
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microwave treatment: a facile method for the solid state modification of potassium-promoted iron on silica Fischer–Tropsch catalysts†
Mbongiseni W. Dlaminia,
Neil J. Covillea and
Michael S. Scurrell*ab
aDST-NRF Centre of Excellence in Catalysis (c*change) and Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg, South Africa. E-mail: scurrms@unisa.ac.za
bDepartment of Civil and Chemical Engineering, University of South Africa, Florida, Johannesburg, South Africa
First published on 18th February 2016
Abstract
Potassium-promoted (0–1.5 wt%) iron–silica catalysts for Fischer–Tropsch synthesis (FTS) have been modified using microwave radiation. Radiation produced few or no modifications in the bulk properties, but surface and catalytic behaviour were markedly changed in K promoted 10 wt% of Fe/SiO2 (10Fe/SiO2) catalysts. The effect of potassium on CO adsorption was relatively insignificant in untreated catalysts, but was large in microwave-modified catalysts. Radiation induced an increase in CH4 formation in CO + H2 temperature programmed surface reactions. Microwave treatment promoted CH4 formation from graphitic carbon in these catalysts, while decreasing CH4 formation from α- and β-carbon species, and overall favoured strong CO adsorption onto the catalyst surface. Microwave effects were catalyst particle size and treatment duration-dependent. At low alkali concentration, microwaved samples showed improved ethene selectivities, higher alpha values and lower methane and light alkene selectivities. When 0.7 wt% K was added to the 10Fe/SiO2 catalyst, the α value increased from 0.59 to 0.66 after treatment of the sample with microwave radiation in the solid state.
1. Introduction
The Fischer–Tropsch (FT) synthesis is a key technology for producing clean fuels and chemicals using syngas derived from coal, shale gas, natural gas or biomass and is proving to be an effective substitute for liquid fossil fuel.1–3 As a consequence of the current unstable crude oil prices and its financial significance, the FT process has attracted renewed interest from researchers both in academia and industry to improve the overall performance of FT catalysts. Of all the transition metals which are active in FT synthesis, iron-based catalysts are preferred for synthesis gas compositions with a low H2/CO ratio due to their ability to produce additional H2 via the water-gas shift (WGS) reaction. Furthermore, iron-based catalysts are cheap and readily available and are highly selective for the production of short-chain olefins and oxygenates, which are key industrial chemical building blocks.4 Iron FT catalysts are typically promoted with potassium to enhance their selectivity towards long-chain hydrocarbons.Recently, the modification of unsupported iron-based catalysts by microwave radiation was reported.5 The essential findings were that microwave treatment enhanced the surface properties of such catalysts, as was clearly revealed by studies using a temperature programmed surface reaction (TPSR) between adsorbed CO and hydrogen. The microwave-induced effects are considered to be the result of localized temperature gradients set up almost instantly when the solids are exposed to radiation. Some preliminary studies on supported potassium-promoted iron have indicated similar beneficial effects.6 The essential changes seen are an increase in specific activity, a decrease in methane selectivity, an increase in chain-growth probability, a higher olefinic content of the product hydrocarbons and an increase in the water gas shift activity, as evidenced by a higher selectivity to carbon dioxide. Most of these changes would normally be considered to be desirable in the context of producing synthetic liquid hydrocarbon fuels from sources with relatively low hydrogen content such as coal and biomass. Indications are that the surface elemental ratios of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :K:Si are altered by exposure to microwave radiation.6 Since potassium loading affects various aspects of iron catalyzed Fischer–Tropsch synthesis, it is likely that microwave induced changes in the surface potassium are responsible for the observed effects, though the exact chemistry is yet to be revealed. In addition, we have noted that iron oxides supported on silica treated with microwave radiation perform better than untreated samples, in part because any (relatively inactive) iron-silicate phases formed in the catalysts are effectively destroyed by microwave treatment.6 This aspect merits further study in its own right, but is consistent with experience gained by using microwave radiation to enhance the metallurgical extraction of various mineral ores which are otherwise difficult to extract.7,8
:K:Si are altered by exposure to microwave radiation.6 Since potassium loading affects various aspects of iron catalyzed Fischer–Tropsch synthesis, it is likely that microwave induced changes in the surface potassium are responsible for the observed effects, though the exact chemistry is yet to be revealed. In addition, we have noted that iron oxides supported on silica treated with microwave radiation perform better than untreated samples, in part because any (relatively inactive) iron-silicate phases formed in the catalysts are effectively destroyed by microwave treatment.6 This aspect merits further study in its own right, but is consistent with experience gained by using microwave radiation to enhance the metallurgical extraction of various mineral ores which are otherwise difficult to extract.7,8
Microwave applications in chemistry usually focus on synthesis in the liquid-phase though some studies of radiation effects seen during heterogeneous catalysis are also available.9–11 Further, as mentioned above we have seen effects for both supported and unsupported iron-based FT catalysts, but the magnitude of the effect seems to be higher for supported materials, for reasons which are not yet clear.5,6 In this work we have therefore investigated supported FT catalysts based on silica and have studied the behaviour of observed microwave effects as a function of potassium loading up to 1.5 wt% with a constant Fe loading of 10 wt%. We have also included actual catalytic studies of the microwave induced synthesis on these Fischer–Tropsch catalysts. Catalysts prepared by incipient wetness impregnation or deposition–precipitation have both been examined because there are significant differences between the two and they have been found to respond differently to microwave treatment. In summary, our catalyst preparation method is conventional in nature but we have used an unconventional microwave radiation treatment to synthesize the catalysts before catalyst reduction and use.
2. Experimental
2.1 Catalyst preparation
For incipient wetness impregnation (IWI), a ferric nitrate solution obtained by dissolving Fe(NO3)3·9H2O in deionized water was added dropwise to the silica support. The slurry was then dried at 120 °C for 12 h to remove moisture. Calcination was performed at 350 °C for 6.5 h using a heating rate of 10 °C min−1. In this study, the loading of iron was maintained at 10 wt%. The 10Fe/SiO2 catalyst base powder was then impregnated with appropriate amounts of K2CO3 solutions to produce the desired catalyst compositions. For this study, potassium loadings of 0.2, 0.5, 0.7, 1.0 and 1.5 wt% were desired. The catalyst was then dried at 120 °C for 12 h followed by calcination at 350 °C for 6.5 h. For brevity, these catalysts are referred to as xK/10Fe/SiO2, where x is the nominal potassium loading (wt%). A precipitation (DP) procedure using ferric nitrate, potassium carbonate and urea was also used to make the catalyst. In situ urea hydrolysis was allowed to proceed for 3 h at 100 °C. The slurry was then dried at 90 °C under vacuum for 40 min followed by another drying period of 12 h at 120 °C. Calcination of the DP-synthesized samples was also done at 350 °C for 6.5 h. A 1.0 wt% K loading was maintained for the DP-prepared catalysts, while the iron loading was kept at 10 wt%. These catalysts are denoted as 1.0K/10Fe/SiO2-DP.2.2 Catalyst characterization, MW treatment and testing
The samples were characterized using various techniques as described in our previous publication.5 Prior to microwave treatment, about 0.5 g catalyst mass was evenly distributed as a thin layer in a microwave-transparent Petri dish. A Defy (900 W, 2.45 GHz) domestic microwave was used to pre-treat the calcined catalysts. All microwave pre-treatment experiments were done with catalysts in the solid state. Microwave pre-treatment was done for 10 s at a power level of 450 W. The effect of the microwave irradiation time was also studied. The pre-treatment time was increased from 10 s to 20, 30, 40, 60, 300 and 600 s, while the power level was maintained at 450 W for all experiments.Thin samples for TEM analysis were prepared by crushing the K/Fe and K/Fe/SiO2 catalysts with a mortar and pestle followed by dispersion in methanol using an ultra-sound bath. An appropriate amount of the sample was then placed onto a carbon coated copper grid, dried and then introduced to the microscope. The samples were studied using a TECNAI G2 SPIRIT transmission electron microscope operating at an accelerating voltage of 120 keV.
