
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSide-chain conjugated polymers for use in the active layers of hybrid semiconducting polymer/quantum dot light emitting diodes†
Ana
Fokina
ab,
Yeonkyung
Lee
c,
Jun Hyuk
Chang
de,
Lydia
Braun
a,
Wan Ki
Bae
d,
Kookheon
Char
e,
Changhee
Lee
c and
Rudolf
Zentel
*a
aInstitute for Organic Chemistry, Johannes Gutenberg University, Duesbergweg 10-14, 55128, Mainz, Germany. E-mail: zentel@uni-mainz.de
bGraduate School for Excellence Materials Science in Mainz, Johannes Gutenberg University Mainz, Staudingerweg 9, 55128 Mainz, Germany
cDepartment of Electrical Engineering and Computer Science, Inter-University Semiconductor Research Center (ISRC), Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-742, Korea
dPhoto-Electronic Hybrids Research Center, National Agenda Research Division, Korea Institute of Science and Technology, 14-gil 5, Hwarang ro, Seongbuk gu, Seoul 136-791, Korea
eSchool of Chemical and Biological Engineering, The National Creative Research Initiative Center for Intelligent Hybrids, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea
First published on 21st October 2015
Abstract
Three monomers, M1–M3, with modified carbazole cores and styrene functionality were prepared for use in the active layers of hybrid polymer/quantum dot light emitting diodes. Utilizing reversible addition fragmentation chain transfer polymerization, side-chain conjugated polymers, P1–P3, with narrow polydispersities and disulfide end groups were obtained. The thermal, optical, and electrochemical properties of the polymers varied depending on the substituents of the carbazole cores. Through the disulfide end groups the polymers were chemically blended with quantum dots to obtain QD/polymer hybrids, which were further used as active layers in light emitting diodes. The fabricated devices retained the superior electroluminescence properties of QDs and showed good device performance with a highest external quantum efficiency of 6.09%. Moreover, a correlation between the HOMO level of the polymer and device performance was identified.
Introduction
Quantum dots (QDs) have been widely studied as promising emitting materials in light emitting diodes (LEDs) due to their high stability, high quantum yield, easily tunable emission wavelengths and particularly narrow emission profiles. Since the first report in 1994 by Colvin et al. a tremendous amount of work has been performed to understand how QD-based LEDs (QLEDs) work as well as how to improve device performance.1–7 In the state-of-the-art QLEDs, a thin active layer composed of QDs is sandwiched between an organic hole transport layer and an inorganic electron transport layer. Recently, the optimization of the charge carrier injection balance in the active layer has been intensively pursued.8–12 Facilitated hole injection as well as suppressed electron injection were shown to lead to enhanced device performance. As commonly used QDs possess low lying valence bands, hole injection from commercially available hole transport materials (HTMs) is rather challenging due to the large energy offset between the highest occupied molecular orbital (HOMO) of HTMs and the valence band of QDs. Kwak et al's. comparative study showed that utilizing HTMs with lower HOMO levels provided a smaller energy barrier for hole injection from HTMs into QDs and led to LED devices with improved performances.8 Additionally, Cho et al. showed that a reduced band offset between the QDs and the hole transport layer could be achieved by an increase of the QD valance band energy via the crosslinking of the QD layer.9 A nearly energy barrier free electron injection into QDs from the electron transport layer leads to the constant presence of an excess of electrons in the active layer (i.e. an electron overcharge problem). An excess of electrons leads to non-emissive exciton recombinations and thus efficiency decay. Bae et al. proposed a new QD structure with a modified shell which provided an energetic barrier between the emissive QD core and the electron transport layer leading to partial elimination of the excess electrons and suppression of non-emissive Auger recombination.10 Additionally, modification of device architecture has proven to be a successful strategy in addressing the problem of electron overcharge. Leck et al. were able to suppress the amount of injected electrons by the incorporation of a QD layer between hole transporting layers.11 Furthermore, Dai et al. showed that the incorporation of an insulating polymer (polymethylmethacrylate) layer between the electron transport layer and the QDs can reduce the quantity of injected electrons thus preventing an excess of electrons.12An interesting alternative to thin QD-only active layers sandwiched between electron and hole transport layers is provided by an active layer consisting of QDs distributed in a semiconducting matrix. A recent study by Bae et al. shows that the distribution of QDs within a semiconducting polymer matrix leads to a better charge balance within the active layer as well as to the efficient distribution of charge carriers and excitons through the active layer leading to devices with a reduced efficiency roll-off.13 Two-component active layers (composed of QDs and a semiconducting polymer) have previously also attracted attention as a pathway to obtain white LEDs with one emission layer.14–17 Fluorescent semiconducting polymers have been employed as encapsulating polymers resulting in combined QD and polymer emissions. The emission intensity of each compound could be tuned by either varying the component ratios or controlling the applied bias. However, if the amount of incorporated QDs is significantly increased, LEDs with pure QD emission can be produced.18–22 For the incorporation of a large amount of QDs within a polymer matrix while maintaining a homogeneous distribution of the QDs the physical blending of the two components is usually insufficient and phase separation typically occurs in physically blended films. Uniform QD distribution can be achieved by chemical blending of QDs and polymers (i.e. polymers grafted onto QDs).19,23 An effective route to the fabrication of chemically blended QD/polymer hybrids is the ligand exchange procedure.24 Polymers used in the ligand exchange must possess functional groups which can coordinate to the QD's surface and replace the initial small molecule ligands. After the ligand exchange procedure, the QD surface is functionalized with polymer chains. The modified QDs may now be easily homogenized with an excess of free polymer chains preventing phase separation in films.
