation factor values (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ = G′′/G′ ≈ 0.15) (Fig. 2A; ESI, Table S1†), indicating a good response of both hydrogels to external forces. Interestingly, the incorporation of small amounts of κ-carrageenan (hydrogel-6) produced an increase in G′ values compared to those obtained for the analogues non-crosslinked hydrogels (i.e., hydrogel-6vs. hydrogel-5; hydrogel-4vs. hydrogel-1) (Fig. 2A).45 Thus, the presence of κ-carrageenan seems to produce highly associated materials due to colloidal forces between the cationic niosomes and the anionic biopolymer. This was also reflected in the oligonucleotide release rates (vide infra). Again, the incorporation of cationic niosomes into hydrogels containing κ-carrageenan (hydrogel-4) did not induce any significant change in the rheological properties when compared to hydrogel-6.
δ = G′′/G′ ≈ 0.15) (Fig. 2A; ESI, Table S1†), indicating a good response of both hydrogels to external forces. Interestingly, the incorporation of small amounts of κ-carrageenan (hydrogel-6) produced an increase in G′ values compared to those obtained for the analogues non-crosslinked hydrogels (i.e., hydrogel-6vs. hydrogel-5; hydrogel-4vs. hydrogel-1) (Fig. 2A).45 Thus, the presence of κ-carrageenan seems to produce highly associated materials due to colloidal forces between the cationic niosomes and the anionic biopolymer. This was also reflected in the oligonucleotide release rates (vide infra). Again, the incorporation of cationic niosomes into hydrogels containing κ-carrageenan (hydrogel-4) did not induce any significant change in the rheological properties when compared to hydrogel-6.
 | ||
| Fig. 2 Comparative rheological DFS (A) and DSS (B) measurements between hydrogel-1 and native hydrogel (hydrogel-5) (left); native hydrogel (hydrogel-5; without cationic vesicles) and physically crosslinked hydrogel with κ-carrageenan (hydrogel-6; without cationic vesicles) (middle); hydrogel-4 and hydrogel-6 (right). The arrows in DSS plots indicate the cross-point between G′ and G′′ (γ at break). The hydrogels were prepared in PBS (pH 7.4) and measured at room temperature (25 °C). | ||
On the other hand, DSS (dynamic strain sweep) measurements confirmed that all hydrogels remained intact while applied an oscillation stress within the linear viscoelastic regime (Fig. 2B; ESI, Fig. S3†). The presence of κ-carrageenan in the formulations produced only slight differences in the critical strain (ESI, Table S1†). Above the critical strain, crosslinked hydrogels induced a slighter resistance to the shear stress (γ) until G′′ exceeded G′ (i.e., γ at break = 68% vs. 43% for hydrogel-6 and hydrogel-5, respectively). The encapsulation of cationic niosomes into the gel matrices also increased slightly the critical strain γc values in comparison to native hydrogels (i.e., γc = 16.6% and 20% for hydrogel-1 and hydrogel-4vs. 13% and 13.4% for hydrogel-5 and hydrogel-6, respectively) (ESI, Fig. S3 and Table S1†). A similar behaviour was observed in the resistance of the hybrid gels to shear stress (e.g., γ at break = 87% vs. 68% for hydrogel-4 and hydrogel-6, respectively). These differences were also observed for hydrogel-1 when compared to the corresponding native material. These results suggest that the presence of κ-carrageenan and cationic niosomes within the supramolecular gel network could induce certain deformations in the systems under specific stress conditions, thus increasing the hydrogel rigidity and the corresponding shear stress resistance.46 Finally, DTS (dynamic time sweep) measurements confirmed the temporal stability of the supramolecular hydrogels within the viscoelastic regime (0.1% strain, 1 Hz frequency) (ESI, Fig. S4†).
2.2 Small-angle X-ray scattering measurements
The behavior of lipid-1 was investigated in aqueous media and their diffraction patterns (SAXS and WAXS experiments) were recorded at several temperatures (Fig. 3). SAXS curves were identical at both 25 °C and 37 °C whereas some increased background was appreciated around q = 2 nm−1 at 45 °C (Fig. 3A). A partial fusion was observed at 55 °C with the presence of a band centered at q = 1.94 nm−1 although the peaks did not show appreciable changes. At 55 °C, a complete fusion was observed and the band only remained in the SAXS domain. Importantly, the SAXS pattern at 50 °C did not change as a function of time (the three consecutive acquisitions were identical within experimental error), suggesting the coexistence of the lamellar and melted phases at this temperature. Therefore, we carried out the ODN encapsulation and release studies below this temperature in order to ensure the integrity of the vesicles during the experiments. | ||
| Fig. 3 (A) Scattered intensity of the fully hydrated lipid as a function of dispersion vector q at different temperatures. (B) WAXS spectra at several temperatures of the fully hydrated lipid as a function of scattering vector modulus q. | ||
Using the same range of differing temperatures, the WAXS region (Fig. 3B) showed a single peak for the samples, which determined a lamellar order. This experiment confirmed a slight shift to smaller q values due to the increased mobility of the hydrophobic chains. This rise showed an interchain distance increase from 0.417 nm to 0.425 nm, which in turn implied a density decrease of about 3.5%.