Energy dispersive X-ray spectroscopy (EDS) EDS is an X-ray technique that was used to identify the elemental composition of the catalysts. When a conducting sample is bombarded with a beam of electrons, X-rays are emitted. The emitted X-rays are unique for a given element and can therefore be used to identify elements present in a given sample. The EDS system used in this study was integrated to the TECNAI G2 SPIRIT transmission electron microscopy instrument.
All the gases used in this study (UHP grade) were supplied by AFROX (African Oxygen) Ltd. A stainless steel fixed-bed micro-reactor was used for the catalytic reactions. All gas lines after the reactor were kept at 150 °C, and a hot trap, also held at 150 °C, was placed immediately after the reactor in order to collect wax. A second trap kept at ambient temperature was used for the collection of the oil and water mixture. A catalyst mass of 0.5 g was loaded to a stainless steel fixed bed reactor, and the catalyst reduction was carried out at 350 °C for 18 h using hydrogen gas and a heating rate of 10 °C min−1. Catalytic reactions were conducted at 275 °C with synthesis gas composition (H2:CO:N2 = 60:30:10) at a flow rate of 20 mL min−1 and the reactor pressure was raised to 1.0 MPa. Analysis of the product spectrum was done online using two GCs equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID), identical to that used before.5 Mass balances were performed on carbon and oxygen and were close to levels of 100 ± 5%.
3. Results and discussion
3.1 Catalyst characterization
The elemental compositions of the samples were determined by XRF and are presented in Table S1.† As inferred from the tabulated data, the calculated and analyzed weight loadings were similar. Table S1† also summarizes the textural properties of the various catalysts prior to and after exposure to microwave radiation (10 s). The BET surface areas of all the silica-supported catalysts are in the range of 220–284 m2 g−1, samples with higher K loadings having lower BET surface areas. A study of Dry and Oosthuizen over a fused iron catalyst gave similar observations which were ascribed to the fact that potassium improves agglomeration of the FeOOH precursor and could further enlarge the crystallite size of α-Fe2O3 on calcination.12 The morphology and particle size of the crystallites was analyzed using TEM and from the images it was noted that the iron crystallites were well dispersed on the silica support (Fig. 1). The average particle size for the non-microwaved iron particles was in the range 40–60 nm for the various loadings of potassium. Microwave pre-treatment did not alter the morphology or the particle sizes of the catalysts. | ||
| Fig. 1 TEM images of 0.2K/10Fe/SiO2 and 0.7K/10Fe/SiO2. | ||
The calcined catalysts were further characterized using H2-TPR prior to microwave pre-treatment (Fig. S1, see also Table S2†). While the reduction of unsupported iron catalysts occurs in two simple steps (α-Fe2O3 to Fe3O4, followed by Fe3O4 to α-Fe), a more complex behaviour is exhibited by silica supported samples. At a low promoter loading (0.2 wt% K) the profile shows four reduction peaks. The small peak at 310 °C could be related to a minor process of reduction of small particles of Fe2O3 occurring on the silica surface i.e. at temperatures <390 °C. This peak increases in intensity and also shifts to higher temperatures as the loading of potassium is increased in the samples. Also, at potassium loadings of 0.7, 1.0 and 1.5 wt% K the peak gradually separated into two distinct peaks. The second reduction peak for the 0.2K/10Fe/SiO2 catalyst occurs at 425 °C and it represents the reduction of bulk Fe2O3 to Fe3O4. Corresponding peaks in the other samples at higher promoter contents have been highlighted using a dotted line (Fig. S1†). Clearly the peak shifts to higher temperatures with an increase in the potassium loading since potassium inhibits the reducibility of iron.13 The reduction of Fe3O4 to Fe occurs at 577 °C for the 0.2K/10Fe/SiO2 sample and is labelled as Peak 3. This peak does not shift on addition of potassium. Peak 4 occurs between 657 and 663 °C for the various samples as tabulated in Table S2.† This peak represents the reduction of iron(II) silicate to α-Fe. This peak is broad and it indicates that large amounts of iron silicates were present in the samples. The large amounts of iron silicates are attributed to a strong interaction between the iron species and silica. A small peak at ∼750 °C is also observed. It could be assigned to the reduction of the K2CO3 phase, in agreement with its growth with the increase of the potassium content and the analogous assignment of this peak by Stobbe et al.14 and Guglielminotti and co-workers.15 Microwave treatment did not introduce any significant changes in the reduction profiles of the silica supported catalysts.
3.2 Temperature programmed surface reactions (TPSR) studies
Fig. S3a† shows TPSR profiles of the non-microwave irradiated catalysts with varying loadings of potassium (0 to 1.5 wt% K) reveal that methane was the main hydrocarbon detected during the experiments. Data for the 0.2K/10Fe/SiO2 sample is shown as an example in Fig. 1a. The CH4 profiles are similar in shape i.e. two sets of peaks appearing at ∼300 and at ∼500 °C are common in the profiles. However, the peaks shift to higher temperatures as the loading of potassium is increased (Fig. S3a†). It is known that K2O on the surface of an iron catalyst increases the Fe–C bond strength for CO bonded to Fe,16–18 leading to a shift in the desorption process to higher temperatures with K loading. Graf and Muhler19 reached the same conclusion on the influence of the potassium promoter after performing CO temperature programmed desorption (TPD) experiments. The peaks recorded in this work are broad and indicate the existence of several overlapping peaks that coexist. The total areas under these peaks were obtained by integration and were used together with methane calibration data to quantify the CH4 evolved during the TPSR experiments. The total amounts of methane evolved from the various catalysts are listed in Table 1 as a function of the potassium content. It can be seen that the CH4 produced from all the samples is in the range 31–32 μmol g−1, indicating that the potassium loading does not have a significant impact on the amount of methane produced from these catalysts. This implies that surface iron crystallites are not affected by the potassium ions. The data of Table 1 also shows the reproducibility of the results obtained from the TPSR technique. It is noted that even though potassium did not affect the total CH4 evolved, it did change the surface carbon species from which CH4 is produced. For example adsorbed atomic carbon (Cα) formation decreased monotonously as the loading of potassium was increased. The rate of reactivity of the C entities obviously has implications for FTS catalysis.