While many semiconducting polymers and small molecules have been intensively investigated as host materials in the active layers of fluorescent and phosphorescent organic LEDs,25–27 only a few polymers have been tested as components of an active layer in hybrid semiconducting polymer/QD-based LEDs.14–16,18,20,22,28,29 In order to address the issue of charge injection balance, polymers with high lying lowest unoccupied molecular orbitals (LUMOs) and low lying HOMOs should be developed. High LUMO levels are important for reducing electron injection into QDs (i.e. electron overcharge problem), while low HOMOs and hole conducting ability should enable sufficient hole injection into QDs. Side-chain conjugated polymers are promising candidates to fulfill the above mentioned criteria and have been previously successfully investigated as host materials in organic LEDs.30–34
Herein, we report the synthesis of three side chain conjugated polymers which are intended for use in the active layers of hybrid polymer/QD LEDs. The three polymers are comprised of a polystyrene backbone, carbazole-based electroactive side chains and a disulfide end group. Depending on the substituents of the carbazole functionality the synthesized polymers exhibit different thermal, optical and electrochemical properties. The disulfide endgroup enables the binding of the polymer chains onto a QD surface during the QD/polymer hybrid synthesis. The obtained QD/polymer hybrids were then studied as the active layers in LEDs.
Experimental
Materials and methods
Solvents used in the reactions were purchased from Sigma Aldrich and used without further purification unless stated otherwise. Dry DMF was purchased from Acros Organics (99.8% extra). THF for polymerization reactions was dried over Na prior to use. Solvents used in column chromatography had technical grade purity and were additionally purified by distillation. Chemicals were purchased from Sigma Aldrich, Acros Organics, Alfa Aesar, or Fluka. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was purchased from Acros Organics, recrystallized from diethylether and stored at −20 °C. S-1-Dodecyl-S′-(α,α′-dimethyl-α′′-pentafluorophenyl acetate)trithiocarbonate, cysteamine methyl disulfide and quantum dots with a CdSe core, a CdxZn1−xS shell (core diameter 4 nm, total diameter 16 nm with oleic acid surface ligands) were synthesized according to the literature.28,35,36All NMR spectra were recorded in CDCl3 (Deutero GmbH) with a Bruker AV 400 or Bruker AC 300 spectrometer at room temperature. Chemical shifts are reported with respect to the residual solvent signal as internal standards CDCl3 (1H, 13C): 7.26 ppm and 77.16 ppm. Polymer molecular weight was determined by gel permeation chromatography (GPC) in THF with polystyrene as the external and toluene as the internal standard. UV–VIS spectra were recorded on a Varian Cary 5000 spectrometer. Emission spectra were recorded on a Varian Cary Eclipse spectrometer. DSC measurements were performed with Perkin Elmer DSC 8500 under a nitrogen flow at a 10 °C min−1 heating/cooling rate with three heating cycles (50 °C–250 °C) and two cooling cycles from (250 °C–50 °C). TGA measurements were performed with Perkin Elmer Pyris 6 TGA under a nitrogen flow at a 10 °C min−1 heating rate from 30 °C until 800 °C. Cyclic voltammetry experiments were performed with a Biologic SP-50 voltammetric analyzer using platinum wire working and counter electrodes and a 0.01 M Ag/AgNO3 reference electrode with a scan rate of 100 mV s−1 with [nBu4N][PF6] as the supporting electrolyte in dichloromethane.