Additionally, the fitting of the experimental scattering data recorded at 25 °C was consistent with a lamellar model (Fig. 4A). It is noteworthy the absence of the expected second peak for the lamellar arrangement. This is related to the electronic profile of the lamellae coupled with the interlamellar distance. This can be appreciated from the single lamellae curve shown in red. The first minimum coincides with the position where the second peak should appear. Fig. 4B shows the corresponding electronic density profile for the lamellae compared with the electronic density profile obtained from the Fourier reconstruction using the three visible peaks. Although both electronic profiles are consistent, we believe that our model profile is more realistic in the sense that there is not a physical reason for the secondary minima around ±1 nm from the bilayer center. From the electronic profile and the known volume and electron composition of the molecule we determined a hydrophobic bilayer half thickness of 1.43 ± 0.1 nm, a total bilayer thickness of 4.8 ± 0.1 nm, an area per molecule of 0.541 ± 0.003 nm2 and 13 ± 2 water molecules per polar head. This amount of water roughly corresponds to a 30% hydration. Furthermore, the sharpness of the peaks suggests a strong correlation between the bilayers, which corresponds to 28 ± 2 correlated bilayers, according to the model.
 | ||
| Fig. 4 (A) SAXS intensity as a function of dispersion vector q of the fully hydrated lipid at 25 °C (open black squares), fitted profile (black line) and bilayer form factor (red line). (B) Electronic density profile corresponding to the fitting in Fig. 3. Black line corresponds to the MCG profile and the red line to the Fourier reconstruction. The Fourier reconstruction was scaled to produce the same integral as the MCG model. | ||
2.3 In vitro release studies
As mentioned above, in order to investigate and validate the efficacy of hydrogels-(1–4) as drug carriers, we studied the release kinetics of physiologically important biomolecules like oligonucleotides, which are involved in well-known gene regulation processes.47,48 The interaction between fluorescently labelled oligodeoxynucleotide FITC–ODN49 and cationic niosomes afforded the corresponding complexes (nioplexes) (Fig. 1A) with an optimal amine to phosphate (N/P) charge ratio of 14 as determined by the electrophoretic mobility shift assay (EMSA) (Fig. 5). | ||
| Fig. 5 Electrophoretic mobility shift assay (EMSA). FITC–ODN was complexed with growing cationic niosomes N/P charge ratios in 100 mM of PBS in a native polyacrylamide gel (20%) at 150 V and 25 °C. Bands were stained with SYBR-Green II and bromophenol blue was used as a marker (mk). The disappearance of the FITC–ODN band indicated the completion of the complex formation (i.e., N/P ratio = 12). | ||
In vitro release studies of the composite hydrogels made of Fmoc–Phe–OH and (FITC–ODN)-loaded cationic nioplexes (1–4) were performed at 37 °C to emulate physiological conditions. As a reservoir for the release, hydrogels-(1–4) were immersed in PBS solution and aliquots were removed at regular intervals and analyzed by fluorescence spectroscopy according to a proper standard curve of FITC–ODN in solution (r2 > 0.99) (ESI, Fig. S5†). The removed volume was always replaced by the same amount of fresh buffer. In the case of hydrogel-1, the release kinetics was controlled by erosion of the polymeric matrix and ca. 90% of the FITC–ODN was rapidly liberated in ca. 1.5 h (Fig. 6).