| Catalyst | Peak parametersa | CH4 total area (μmol g−1) | |||
|---|---|---|---|---|---|
| Carbidic α | Amorphous β | Carbide γ | Graphitic δ | ||
| a Peak parameters are presented as: deconvoluted peak area (%)/peak position (°C). | |||||
| 0.2K/10Fe/SiO2 | 18.9/288 | 35.5/348 | 16.7/493 | 28.9/528 | 32 |
| 0.2K/10Fe/SiO2-MW | 14.5/289 | 24.2/344 | 23.9/511 | 37.4/563 | 36 |
| 0.5K/10Fe/SiO2 | 12.7/296 | 35.4/357 | 29.6/468 | 22.3/594 | 32 |
| 0.5K/10Fe/SiO2-MW | 12.7/354 | 13.8/413 | 46.2/509 | 27.3/586 | 40 |
| 0.7K/10Fe/SiO2 | 14.1/314 | 39.4/381 | 35.6/507 | 10.9/601 | 31 |
| 0.7K/10Fe/SiO2-MW | 3.8/288 | 32.1/359 | 31.3/500 | 32.8/559 | 40 |
| 1.0K/10Fe/SiO2 | 9.4/328 | 55.7/386 | 26.5/512 | 8.4/594 | 32 |
| 1.0K/10Fe/SiO2-MW | 1.6/280 | 26.2/368 | 27.2/510 | 45.0/553 | 53 |
| 1.5K/10Fe/SiO2 | 7.0/304 | 32.1/360 | 36.1/439 | 24.8/540 | 31 |
| 1.5K/10Fe/SiO2-MW | 3.6/297 | 16.1/358 | 8.0/426 | 72.3/541 | 51 |
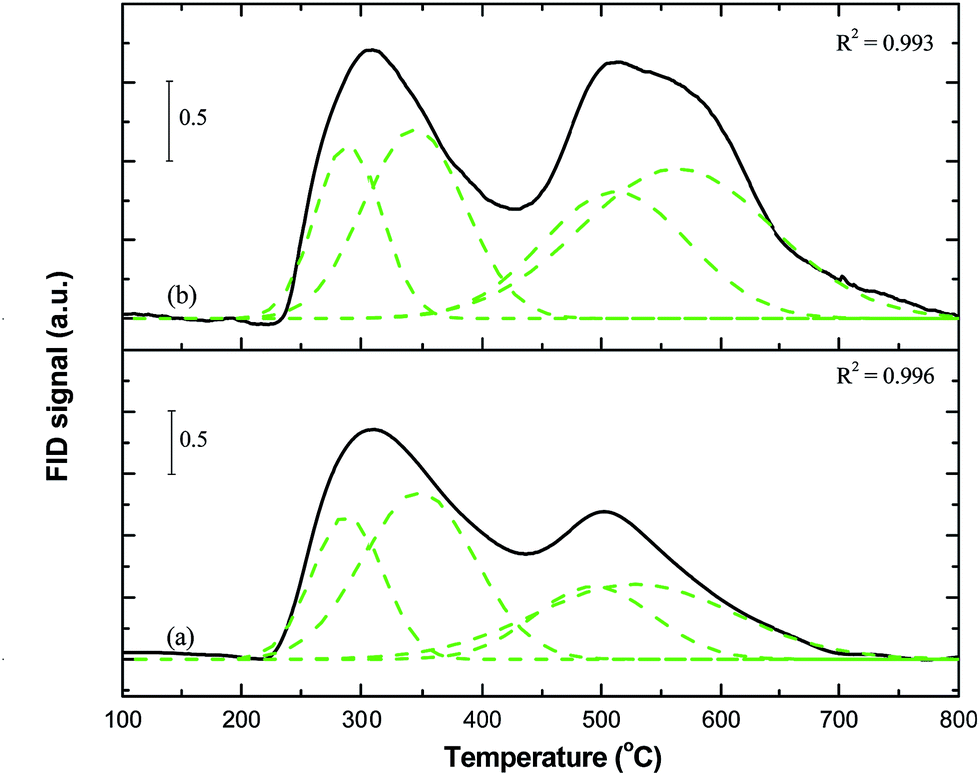 | ||
| Fig. 2 TPSR profiles of 0.2K/10Fe/SiO2 (unmodified) and 0.2K/10Fe/SiO2-MW (modified using microwave radiation) catalysts displaying the evolution of methane as a function of temperature. Data on the individual contributions of the carbon species are tabulated in Table 1. | ||
From the data in Table 1 and Fig. 2 it is evident that microwave pre-treatment induces a small increase in the overall CH4 evolved. For example, the total area for the CH4 peaks in the 0.2K/10Fe/SiO2-MW catalyst corresponds to 36 μmol g−1, whereas 32 μmol g−1 CH4 (i.e. 13% increase) was evolved from the untreated catalyst. An analysis of the contributions from the various carbon species shows that the α- and the β-carbon species appearing at 289 and 344 °C, respectively, decreases after microwave pre-treatment. However, Fig. 2 and Table 1 show that CH4 formation in the microwaved sample is greatly enhanced from the γ- and δ-carbon species. As a result the γ- and the δ-species contribute a combined 61% of the 36 μmol g−1 produced from the 0.2K/10Fe/SiO2-MW catalyst. Upon increasing the loading of potassium from 0.2 to 0.7 wt%, a significant change in the TPSR profile for the microwave modified catalyst was noted. The methanation plots obtained from 0.7K/10Fe/SiO2 and the 0.7K/10Fe/SiO2-MW catalysts are displayed in Fig. S5a and b† respectively and peak parameters for the individual carbon species are listed in Table 1. The microwaved catalyst displays an overall increase in the rate of methanation of 23% relative to the non-microwaved catalyst. This increase in methane production is attributed to the microwave effect as all other parameters were kept constant. Fig. S5b† shows that the change in the profile is due to CH4 being produced from δ-carbon species at 559 °C.