Synthesis of 9-(4-methoxyphenyl)carbazole 1. Carbazole (1.00 g, 5.98 mmol), 4-iodoanisole (1.68 g, 7.18 mmol), pulverized copper (1.52 g, 23.92 mmol), potassium carbonate (6.61 g, 47.84 mmol) and 18-crown-6 ether (0.32 g, 1.20 mmol) were added to a Schlenk flask under a nitrogen atmosphere. ortho-Dichlorobenzene (30 mL) was added and the reaction mixture was heated to 200 °C for 18 hours. The hot mixture was then vacuum filtered. The filtrate was distilled in vacuo (20 mbar, 70 °C) to remove the solvent. The residue was purified via column chromatography (eluent hexanes/ethyl acetate 9
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to yield 9-(4-methoxyphenyl)carbazole as a colorless powder (1.20 g, 4.39 mmol, 73%). 1H-NMR (300 MHz, CDCl3): δ/ppm 8.16 (dt, J = 7.7, 1.0 Hz, 2H), 7.51–7.27 (m, 8H), 7.16–7.08 (m, 2H), 3.93 (s, 3H). 13C NMR (75.4 MHz, CDCl3) δ/ppm 158.97, 141.48, 130.42, 128.70, 125.96, 123.21, 120.38, 119.76, 115.18, 109.82, 55.74. HRMS (ESI+) m/z calcd 274.1232 [M + H] found 274.1226.
:1) to yield 9-(4-methoxyphenyl)carbazole as a colorless powder (1.20 g, 4.39 mmol, 73%). 1H-NMR (300 MHz, CDCl3): δ/ppm 8.16 (dt, J = 7.7, 1.0 Hz, 2H), 7.51–7.27 (m, 8H), 7.16–7.08 (m, 2H), 3.93 (s, 3H). 13C NMR (75.4 MHz, CDCl3) δ/ppm 158.97, 141.48, 130.42, 128.70, 125.96, 123.21, 120.38, 119.76, 115.18, 109.82, 55.74. HRMS (ESI+) m/z calcd 274.1232 [M + H] found 274.1226.
Synthesis of 3,6-dibromo-9-(4-methoxyphenyl)carbazole 2. 9-(4-Methoxyphenyl)carbazole (1.00 g, 3.66 mmol) was dissolved in dry DMF (30 mL) in a Schlenk flask under a nitrogen atmosphere. Recrystallized N-bromosuccinimide (2.61 g, 14.64 mmol) was dissolved in a separate Schlenk flask in dry DMF (30 mL) under a nitrogen atmosphere. Both solutions were cooled in an ice bath for 10 minutes and the NBS solution was slowly added to the 9-(4-methoxyphenyl)carbazole solution. The mixture was left under stirring at room temperature for 48 hours after which it was poured onto an ice/water mixture leading to the precipitation of 3,6-dibromo-9-(4-methoxyphenyl)carbazole. The obtained 3,6-dibromo-9-(4-methoxyphenyl)carbazole was collected via vacuum filtration, washed several times with cold acetone and dried under vacuum for 24 hours (1.48 g, 3.43 mmol, 94%). 1H-NMR (300 MHz, CDCl3): δ/ppm 8.19 (dd, J = 2.0, 0.6 Hz, 2H), 7.49 (dd, J = 8.7, 2.0 Hz, 2H), 7.41–7.33 (m, 2H), 7.17 (dd, J = 8.7, 0.6 Hz, 2H), 7.14–7.08 (m, 2H), 3.92 (s, 3H). 13C NMR (75.4 MHz, CDCl3) δ/ppm 159.42, 140.46, 129.43, 129.39, 128.53, 123.81, 123.28, 115.40, 112.93, 111.59, 55.78. HRMS (ESI+) m/z calcd 428.9364 found 428.9345.
Synthesis of 3,6-bis(4-methylphenyl)-9(4-methoxyphenyl)carbazole 3. 3,6-Dibromo-9-(4-methoxyphenyl)carbazole (1.21 g, 2.81 mmol), p-tolylboronic acid (985 mg, 7.24 mmol) and tetrakis(triphenylphosphine)palladium(0) (162 mg, 0.14 mmol) were added to a Schlenk flask. Toluene (35 mL) was added to the mixture, followed by the addition of a 2 M aqueous sodium carbonate solution (8.5 mL) and ethanol (4.25 mL). The biphasic mixture was freeze–pump–thawed (3×) and left under a nitrogen atmosphere. A condenser was attached and the mixture was heated at 90 °C for 20 hours under a nitrogen atmosphere. The mixture was then cooled to room temperature and the organic phase was separated from the aqueous. The aqueous phase was extracted twice with ethyl acetate and the combined organic phases were washed with water and brine. After drying with magnesium sulfate the solvents were removed by rotary evaporation. The residue was purified via column chromatography (eluent hexanes/dichloromethane) to yield 3,6-bis(4-methylphenyl)-9(4-methoxyphenyl)carbazole (820 mg, 1.81 mmol, 64%). 1H-NMR (300 MHz, CDCl3): δ/ppm 8.38 (dd, J = 1.8, 0.6 Hz, 2H), 7.71–7.59 (m, 6H), 7.55–7.46 (m, 2H), 7.39 (dd, J = 8.5, 0.6 Hz, 2H), 7.34–7.27 (m, 4H), 7.19–7.10 (m, 2H), 3.94 (s, 3H), 2.43 (s, 6H). 13C NMR (75.4 MHz, CDCl3) δ/ppm 159.02, 141.25, 139.25, 136.35, 133.41, 130.40, 129.64, 128.55, 127.29, 125.57, 123.89, 118.72, 115.25, 110.14, 110.11, 55.76, 21.24. HRMS (ESI+) m/z calcd 453.2092 found 453.2103.