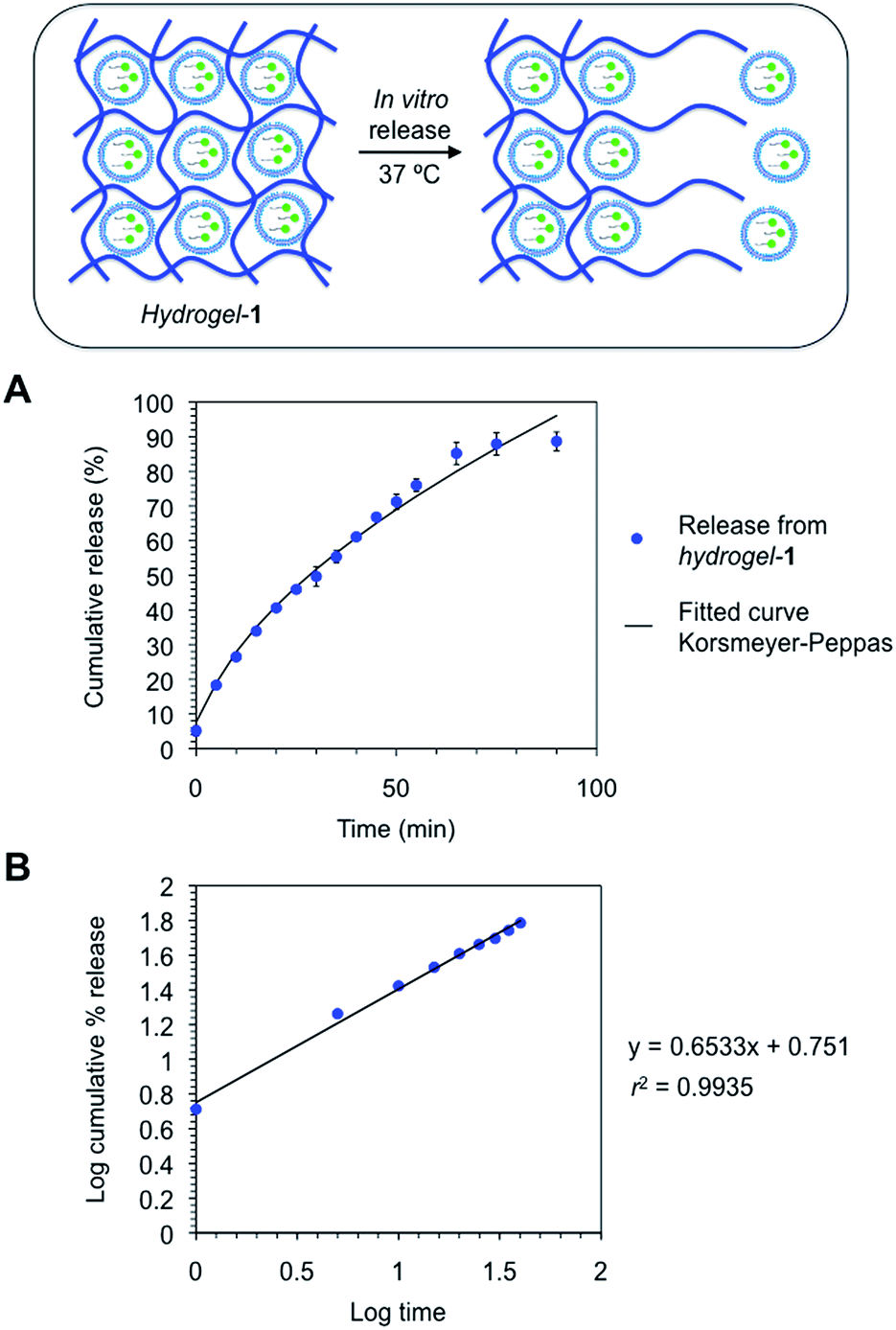 | ||
| Fig. 6 (A) Cumulative release profile of the niosomal formulation containing FITC–ODN from hydrogel-1 in PBS solution at 37 °C, along with the fitted curve (n = 2) according to Korsmeyer–Peppas equation [eqn (4)]. (B) log–log plot showing the monomial relationship between FITC–ODN concentration and time. Model fitting parameters are given in the ESI (Table S3†). | ||
It is well-accepted that the rate of drug release can be modulated by adding additional ligands to the gel networks like synthetic molecules50 and biopolymers51,52 as crosslinking agents. This convenient strategy has helped to improve composite formulations and prolong the release of both small therapeutic drugs and macromolecules.53 This encouraged us to incorporate in our hydrogel formulation growing concentrations of κ-carrageenan (i.e., 0.5, 0.8 and 1%; hydrogel-2, hydrogel-3 and hydrogel-4, respectively)54 that would act as a physical crosslinking agent. The total cumulative release profiles of niosomal FITC–ODN complexes from these hydrogels crosslinked with κ-carrageenan are shown in Fig. 7. In general, the incorporation of the biopolymer within the supramolecular gel network made of Fmoc–Phe–OH prolonged considerably the FITC–ODN releasing time compared to that observed for hydrogel-1. Specifically, ca. 80–90% of FITC–ODN was available in the receptor phase after 5 h of incubation of hydrogels-(2–4). This result suggests a fall in hydrogel erosion processes induced by the presence of the biopolymer.55 Furthermore, it is worth mentioning that the presence of κ-carrageenan in the formulation was able to reduce the initial burst release during the first few minutes of the experiment (19% vs. 8% for hydrogel-1 and hydrogels-(2–4), respectively) (Fig. 6A and 7). This was probably due to the increase of the entanglement density of the non-covalently crosslinked hydrogels.
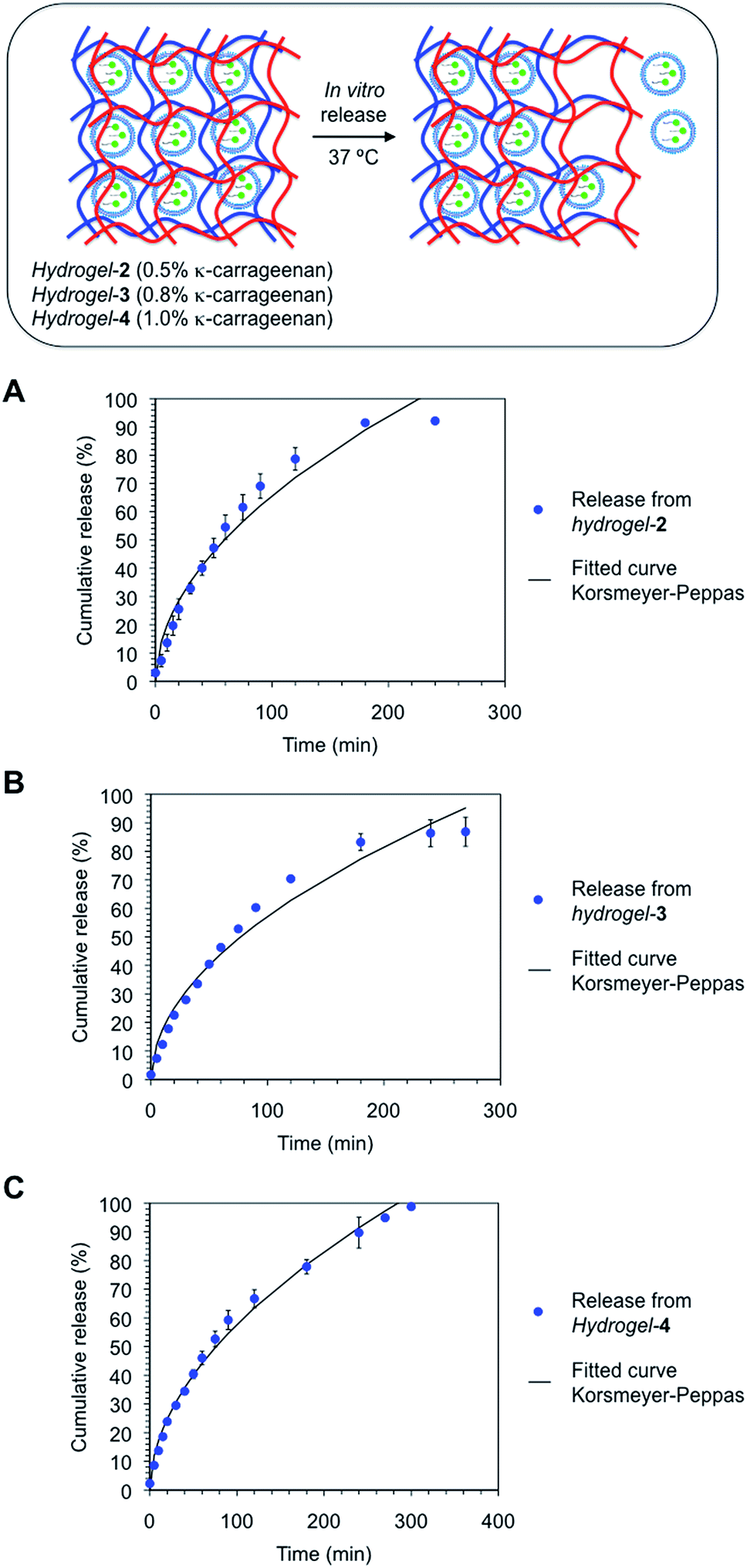 | ||
| Fig. 7 Cumulative release profiles and corresponding fitted curves (n = 2; according to Korsmeyer–Peppas equation [eqn (4)]) of the niosomal formulation containing FITC–ODN from (A) hydrogel-2, (B) hydrogel-3 and (C) hydrogel-4 in PBS solutions at 37 °C. Hydrogels were crosslinked with growing concentrations of κ-carrageenan (0.5, 0.8 and 1% w/v, respectively). Model fitting parameters and log–log plots are given in the ESI (Table S3 and Fig. S8†). The combination of the three release curves is displayed in the ESI (Fig. S6†). | ||
Although the obtained niosomal FITC–ODN release profiles from physically crosslinked hydrogels-(2–4) were similar, the faster release was observed for hydrogel-4 having the highest concentration of κ-carrageenan (1% w/v). This could be explained by the increase of the hydrophilic character of the hydrogel-4 network in which additional biopolymer may contribute to a better dissolution of the swollen supramolecular matrix and thus promoting hydrogel erosion processes and thereby accelerating the drug release.