Fig. S6 and S7† compares TPSR profiles for the 1.0K/10Fe/SiO2 and the 1.5K/10Fe/SiO2 catalysts before and after microwave radiation (Table 1). The TPSR profiles for the modified catalysts (1.0K/10Fe/SiO2-MW and 1.5K/10Fe/SiO2-MW) clearly differ from those recorded for the unmodified catalysts. Both these catalysts display a large signal at ca. 540 °C which is due to CH4 being formed from carbide and graphitic carbon. These signals account for a combined 72 and 80% of the total hydrocarbons produced from the 1.0K/10Fe/SiO2-MW and 1.5K/10Fe/SiO2-MW samples, respectively. This is to be compared with the 35 and 61% combined contributions from carbide and graphite-like carbons recorded from the non-microwaved catalysts with the same composition. The total methane evolved when using the 1.0K/10Fe/SiO2-MW catalyst was 53 μmol g−1, which shows a 66% increase in CH4 relative to the 1.0K/10Fe/SiO2 catalyst which produced 32 μmol g−1. An increase of 64% was induced by microwave pre-treatment for the catalysts with 1.5 wt% potassium. Methanation of the α- and β-carbon species was found to decrease after modification of both the 1.0 and 1.5 wt% K samples, hence the decline in their percentage contribution to the total CH4 evolved.
A comparison of the total CH4 content produced in both the microwaved and non-microwaved catalysts during TPSR experiments is summarized in Fig. 3 as a function of the potassium loading. Clearly the increases seen were induced by microwave pre-treatment since all other variables were kept constant. From this plot it can be seen that the microwave effect increases with the loading of potassium and attains an optimum value at 1.0 wt% potassium loading for which case an increase of 66% in the total hydrocarbon content results. At low K loadings, microwave induced effects were seen to promote methanation from mainly γ- and δ-carbon species, while involvement of α- and β-carbon decreased. At high K loading, methanation from the α-, β- and γ-species was seen to decline after microwave pretreatment. In contrast, CH4 produced from graphitic carbon increases upon MW exposure. Unmodified catalysts display the opposite behaviour since methanation is generally favoured from α- and β-carbon species.
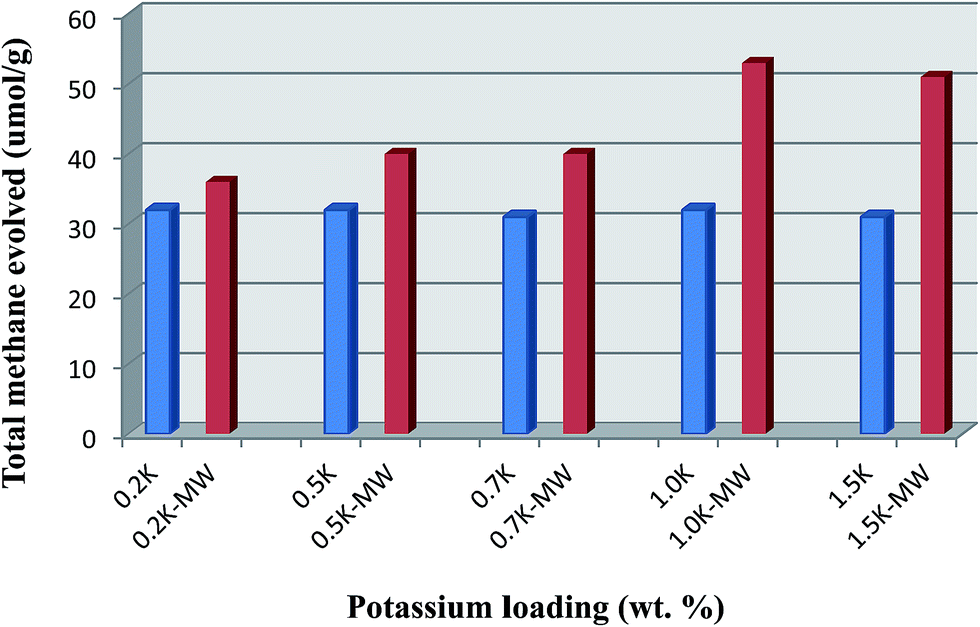 | ||
| Fig. 3 Comparison of methane formed in microwaved and non-microwaved samples as a function of potassium loading. All the catalysts are supported on silica and contain 10 wt% Fe. | ||
The general increase in microwave-induced effects as a function of potassium can be explained by considering the known effects of potassium in Fischer–Tropsch synthesis. With ensembles of iron atoms it is difficult to cleave the C–O bond. The addition of K enhances bond-breaking. The effect of microwave pre-treatment on the catalysts is believed to result in the migration of potassium ions to the surface of the catalyst.6 Thus surface iron atoms in a microwave modified catalyst are surrounded by potassium ions and become “electron rich”. The C–O bond on such iron atoms is easily cleaved and hydrocarbons are more readily produced. It is well known that surface basicity can promote CO adsorption and suppress H2 adsorption.22
3.3 Effect of the MW treatment time
The microwave pre-treatment effect reported thus far has been accomplished by exposing the catalyst to microwave radiation that is at 450 W power level for only 10 s. To investigate the effect of longer duration times, temperature programmed reduction (TPR) studies were performed with the 1.0 wt% K sample since it was shown to be optimal for the microwave effect (Fig. 3). Reduction profiles of iron catalysts that were irradiated for varying periods of time (10–40 s) with microwaves did not change significantly (Fig. S8, Table S3†). As demonstrated in the table, the peaks from the microwaved samples show a marginal shift to lower temperatures and illustrate that the irradiation time does not affect the reduction behaviour of the catalyst. Further, profiles measured after 40 s of microwave pre-treatment were similar to those recorded after 20 s of treatment.TPSR measurements were done to investigate the effect of the MW irradiation duration. It was found that the profiles followed the general pattern observed in Fig. 2 with the display of two broad peaks before deconvolution. Data calculated from the peak areas are tabulated in Table 2. The total methane production increased as the microwave irradiation time was increased from 0 s (i.e. no pre-treatment) to 10 min. To identify the origin of the increases in methane production after microwave pre-treatment, a plot of the CH4 from the two broad peaks is shown in Fig. 4. As seen in preceding sections, the low temperature broad peak accounts mainly for methane produced from α- and β-carbon species, while the high temperature broad peak accounts for methane evolved from γ- and δ-carbon species. As illustrated by this figure, the broad low temperature peak area is fairly constant throughout the irradiation period. It is not affected by the irradiation time. In contrast, the high temperature peaks displays significant increases as the irradiation time is increased, especially between 40 and 120 s of MW irradiation.
| Microwave pre-treatment time | Total CH4 evolved (μmol g−1) | |
|---|---|---|
| IWI | DP | |
| 0 s | 32 | 41 |
| 10 s | 53 | 46 |
| 20 s | 50 | — |
| 30 s | 59 | — |
| 40 s | 57 | — |
| 120 s | 188 | 49 |
| 300 s | 194 | 88 |
| 600 s | 232 | 117 |
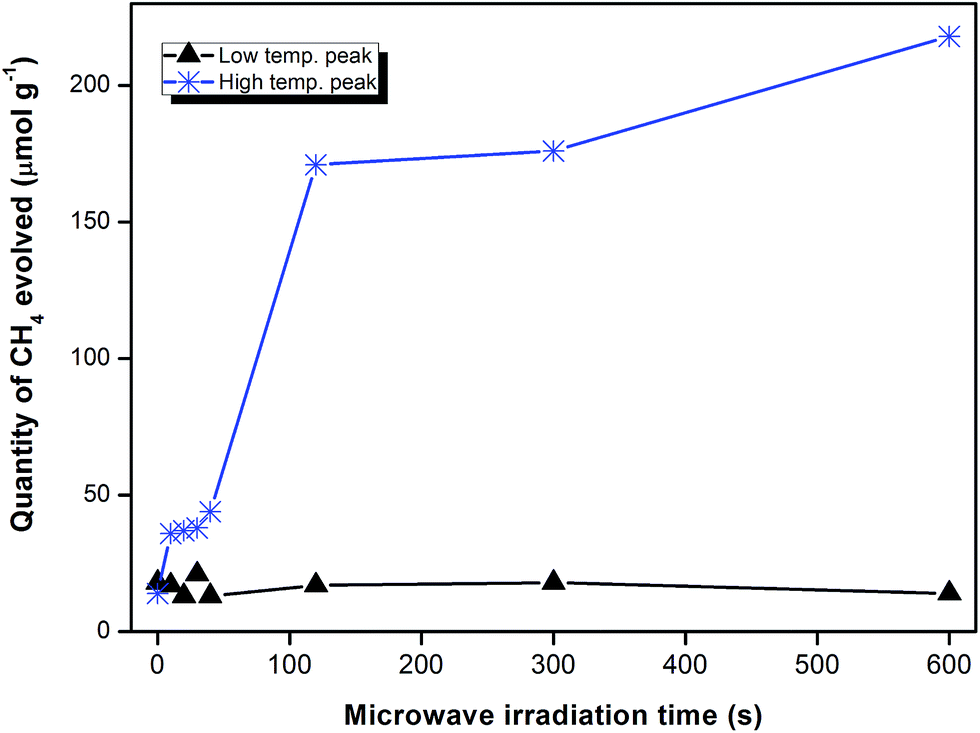 | ||
| Fig. 4 Variation in the methane content produced by the low and high temperature TPSR peaks as a function of the microwave irradiation time. Data shown is for the 1.0K/10Fe/SiO2 catalyst. | ||
The most probable effects of microwave radiation are due to the development of local high temperature spots. The silica support will tend to be microwave transparent but iron oxide, and especially magnetite, is a good microwave absorber. We believe that solid-state modifications, albeit at a local level, may well result from local temperature excursions. We have seen that microwave effects are dependent on the power levels used, the duration of irradiation5 and the shape of the sample.9 We have also seen that Fe:K surface atomic ratios in potassium–iron–silica catalysts can be significantly modified by microwave action6 The magnetite/haematite ratio in the initial sample is likely to determine the overall microwave absorption realized. The water content of the sample may also play a role, but none of these factors have yet been systematically examined in our work. Some, and perhaps all of the effects seen using microwave radiation, might be seen by using conventional heating, though again, this has not been examined in any critical way to date. What we do propose is that microwave irradiation is a simple and convenient method for modifying solids catalysts and that the action is clearly demonstrated in a modification of surface properties, including elemental ratios and chemisorptive properties.6
3.4 The effect of particle size on microwave treatment
The microwave pre-treatment effect on catalysts with different crystallite sizes was examined by preparing catalysts of the same composition using two techniques; deposition–precipitation (DP) and incipient wetness impregnation (IWI). Iron particles in a catalyst prepared using deposition–precipitation generally have much smaller particle sizes than those prepared using the incipient wetness impregnation technique.23,24 Consistent with this finding, our 1.0K/10Fe/SiO2-DP catalysts had an average particle size in the range 10–25 nm, whereas the catalyst prepared by incipient wetness impregnation (1.0K/10Fe/SiO2) had an average particle size in the range 40–60 nm. A plot of the total CH4 evolved for the two types of catalysts is shown in Fig. 5. As expected, the catalyst prepared using the deposition–precipitation technique produced more methane relative to its incipient wetness impregnation counterpart for the unmodified catalysts (i.e. at 0 s treatment) (Table 2). | ||
| Fig. 5 Total CH4 produced in the TPSR by the 1.0K/10Fe/SiO2-DP and the 1.0K/10Fe/SiO2 catalysts that were irradiated for various periods. | ||
This is because catalysts prepared using the DP procedure have small crystallites and are therefore highly active. As the pre-treatment time was increased to 10 s, 2 min, 5 min and then 10 min, the total methane produced is seen to increase for both samples. The DP-prepared catalyst showed lower TPSR activity relative to the catalyst prepared by the IWI method. This shows that microwave pre-treatment is favoured for larger particle sizes than for smaller ones. As seen in the preceding sections, MW pre-treatment also favours CH4 formation from γ- and δ-carbon species in these samples.
3.5 Methanation studies
Methanation studies were performed to study the microwave pre-treatment effect on the CO chemisorption strength at the catalyst surface. Fig. 6 shows the methane profiles resulting from TPSR experiments performed in which a methanator was used for the 0.7K/10Fe/SiO2 and 1.0K/10Fe/SiO2 catalysts before and after microwave treatment. The profiles depict several CH4 peaks as a result of the hydrogenation of pre-adsorbed CO. The low T peaks occur at 116 and 216 °C while a broad peak was observed at T > 500 °C. Both low temperature peaks are attributed to weak CO chemisorption in the molecular state. The peak at 116 °C is due to catalytically active sites that are not influenced by the potassium promoter, while the signal at 211 °C is due to sites that are influenced by K. A similar classification has also been used by Pour et al. when copper was used to promote an Fe catalyst.25,26 CH4 signals produced from strongly chemisorbed CO shifts to higher temperatures when a methanator is used (see peak at T > 510 °C). | ||
| Fig. 6 TPSR profiles recorded from methanation studies using the (a) 0.7K/10Fe/SiO2 and (b) 1.0K/10Fe/SiO2 catalysts. | ||
Analysis of data for the 0.7K/10Fe/SiO2 and 0.7K/10Fe/SiO2-MW catalysts (Fig. 6a) showed that there is a slight increase in the total CH4 produced (6%) from the pre-treated sample. Examination of the peak areas shows that Peak 1 and Peak 2 decreased slightly upon microwave treatment while Peak 3 increased (Table S4†). Since Peak 3 signal is attributed to strongly (dissociatively) chemisorbed CO, it is clear that microwave pre-treatment favours the strong chemisorption of CO. Fig. 6b shows methane profiles for 1.0K/10Fe/SiO2 and 1.0K/10Fe/SiO2-MW done via the methanator (data in Table S4†). The microwave effect at this potassium loading was more pronounced. Peaks due to weakly chemisorbed CO (116 and 216 °C) resulted in 2142 and 1303 units of CH4 for the non-microwaved sample, whereas the microwaved sample had 789 and 1716 peak area units, respectively. However, Peak 3 resulted in a larger peak area of 5794 units in the microwaved sample relative to its non-microwaved counterpart (2750 units). This data confirms earlier findings that microwave pre-treatment promotes the formation of strong CO chemisorption sites while suppressing weak CO chemisorption sites on iron catalysts. This indicates that the MW effect increases the binding of CO to iron. Microwave pre-treatment resulted in a 34% overall increase in the total CH4 evolved at a 1% K loading, illustrating that microwave modification of the catalysts does affect the production of hydrocarbons and the effect is dependent on the loading of potassium.