The analysis of the niosomal release was also studied by native polyacrylamide gel electrophoresis (PAGE) in order to clarify whether the presence of the cationic niosomes loaded in the supramolecular hydrogel could delay and control the oligonucleotide release from the supramolecular hydrogel.
The in vitro control release was carried out as described above and the collected aliquots in PBS were analyzed by PAGE (Fig. S7†). As a control, the same release experiment was also studied in the presence of 100 mM Triton-X100 (Fig. S7B†). As displayed in Fig. S7A,† the oligonucleotide is partially released from the hydrogel at different times which suggests the presence of cationic niosomes might control the FITC–ODN liberation through the hydrogel. To corroborate these findings, Triton-X100 solution was added at the beginning of the experiment in order to solubilize the niosomal component and thus generate the unformulated FITC–ODN release in the receptor phase.56 As expected, native PAGE gels confirmed the use of the surfactant generated a faster oligonucleotide release at the first times of the release experiment (at t = 15 min et seq). This effect was also confirmed from the analyses of the anticipated band intensities. However, it was very difficult to visualize and confirm the presence of intact niosomal particles in the receptor phase by monitoring the release by native PAGE.
2.4 Drug release mathematical models
The release data obtained for hydrogels-(1–4) was fitted according to three theoretical models that describe the diffusion-related drug release from polymeric matrices. The selected models are defined by the Higuchi equation [eqn (3)], Korsmeyer–Peppas equation for burst release [eqn (4)] and Weibull equations for delayed release [eqn (5)].57–59 The first two models are short time approximations and generally limited to the first 60% of the release.60The total FITC–ODN cumulative release from all hydrogel matrices was found to follow a Korsmeyer–Peppas kinetic model (i.e., their regression coefficients were greater (r2 > 0.99) than the rest of theoretical models (ESI, Tables S2–S4†)). According to Peppas' equation, the FITC–ODN release afforded n (diffusion coefficient) values ranging from 0.51 to 0.56. These exponent values are in close agreement with those predicted for non-Fickian diffusions.57 This anomalous transport suggests the presence of specific interactions and/or erosion mechanisms between cationic niosomes and the fibrous gel matrices. Overall, the results of this study suggest a diffusion-controlled release as the main process that govern the liberation of FITC–ODN from the hydrogel matrices.61
2.5 MTT assay
To evaluate the cytocompatibility of Fmoc–Phe hydrogels crosslinked with κ-carrageenan in combination with cationic niosomes and nioplexes, a MTT reductase assay was carried out.62 This assay is based on the tetrazolium salt reduction to formazan crystals by a dehydrogenase that is present in mitochondria. Then, untreated cells (Blank) and HeLa cells only treated with hydrogel-3 (Mock) were selected as our negative controls. Furthermore, cationic niosomes-loaded hydrogel-3 without forming complexes at two concentrations (1.7 μM and 4.2 μM, referred to lipid concentration) besides hydrogel-3 containing cationic nioplexes (120 nM and 300 nM, referred to oligonucleotide concentration) were also evaluated.When cultured for 24 h, hydrogel-3 was totally degraded whereas both cationic niosomes and nioplexes were released from the hydrogel. As displayed in Fig. 8A, the anticipated hydrogel and cationic niosome hybrids did not show any evidence of toxicity obtaining good cellular viabilities (up to ca. 80%). These results also demonstrated normal cell morphology as illustrated in Fig. 8B–D which confirmed the suitability of the hydrogel-3 to be used in cell culture.
 | ||
| Fig. 8 (A) Cell viability studies according to the MTT assay in HeLa cells in the presence of hydrogel-3 (Mock) and hydrogel-3 containing both cationic niosomes (at 1.7 μM and 4.2 μM) and cationic nioplexes (at 120 nM and 300 nM). Nioplexes were obtained from a N/P ratio of 14 according to EMSA assays. Blank corresponds to untreated HeLa cells. Data were means ± SD of six independent experiments and the normalized cellular viability was expressed as a percentage to the untreated control cells. A phosphorothioate oligonucleotide complementary to the mRNA luciferase gene (5′-CGT TTC CTT TGT TCT GGA-3′) was used as an antisense oligonucleotide model. (B) HeLa cell morphology images of untreated cells (blank). (C) HeLa cell morphology images in the presence of cationic niosomes. (D) HeLa cell morphology images in the presence of cationic nioplexes. HeLa cell images in the presence of mock (only hydrogel) are displayed in Fig. S9.† | ||
2.6 Preliminary in vitro drug delivery studies
Firstly, we selected a phosphorothioate oligonucleotide complementary to the mRNA luciferase gene63 to evaluate the gene silencing properties and therefore confirm the niosomal release from hydrogels but unfortunately the presence of Fmoc–Phe inhibited the luciferase gene, as described for other Phe analogues.64For this reason, we moved forward and selected pMax-GFP plasmid as a model to carry out transfection studies. For the preparation of the niosomal formulation, we used the same protocol as described by our research group previously.30 After forming GFP nioplexes at two N/P ratio (4 and 6), particles were entrapped in hydrogel-3 and placed in a transwell insert system. As a control experiment, GFP plasmid, in the absence of cationic niosomes, was encapsulated within the hydrogel. After 24 h of incubation, hydrogel was practically degraded and cellular uptake was analyzed by flow cytometry (Fig. 9). Firstly, we selected a normal gate (R1) for HeLa cells based on their light scatter properties, which distinguished single cells by relative size (Fig. 9A). This allowed us to analyze specific populations and selectively focus on the relationship between fluorescently labeled and unlabeled cell populations.