3.6 Fischer–Tropsch catalysis
The effect of potassium ions on FT activity was evaluated at 275 °C in a stainless steel fixed-bed reactor. Carbon monoxide (CO) conversions with respect to time on stream (TOS) for the 0.2K/10Fe/SiO2, 0.7K/10Fe/SiO2 and 1.5K/10Fe/SiO2 catalysts are presented in Fig. 7. The steady-state CO conversions were found to decline with an increase in the promoter content, with averages of 24.6, 17.8 and 12.4% for catalysts with K loadings of 0.2, 0.7 and 1.5 wt%, respectively (Fig. 7). The effect of potassium on FT synthesis activity has been investigated by several researchers,27–30 and the activity was reported to increase,31 passes through a maximum,22,32 had little effect or suppressed it as a function of the presence of K.22,33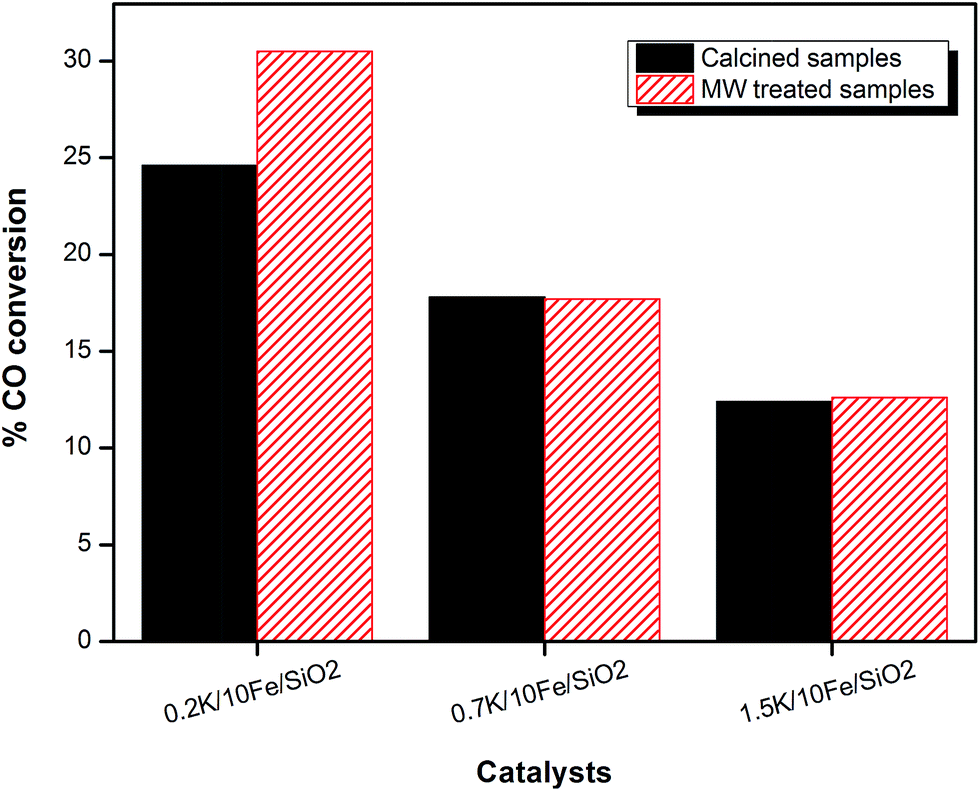 | ||
| Fig. 7 A comparison of CO conversions for microwaved and non-microwaved samples obtained at steady state conditions. | ||
Table 3 summarizes the steady state performance of the catalysts in the FT synthesis. From the table it is evident that increasing the promoter loading decreases the CO consumption rate, the FT reaction rate and the activity. The activity decreases from 21.4 to 3.96 μmol s−1 gFe−1 as the potassium content is increased from 0.2 to 1.5 wt%. Selectivity towards different hydrocarbon fractions was studied using an online GC fitted with an FID and they are reported as percentages. Selectivity towards methane was observed to decline as the promoter content increased. The 1.5K/10Fe/SiO2 catalyst had the highest C5–C8 selectivity (44.9 wt%), followed by the 0.7K/10Fe/SiO2 catalyst (28.0%). This indicates that as the K/Fe ratio increases, the proportion of methane in the products decreases and the fraction of C2+ products increases.
| Catalyst | 0.2K/SiO2 | 0.2K/SiO2-MW | 0.7K/SiO2 | 0.7K/SiO2-MW | 1.5K/SiO2 | 1.5K/SiO2-MW |
|---|---|---|---|---|---|---|
| CO conversion (%) | 24.6 | 30.5 | 17.8 | 17.7 | 12.4 | 14.6 |
| Rate CO (mol s−1) | −5.34 × 10−7 | −3.71 × 10−7 | −3.84 × 10−7 | −3.50 × 10−7 | −9.90 × 10−8 | −2.13 × 10−7 |
| Rate CO2 (WGS) | 2.08 × 10−7 | 1.16 × 10−7 | 1.25 × 10−7 | 1.16 × 10−7 | 2.33 × 10−8 | 7.34 × 10−8 |
| Rate FT | 3.26 × 10−7 | 2.55 × 10−7 | 2.59 × 10−7 | 2.34 × 10−7 | 7.57 × 10−8 | 1.39 × 10−7 |
| Activity (μmol s−1 gFe−1) | 21.4 | 14.8 | 15.4 | 14.0 | 3.96 | 8.50 |
| α | 0.55 | 0.56 | 0.59 | 0.66 | 0.79 | 0.77 |
| C2 olefin % | 7.69 | 16.8 | 39.3 | 41.5 | 61.9 | 55.6 |
|
||||||
| Selectivity | ||||||
| C1 | 28.2 | 26.2 | 14.6 | 11.4 | 3.8 | 12.4 |
| C2–C4 | 38.6 | 39.9 | 42.4 | 38.3 | 36.2 | 35.8 |
| C5–C8 | 18.2 | 18.9 | 28.0 | 35.3 | 44.9 | 36.7 |
| CO2 | 8.55 | 4.77 | 5.11 | 4.74 | 1.27 | 3.09 |
Addition of the alkali metal ions is also seen to increase ethylene selectivity in the C2 hydrocarbons. The increase is from 7.7 to 62%. The observed trends were expected and they are consistent with the tendency of the alkali metal ions to decrease the hydrogenation ability of a catalyst and thus accelerate chain growth probability and the olefin selectivity, possibly as a result of the increased catalyst basicity. The chain growth probability was seen to increase from 0.55 to 0.79 as the loading of potassium was increased.
The prepared samples were subsequently microwaved to modify their catalytic properties, average CO conversions measured at steady-state for the microwaved and non-microwaved samples were compared (Fig. 7). The CO conversions of the catalysts declined from 30.5 to 14.6% as the promoter content increased from 0.2 to 1.5 wt%. It is evident that both microwaved and non-microwaved samples display a similar trend: as the K/Fe ratio increases; the activity of the catalyst declines (Fig. S9 and S10†). It was observed, however, that the 0.2K/10Fe/SiO2-MW sample gave a significantly higher CO conversion (30.5%) relative to its non-microwaved counterpart (24.6%) (Fig. 7).
Data comparing the steady state performances of the microwave pre-treated and the untreated samples are displayed in Table 3. Addition of the promoter to the microwaved samples lowered the selectivity towards methane and the very light hydrocarbons (C2–C4), while the selectivity towards the heavier hydrocarbons (C5–C8) was enhanced. Analysis of the microwave effect on samples of the same composition indicates that selectivity towards methane is suppressed in the microwave modified catalysts at low potassium loadings (0.2 and 0.7 wt%). Microwave pre-treatment appears to enhance methane selectivity at high potassium loadings. The modified samples also displayed improved chain growth probabilities at low alkali loadings, while a slight decrease in the alpha value is observed on the sample with a high potassium loading. For simplicity the olefin content was analyzed by measuring the C2 olefin proportion relative to the total C2 fraction in the products. Potassium addition clearly favours the formation of olefins for both microwaved and non-microwaved samples. Reasons for this observation are well documented in the literature.32–34 Comparing catalysts of similar compositions shows that microwave pre-treatment enhances olefin formation for the samples with 0.2 and the 0.7 wt% potassium. Ethylene content increased from 7.69 to 16.8 wt% for the 0.2K/10Fe/SiO2-MW catalyst, while for the 0.7K/10Fe/SiO2-MW catalyst increased from 39.3 to 41.5 wt%. The increase in the olefin content is attributed to the effect of microwave pre-treatment, since all other variables were kept constant. The ethylene fraction declined from 61.9 to 55.6 wt% for the 1.5K/10Fe/SiO2-MW, suggesting that microwave modification only enhances olefin formation when low alkali metal loadings (0.2 to 0.7 wt%) are used, otherwise olefin formation is suppressed. Using iron catalysts instead of cobalt catalysts in FTS promotes the water gas shift (WGS) reaction and CO2 selectivity is normally used as a measure of the WGS activity. Selectivity towards CO2 was noted to decline relatively at low promoter loadings while it was enhanced in the sample with the highest potassium content. A decline in the WGS reaction is undesirable since it results in excess water in the reactor and this can lead to catalyst deactivation, particularly for medium to high conversions with iron catalysts.35 The 1.5K/10Fe/SiO2-MW catalyst displayed the largest improvement in activity (3.96 to 8.5 μmol s−1 gFe−1) which was induced by microwave modification of the catalyst.
FT performance data measured when the irradiation time was increased from 10 s to 10 min is displayed in Table 4. No significant influence on CO conversion was noted. However, the data confirmed that microwave induced effects are generated immediately after a short radiation period. This duration effect did however reduce the CH4 selectivity. Also the alpha value dropped from 0.79 to 0.63% as the irradiation period was increased to 10 min. Although the percentage CO conversions are similar, the rate of CO consumption increased from 9.90 × 10−8 to 2.25 × 10−7 mol s−1 as the irradiation time increased. This is in line with the considerably higher CO2 levels produced from the samples that were irradiated for 10 s and 10 min. The WGS reaction was also enhanced. Ethylene selectivity was also enhanced by increasing the duration of the pre-treatment period.
| Catalyst | 1.5K/SiO2 | 1.5K/SiO2-MW (10 seconds) | 1.5K/SiO2-MW (10 minutes) |
|---|---|---|---|
| CO conversion (%) | 12.4 | 14.6 | 12.4 |
| Rate CO (mol s−1) | −9.90 × 10−8 | −2.13 × 10−7 | −2.25 × 10−7 |
| Rate CO2 (WGS) | 2.33 × 10−8 | 7.34 × 10−8 | 5.46 × 10−8 |
| Rate FT | 7.57 × 10−8 | 1.39 × 10−7 | 1.70 × 10−7 |
| Activity (μmol s−1 gFe−1) | 3.96 | 8.50 | 8.99 |
| α | 0.79 | 0.77 | 0.63 |
| C2 olefin % | 61.9 | 55.6 | 67.0 |
|
|||
| Selectivity | |||
| C1 | 3.8 | 12.4 | 9.33 |
| C2–C4 | 36.2 | 35.8 | 39.3 |
| C5–C8 | 44.9 | 36.7 | 36.3 |
| CO2 | 1.27 | 3.09 | 2.25 |
It was observed that certain features such as the ethylene and CO2 selectivities were enhanced by microwave pre-treatment (10 s) at low alkali ion concentration. These features were suppressed by high alkali ion concentration on the surface of the metal if pre-treatment was performed for only 10 s. Pre-treatment improved both the FT reaction rate and the activity. It is therefore believed that microwave pre-treatment can enhance FT performance at all potassium loadings, but different K loadings require different irradiation times. The absolute rate of the FTS reaction corresponds to an apparent TOF values in the range 0.035–0.085 s−1 for the IWI catalysts (computed on surface Fe atoms). There is a close correspondence with the range of TOFs (based on surface Fe atoms) of 0.033–0.065 s−1 reported by Linganiso et al.6 for similar silica supported iron catalysts, though that work used a lower temperature (250 °C rather than 275 °C) but higher pressure (2.0 instead of 1.0 MPa).