 | ||
| Fig. 9 Flow cytometry analysis of GFP expression in HeLa cells. (A) Normal cell population (negative control). (B) Control. (C) Flow cytometry analysis for GFP released from hydrogel-3. (D) Flow cytometry analysis for niosomal GFP complexes (N/P ratio of 4). (E) Flow cytometry analysis for niosomal GFP complexes (N/P ratio of 6). A flow cytometry histogram shows a slight shift between cell population containing GFP and untreated cells after 24 h incubation. | ||
As displayed in Fig. 9E, our results suggest that GFP was taken more efficiently by HeLa cells when GFP-forming complexes with the lipid-1 at N/P ratio of 6 were entrapped in hydrogel-3 (17%) according to the gate positive population (Fig. 9E). When we compare this result with the transfection promoted by the unformulated GFP (Fig. 9C), the cell population was transfected roughly 4.6 per cent. Similar results were obtained in the case of GFP-forming complexes at N/P ratio of 4 (3.6%) (Fig. 9D). This difference can be also observed in the flow cytometry histogram, which shows a slight difference shift between fluorescent labelled and unlabelled cell populations.
This result suggests that part of the nioplexes integrity were unaltered when released from the hydrogel and continued to be active during the transfection process took place. These preliminary observations look promising and further studies involving hydrogelator concentration, optimized formulations and use of alternative crosslinking agents are currently investigated.
3. Experimental section
3.1 Materials
All reagents employed in this work were used as received without further purification. Polysorbate-80 (Tween-80), Fmoc-L-phenylalanine (Fmoc–Phe–OH), κ-carrageenan and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT reagent) were purchased from Sigma-Aldrich. A fluorescently labelled oligodeoxynucleotide sequence (5′-FITC-CGG AGG TAC ATT CGA CTT GA-3′; FITC–ODN) was synthesized in house in a DNA/RNA synthesizer ABI 3400 and purified by semi-preparative HPLC according to DMT on-based protocols. FITC (fluorescein isothiocyanate) was automatically coupled at the 5′-termini via phosphoramidite chemistry. Standard phosphoramidites and ancillary reagents used for the synthesis of FITC–ODN were purchased from Link Technologies. Phosphorothioate oligonucleotide of sequence 5′-CGT TTC CTT TGT TCT GGA-3′ was purchased from Proligo (Sigma-Aldrich). PMax-GFP was purchased from Lonza. The amino lipid 1 was synthesized according to the literature (ESI, Fig. S1†).30,31 UV-vis and fluorescence measurements were recorded on a Varian Cary BIO 50 UV/VIS/NIR Spectrometer and a Fluoromax-4 spectrofluorometer, respectively. Flow cytometer analyses were carried out in a Guava® easyCyte 8HT instrument (Millipore).3.2 Methods
000 dynes cm−2 at 25 °C were used for the measurements. After obtaining the supramolecular hydrogels in the absence and in the presence of nioplexes (total gel volume = 2 mL), three different experiments were carried: (1) Dynamic Frequency Sweep (DFS): variation of G′ and G′′ with frequency (from 0.1 to 10 Hz at 0.1% strain); (2) Dynamic Strain Sweep (DSS): variation of G′ and G′′ with strain (from 0.01 to 100%) and (3) Dynamic Time Sweep (DTS): variation of G′ and G′′ with time maintaining both the strain and frequency values constant within the linear viscoelastic regime (strain = 0.1% strain; frequency = 1 Hz).
3.3 General procedures
Hydrogels-(2–4) were obtained as follows: Fmoc–Phe–OH (10 mg; 0.026 mmol) and the corresponding amount of κ-carregeenan (0.5, 0.8 or 1% w/v, respectively) were placed into a screw-capped glass vial (4 cm length × 1 cm diameter) and gently dissolved in 100 mM PBS (3 mL, pH 7.4) at 70 °C. When the three solutions reached 45 °C, the mixture of hydrogelator and κ-carregeenan (500 μL) together with cationic nioplexes:FITC–ODN nioplexes (0.08–0.1 μM) were carefully mixed for 5 min. Finally, the three solutions were cooled down to room temperature affording the supramolecular hybrid hydrogels containing (FITC–ODN)-loaded cationic nioplexes (hydrogel-2, hydrogel-3 and hydrogel-4 for 0.5, 0.8 or 1% w/v, respectively).
The encapsulation efficiency (EE) of the supramolecular hybrid materials was calculated analyzing the initial FITC–ODN amount (starting material) and the remaining mass of FITC–ODN in the receptor phase (at t = 0) [eqn (1)].
 | (1) |
sin(θ/2) is the modulus of the scattering wave vector, θ the scattering angle and λ the wavelength. SAXS patterns were analyzed in terms of a global model using the MCG model of Pabst and coworkers.65 The analysis technique models the full q-range in the SAXS regime, including Bragg peaks and diffuse scattering. The bilayer electronic density profile was calculated as the sum of three Gaussian, one corresponding to the methyl contributions and the two others corresponding to the polar heads, as a difference with respect to the model of Pabst and co-workers.65,66 We also incorporated a constant term through the hydrophobic part of the bilayer to give account of the methylene contribution.67 The interbilayer ordering was taken into account considering the modified Caillé model as in the work of Pabst co-workers.
The volume of the lipid has been estimated to be 0.911 nm3 by using 0.812 nm3 for the hydrocarbon chains and 0.099 nm3 both obtained from partial group volumes: +0.009 per –O– (calculated from the density of 1,1-diethoxy butane); 0.0027 per NH2 and 0 for trisubstituted C.68 To properly scale the electronic density, we have considered the volume as well as the total number of electrons. From this calculation also the number of water molecules per polar head was obtained.
The Fourier reconstruction was performed using the same procedure as J. Pereira-Lachataignerais and co-workers69 and using (- nil + -) for the phasing of the first 4 reflections. This phasing gives a sensible result and coincides with the results obtained by Pereira-Lachataignerais and co-workers.
In vitro control release experiment was also studied by native polyacrylamide electrophoresis (PAGE) gel (20%) (for further details, see ESI†). Cationic nioplexes (1 μM FITC–ODN) were embedded and aliquots (50 μL) were taken at different times. Aliquots were loaded in the presence of 50% glycerol (2 μL) in 1× TBE and gels were run at 150 V and at 25 °C for six h. PAGE gel was stained with SYBR-Green (20 μL) and gel was stirred for 15 minutes. Images were analyzed using ImageJ software (US National Institute of Health)
 | (2) |
3.4 Mathematical models
Release curves were fitted according to Higuchi [eqn (3)], Korsmeyer–Peppas [eqn (4)] and Weibull [eqn (5)] empirical equations. In Higuchi model, the fraction of drug released is proportional to the square root of time; where Mt and M∞ are the cumulative and the maximal amounts of drug released at time t, respectively. k is a constant related to the formulation. In the case of Korsmeyer–Peppas equation, k is a rate constant and n is the release exponent that characterizes the release mechanism (e.g., n = 0.5 for Fickian diffusions and [1/2 < n < 1]) for non-Fickian diffusions. For the Weibull model, b parameter describes the diffusional mechanism of the drug release. When b ≤ 0.75 the Fickian diffusion is the main mechanism while for b values 0.75 < b < 1 diffusion mechanism together with a contribution of another complex release processes can govern the liberation of the molecule. | (3) |
 | (4) |
 | (5) |
3.5 MTT assay
HeLa cells (6000 cells per well) were cultured in a Transwell HTS 96-well plate at 37 °C with a humidified incubator, 5% CO2 overnight to achieve 60% confluence in DMEM supplemented with 10% FBS. Cells only treated with DMEM served as a negative control group (Blank). Hydrogel-3 was previously heated up at 80 °C and filtered through 0.2 μm membrane (15 μL of hydrogel-3; Mock), hydrogel-3 containing only cationic niosomes at two cationic lipid concentrations (1.7 μM and 4.2 μM) and cationic nioplexes-loaded hydrogel-3 at two oligonucleotide concentration (120 nM and 300 nM) were placed on top of the transwell filters and were exposed to HeLa cells for 24 h (total volume = 200 μL). Cells only treated with DMEM served as a negative control group. After removing the supernatant and washed with PBS (200 μL), HeLa cells were incubated at 37 °C with fresh medium (200 μL) for 12 h. Then, 25 μL of MTT solution (5 g L−1 in PBS) were added and HeLa cells were incubated for another 2 h at 37 °C. The resultant formazan crystals were dissolved in DMSO (200 μL) and the absorbance was measured at 560 nm. All experiments were carried out in sextuplicate and normalized cellular viability was expressed as a percentage of the untreated control cells.3.6 In vitro delivery studies and flow cytometry analysis
HeLa cells (105 cells per well) were seeded in a 24 well-plate in a humidified incubator, 5% CO2 in DMEM supplemented with 10% FBS (1 mL) to achieve a 60% confluence. 24 h after seeding, fresh medium without antibiotic (600 μL) was replaced. Hydrogelator and κ-carrageenan at specific concentrations were dissolved in PBS as described previously and filtered through 0.2 μm membrane. GFP plasmid (negative control) and cationic nioplexes (at N/P ratio of 4 and 6) containing GFP were entrapped within the preformed hydrogel (50 μL) the day before. A transwell insert system was introduced and the three hydrogels were placed in. After 24 hours of incubation, DMEM was discharged and cells were washed twice with PBS (500 μL). Cells were collected by trypsinization, centrifugated and resuspended in PBS (300 μL). Finally, transfection efficiencies of the three supramolecular hydrogels was analyzed by flow cytometry.4. Conclusions
In conclusion, this work demonstrates the possibility of using supramolecular hydrogels as versatile carriers for oligonucleotide-loaded nioplexes. As a proof-of-concept, a fluorescently labelled oligodeoxynucleotide (FITC–ODN) was incorporated into cationic niosomes based on an amphiphilic lipid (i.e., lipid-1) and subsequently embedded into biocompatible 3D supramolecular hydrogels based on Fmoc–Phe–OH (pH 7.4). In some cases, small amounts of κ-carrageenan were also added into the gel formulations in order to investigate the crosslinking effect on the drug diffusion through the matrix. Small-angle X-ray scattering (SAXS) measurements showed the formation of lamellar structures for the glycerol-based lipid-1, which remained practically constant in a temperature range between 25 °C and 45 °C. Furthermore, the strong correlation and low flexibility of the bilayers deduced from the SAXS curves supported the stability of the nioplexes under the conditions used in this work. It is worth mentioning that a complete fusion of the lipid-1 was observed in the SAXS pattern when the temperature was increased at 55 °C. Interestingly, this property was previously not detected for the serinol-based lipid analogue.57The results of this investigation showed that Fmoc–Phe–OH–based supramolecular hydrogels (without κ-carrageenan additive) liberated almost the total amount of FITC–ODN in ca. 1.5 h. Nevertheless, physically crosslinked hydrogels with κ-carrageenan were able to prolong the release time of the niosomal oligonucleotide up to 5 h. This increase was presumably due to a reduction of both erosion processes and burst release effects by the presence of the hydrophilic biopolymer. In all cases, the niosomal oligonucleotide release was well fitted by using the Korsmeyer–Peppas kinetics model. The liberation of FITC–ODN was governed by non-Fickian diffusion, which suggested specific interactions between the cationic nioplexes and the gel networks. Moreover, these hybrid materials were found to be non-toxic in cell culture (i.e., HeLa cells). To demonstrate whether nioplexes were still active when released from hydrogels, niosomal formulations containing GFP plasmid at their optimal N/P ratio were loaded within hydrogels and transfection efficiencies were evaluated by flow cytometry. These preliminary results showed better efficiencies for niosomal-loaded hydrogels than unformulated GFP-hydrogel materials, which indicated the presence of intact particles upon the release. We are currently investigating alternative approaches to improve this transfection efficiency by adding new crosslinking agents and the use of other peptide-based supramolecular gelators in order to obtain more robust and efficient vehicles for biomolecules.