4. Conclusions
In summary, this study shows that the behaviour of potassium-promoted supported iron catalysts (K = 0–1.5) can be modified by using microwave radiation. Microwave pre-treatment was found not to induce any modifications on the bulk properties of these catalysts, but surface and catalytic properties were markedly changed. Microwave solid-state modification of the catalysts had a significant impact on the CO adsorption characteristics of these catalysts. While the effect of potassium was relatively minor in untreated catalysts, the effect was much larger for microwave modified catalysts. Microwave pre-treatment induced an overall increase of 13% in CH4 formation for a 0.2 wt% K catalyst, and this continued to rise with the potassium loading up to a 1.0 wt% K catalyst where an increase of 65% in CH4 evolution was recorded.Microwave treatment was found to promote CH4 formation from “graphitic” carbon in these catalysts, while decreasing CH4 formation from α- and β-carbon species. CO adsorption investigations carried out using a methanator also showed that microwave treatment generated catalysts with a strong Fe–CO interaction. The microwave effect on the silica-supported catalysts was seen to increase dramatically as the irradiation time was increased from 10 s up to 10 min. Comparing catalysts composed of small and large crystallites with the same potassium loading showed that the microwave effect is amplified in catalysts with larger particles compared to those containing smaller crystallites. FTS performance was also improved by microwave modification. At low alkali concentration, microwaved samples showed improved (higher) ethene selectivities, higher alpha values and lower (improved) methane and C2–C4 selectivities. For the 0.7K/10Fe/SiO2 catalyst, the alpha value increased from 0.59 to 0.66. Microwave modification of catalysts with high alkali loadings resulted in a decline in the chain growth probability, an increase in olefin selectivity and a decrease methane selectivity. Overall, the microwave radiation-induced changes can be considered to be desirable for FTS catalysis.
Acknowledgements
The authors express gratitude for financial assistance awarded by the DST-NRF Centre of Excellence in Catalysis (c*change) and the National Research Foundation. They are also grateful for further funding awarded by the University of the Witwatersrand.References
- Z. Li, R. Liu, Y. Xu and X. Ma, Appl. Surf. Sci., 2015, 347, 643–650 CrossRef CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- M. W. Dlamini, D. O. Kumi, T. N. Phaahlamohlaka, A. S. Lyadov, D. G. Billing, L. L. Jewell and N. J. Coville, ChemCatChem, 2015, 7, 3000–3011 CrossRef CAS.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich, H.-U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- W. M. Dlamini, N. J. Coville and M. S. Scurrell, J. Mol. Catal. A: Chem., 2015, 409, 19–25 CrossRef CAS.
- L. L. Linganiso and M. S. Scurrell, Curr. Microwave Chem., 2016, 3 Search PubMed , in press.
- R. S. John, A. R. Batchelor, D. Ivanov, O. B. Udoudo, D. A. Jones, C. Dodds and S. W. Kingman, Miner. Eng., 2015, 84, 77–87 CrossRef CAS.
- R. K. Amankwah and G. Ofori-Sarpong, Miner. Eng., 2011, 24, 541–544 CrossRef CAS.
- M. S. Scurrell, unpublished observations.
- X. Zhang, D. Hayward and D. M. Mingos, Catal. Lett., 2003, 88, 33–38 CrossRef CAS.
- D. M. P. Mingos, in Microwave Assisted Organic Synthesis, Blackwell Publishing Ltd., 2009, pp. 1–22, DOI:10.1002/9781444305548.ch1.
- M. E. Dry and G. J. Oosthuizen, J. Catal., 1968, 11, 18–24 CrossRef CAS.
- Y. Yang, H.-W. Xiang, Y.-Y. Xu, L. Bai and Y.-W. Li, Appl. Catal., A, 2004, 266, 181–194 CrossRef CAS.
- D. E. Stobbe, F. R. van Buren, A. J. van Dillen and J. W. Geus, J. Catal., 1992, 135, 533–547 CrossRef CAS.
- E. Guglielminotti, F. Boccuzzi, F. Pinna and G. Strukul, J. Catal., 1997, 167, 153–163 CrossRef CAS.
- M. E. Dry, T. Shingles, L. J. Boshoff and G. J. Oosthuizen, J. Catal., 1969, 15, 190–199 CrossRef CAS.
- C. Li, I. Sayaka, F. Chisato and K. Fujimoto, Appl. Catal., A, 2016, 509, 123–129 CrossRef CAS.
- L. Gonzalo-Chacón, M. Almohalla, E. Gallegos-Suarez, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Appl. Catal., A, 2014, 480, 86–92 CrossRef.
- B. Graf and M. Muhler, Phys. Chem. Chem. Phys., 2011, 13, 3701–3710 RSC.
- J. Xu and C. H. Bartholomew, J. Phys. Chem. B, 2005, 109, 2392–2403 CrossRef CAS PubMed.
- C. Zhang, G. Zhao, K. Liu, Y. Yang, H. Xiang and Y. Li, J. Mol. Catal. A: Chem., 2010, 328, 35–43 CrossRef CAS.
- D. G. Miller and M. Moskovits, J. Phys. Chem., 1988, 92, 6081–6085 CrossRef CAS.
- E. van Steen and F. F. Prinsloo, Catal. Today, 2002, 71, 327–334 CrossRef CAS.
- N. Lohitharn and J. G. Goodwin Jr, J. Catal., 2008, 260, 7–16 CrossRef CAS.
- A. N. Pour, S. M. K. Shahri, H. R. Bozorgzadeh, Y. Zamani, A. Tavasoli and M. A. Marvast, Appl. Catal., A, 2008, 348, 201–208 CrossRef.
- A. N. Pour, M. R. Housaindokht, S. F. Tayyari and J. Zarkesh, J. Nat. Gas Chem., 2010, 19, 284–292 CrossRef CAS.
- R. B. Anderson, B. Seligman, J. F. Shultz, R. Kelly and M. A. Elliott, Ind. Eng. Chem., 1952, 44, 391–397 CrossRef CAS.
- H. Kölbel and H. Giehring, Brennst.–Chem., 1963, 44, 343–369 Search PubMed.
- H. Arakawa and A. T. Bell, Ind. Eng. Chem. Process Des. Dev., 1983, 22, 97–103 CAS.
- J. L. Rankin and C. H. Bartholomew, J. Catal., 1986, 100, 533–540 CrossRef CAS.
- D. B. Bukur, D. Mukesh and S. A. Patel, Ind. Eng. Chem. Res., 1990, 29, 194–204 CrossRef CAS.
- M. E. Dry, in Catalysis Science and Technology, ed. J. R. Anderson and M. Boudart, Springer-Verlag, New York, 1981, vol. 1, p. 159 Search PubMed.
- S. Soled, E. Iglesia, S. Miseo, B. DeRites and R. Fiato, Top. Catal., 1995, 2, 193–205 CrossRef CAS.
- R. A. Dictor and A. T. Bell, J. Catal., 1986, 97, 121–136 CrossRef CAS.
- A. Nakhaei Pour, M. R. Housaindokht, J. Zarkesh and S. F. Tayyari, J. Nat. Gas Sci. Eng., 2010, 2, 79–85 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26628a |
| This journal is © The Royal Society of Chemistry 2016 |