Acknowledgements
This work was supported by the Spanish Ministry of Education (Grants CTQ2013-41514-P, CTQ2014-52588 R, RTC-2014-2038-1), the University of Regensburg, the DFG (DI 1748/3-1), the Generalitat de Catalunya (2014/SGR/624) and the Instituto de Salud Carlos III (CB06_01_0019). S. G. thanks the Spanish Ministry of Education, Culture and Sports for a “Jose Castillejo” grant within the researcher mobility program (Programa Estatal de Promoción del Talento y su Empleabilidad en I+D+i, Subprograma Estatal de Movilidad, del Plan Estatal de Investigación Científica y Técnica y de Innovación 2013–2016). D. D. D. thanks the Deutsche Forschungsgemeinschaft (DFG) for the Heisenberg Professorship Award. We thank the Göpferich research group for assistance and granting us access to the rheometer. We would like to thank Dr Gemma Fabriàs and Mireia Casasampere for assistance and access to the cytometer and Dr Cristina Garrido for her help in the cytometer data processing. The authors also thank Jaume Caelles, at the SAXS-WAXS service at IQAC, for X-ray measurements.References
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- E. Bakaic, N. M. B. Smeets and T. Hoare, RSC Adv., 2015, 5, 35469–35486 RSC.
- H. Kamata, X. Li, U.-I. Chung and T. Sakai, Adv. Healthcare Mater., 2015, 4, 2360–2374 CrossRef CAS PubMed.
- H. K. Lau and K. L. Kiick, Biomacromolecules, 2014, 16, 28–42 CrossRef PubMed.
- S. Van Vlierberghe, P. Dubruel and E. Schacht, Biomacromolecules, 2011, 12, 1387–1408 CrossRef CAS PubMed.
- J. M. Knipe and N. A. Peppas, Regenerative Biomaterials, 2014, 1, 57–65 CrossRef PubMed.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- C. Ma, Y. Shi, D. A. Pena, L. Peng and G. Yu, Angew. Chem., 2015, 127, 7484–7488 CrossRef.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- S.-k. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38–50 RSC.
- Polymer Gels and Networks, ed. Y. Osada and A. R. Khokhlov, Marcel Dekker, New York, 2002 Search PubMed.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- J. Yu, W. Ha, J. Chen and Y. Shi, RSC Adv., 2014, 4, 58982–58989 RSC.
- F. Li, J. He, M. Zhang, K. C. Tam and P. Ni, RSC Adv., 2015, 5, 54658–54666 RSC.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- J. Li, Y. Gao, Y. Kuang, J. Shi, X. Du, J. Zhou, H. Wang, Z. Yang and B. Xu, J. Am. Chem. Soc., 2013, 135, 9907–9914 CrossRef CAS PubMed.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- T. Luo and K. L. Kiick, Eur. Polym. J., 2013, 49, 2998–3009 CrossRef CAS PubMed.
- L. Chen, K. Morris, A. Layborn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015–2022 CrossRef CAS PubMed.
- Y. Zhang, Z. Yang, F. Yuan, H. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028–15039 CrossRef CAS PubMed.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- V. Jayawarna, M. Ali, T. A. Jowit, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- S. Roy and A. Banerjee, Soft Matter, 2011, 7, 5300–5308 RSC.
- B. J. Boyd, Expert Opin. Drug Delivery, 2008, 5, 69–85 CrossRef CAS PubMed.
- I. F. Uchegbu and A. T. Florence, Adv. Colloid Interface Sci., 1995, 58, 1–55 CrossRef CAS.
- H. Abdelkader, A. W. G. Alani and R. G. Alany, Drug Delivery, 2014, 21, 87–100 CrossRef CAS PubMed.
- G. P. Ochoa, J. Z. Sesma, M. A. Díez, A. Díaz-Tahoces, M. Avilés-Trigueros, S. Grijalvo, R. Eritja, E. Fernández and J. L. Pedraz, Pharm. Res., 2014, 31, 1665–1675 CrossRef CAS PubMed.
- S. Grijalvo, A. Alagia, G. Puras, J. Zárate, J. L. Pedraz and R. Eritja, Colloids Surf., B, 2014, 119, 30–37 CrossRef CAS PubMed.
- T. Lian and R. J. Ho, J. Pharm. Sci., 2001, 90, 667–680 CrossRef CAS PubMed.
- J. Parmentier, M. M. M. Becker, U. Heintz and G. Fricker, Int. J. Pharm., 2011, 405, 210–217 CrossRef CAS PubMed.
- M. T. Popescu, S. Mourtas, G. Pampalakis, S. G. Antimisiaris and C. Tsitsilianis, Biomacromolecules, 2011, 12, 3023–3030 CrossRef CAS PubMed.
- S. Grijalvo, J. Mayr, R. Eritja and D. Díaz Díaz, Biomater. Sci., 2016, 4, 555–574 RSC.
- M. Glavas-Dodov, K. Goracinova, K. Mladenovska and E. Fredro-Kumbaradzi, Int. J. Pharm., 2002, 1–2, 381–384 CrossRef.
- W. W. Gao, D. Vecchio, J. M. Li, J. Y. Zhu, Q. Z. Zhang, V. Fu, J. Y. Li, S. Thanphiwatana, D. N. Lu and L. F. Zhang, ACS Nano, 2014, 8, 2900–2907 CrossRef CAS PubMed.
- Y. Liang and K. L. Kiick, Biomacromolecules, 2016, 17, 601–614 CrossRef CAS PubMed.
- S. Mourtas, S. Fotopoulou, S. Duraj, V. Sfika, C. Tsakiroglou and S. G. Antimisiaris, Colloids Surf., B, 2007, 55, 212–221 CrossRef CAS PubMed.
- N. El Kechai, A. Bochot, N. Huang, Y. Nguyen, E. Ferrary and F. Agnely, Int. J. Pharm., 2015, 487, 187–196 CrossRef CAS PubMed.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295–6304 CrossRef CAS PubMed.
- Y. Mao, X. Li, G. Chen and S. Wang, J. Pharm. Sci., 2016, 105, 194–204 CrossRef CAS PubMed.
- N. C. Wickremasinghe, V. A. Kumar and J. D. Hartgerink, Biomacromolecules, 2014, 15, 3587–3595 CrossRef CAS PubMed.
- N. C. Wickremasinghe, V. A. Kumar, S. Shi and J. D. Hartgerink, ACS Biomater. Sci. Eng., 2015, 1, 845–854 CrossRef CAS PubMed.
- M. A. Khalily, M. Goktas and M. O. Guler, Org. Biomol. Chem., 2015, 13, 1983–1987 CAS.
- S. Mourtas, M. Haikou, M. Theodoropoulou, C. Tsakiroglou and S. G. Antimisiaris, J. Colloid Interface Sci., 2008, 317, 611–619 CrossRef CAS PubMed.
- V. Baumann and J. Winkler, Future Med. Chem., 2014, 6, 1967–1984 CrossRef CAS PubMed.
- M. Wood, H. Yin and G. McClorey, PLoS Genet., 2007, 3, 845–854 CAS.
- S. Pérez-Rentero, S. Grijalvo, G. Peñuelas, C. Fàbrega and R. Eritja, Molecules, 2014, 19, 10495–10523 CrossRef PubMed.
- M. Delgado, C. Spanka, L. D. Kerwin, P. Wentworth and K. D. A. Janda, Biomacromolecules, 2002, 3, 262–271 CrossRef CAS PubMed.
- D. Das and S. Pal, RSC Adv., 2015, 5, 25014–25050 RSC.
- J. Wang, Z. Wang, J. Gao, L. Wang, Z. Yang, D. Kong and Z. Yang, J. Mater. Chem., 2009, 19, 7892–7896 RSC.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- Y. Liu, Y. Zhu, G. Wei and W. Lu, Eur. J. Pharm. Sci., 2009, 37, 306–312 CrossRef CAS PubMed.
- E. Ruel-Gariépy, G. Leclair, P. Hildgen, A. Gupta and J. C. Leroux, J. Controlled Release, 2002, 82, 373–383 CrossRef.
- M. Sila, S. Au and N. Weiner, Biochim. Biophys. Acta, Biomembr., 1986, 2, 165–170 CrossRef.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- N. A. Peppas and J. J. Sahlin, Int. J. Pharm., 1989, 57, 169–172 CrossRef CAS.
- P. Costa and J. M. Sousa Lobo, Eur. J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS PubMed.
- A. Karewicz, K. Zasada, K. Szczubialka, S. Zapotoczny, R. Lach and M. Nowakowska, Int. J. Pharm., 2010, 385, 163–169 CrossRef CAS PubMed.
- A. K. Pannier and L. D. Shea, Mol. Ther., 2004, 10, 19–26 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- H.-Y. Zhang, J. Mao, D. Zhou, Y. Xu, H. Thonberg, Z. Liang and C. Wahlestedt, Nucleic Acids Res., 2003, 31, e72 CrossRef PubMed.
- R. Lee and W. D. McElroy, Biochemistry, 1969, 8, 130–136 CrossRef CAS PubMed.
- G. Pabst, M. Rappolt, H. Amenitsch and P. Laggner, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 4000–4009 CrossRef CAS.
- E. Ojeda, G. Puras, M. Agirre, J. Zárate, S. Grijalvo, R. Pons and R. Eritja, Org. Biomol. Chem., 2015, 13, 1068–1081 CAS.
- N. Kucerka, J. F. Nagle, J. N. Sachs, S. E. Feller, J. Pencer, A. Jackson and J. Katsaras, Biophys. J., 2008, 95, 2356–2367 CrossRef CAS PubMed.
- A. Pinazo, L. Pérez, M. Lozano, M. Angelet, M. R. Infante, M. P. Vinardell and R. Pons, J. Phys. Chem. B, 2008, 112, 8578–8585 CrossRef CAS PubMed.
- J. Pereira-Lachataignerais, R. Pons, H. Amenitsch, M. Rappolt, B. Sartori and O. López, Langmuir, 2006, 22, 5256–5260 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Digital pictures, NMR spectroscopy, rheological measurements, fluorescence calibration curve, control experiments, fitting models, native PAGE gels and cell morphology images. See DOI: 10.1039/c6ra01005a |
| This journal is © The Royal Society of Chemistry 2016 |