Synthesis of 3,6-bis(4-methylphenyl)-9(4-hydroxyphenyl)carbazole 4. 3,6-Bis(4-methylphenyl)-9(4-methoxyphenyl)carbazole (490 mg, 1.08 mmol) was dissolved in dry dichloromethane (12 mL) and the solution was cooled in an acetone/dry ice bath. Boron tribromide solution (1.5 mL, 1.51 mmol, 1 M in dichloromethane) was added dropwise, and the solution was slowly warmed after 30 minutes and stirred for 22 hours at room temperature. Then water (2 mL) was added and the mixture was stirred for additional 30 minutes. The solution was washed twice with water and once with brine and dried with magnesium sulfate. After the removal of dichloromethane by rotary evaporation 3,6-bis(4-methylphenyl)-9(4-hydroxyphenyl)carbazole was obtained as a grey powder and was used in the next step without further purification (458 mg, 1.04 mmol, 96%).1H-NMR (400 MHz, CDCl3): δ/ppm 8.37 (dd, J = 1.8, 0.6 Hz, 2H), 7.70–7.59 (m, 6H), 7.48–7.43 (m, 2H), 7.41–7.36 (m, 2H), 7.10–7.04 (m, 2H), 2.43 (s, 6H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 155.07, 141.24, 139.25, 136.38, 133.47, 130.66, 129.65, 128.82, 127.30, 125.60, 123.92, 118.74, 116.78, 110.12, 21.24.
Synthesis of monomer M1. 3,6-Bis(4-methylphenyl)-9(4-hydroxyphenyl)carbazole (770 mg, 1.75 mmol), 4-vinylbenzyl chloride (0.41 mL, 446 mg, 2.92 mmol), potassium carbonate (1.21 g, 8.76 mmol) and a catalytic amount of potassium iodide were added to a flask. A mixture of acetonitrile (12 mL) and DMF (24 mL) was added. The mixture was heated at 60 °C for 3 hours. The mixture was then cooled to room temperature and poured into an ice/water mixture (250 g). After the ice melted dichloromethane (250 mL) was added to the mixture to extract the organic compounds. The aqueous phase was extracted with dichloromethane two more times and the combined organic phases were washed with brine and dried with magnesium sulfate. Dichloromethane was removed by rotary evaporation and the residue was purified via column chromatography (eluent hexanes/dichloromethane) to yield monomer M1 (795 mg, 1.44 mmol, 82%). 1H-NMR (400 MHz, CDCl3): δ/ppm 8.37 (dd, J = 1.8, 0.6 Hz, 2H), 7.69–7.60 (m, 6H), 7.53–7.45 (m, 6H), 7.39 (d, J = 8.5 Hz, 2H), 7.35–7.27 (m, 4H), 7.24–7.17 (m, 2H), 6.77 (dd, J = 17.6, 10.9 Hz, 1H), 5.81 (dd, J = 17.6, 0.9 Hz, 1H), 5.30 (dd, J = 10.9, 0.8 Hz, 1H), 5.17 (s, 2H), 2.43 (s, 6H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 158.16, 141.19, 139.24, 137.67, 136.53, 136.35, 136.31, 133.43, 130.65, 129.65, 128.53, 127.91, 127.28, 126.67, 125.57, 123.91, 118.71, 116.17, 114.43, 110.15, 70.29, 21.24. HRMS (ESI+) m/z calcd 555.2562 found 555.2558.
Synthesis of monomer M2. Carbazole (8.5 g, 0.051 mol), potassium hydroxide (4.28 g, 0.076 mol) and DMF (100 mL) were added to a flask and stirred at room temperature for two hours. Then 4-vinylbenzyl chloride (7.9 mL, 8.53 g, 0.056 mol) was added and the ice bath was removed. The mixture was stirred at room temperature for 24 hours and then was poured into an ice/water mixture (400 g) and a white precipitate formed. After the ice melted the precipitate was filtered and washed with methanol and hexanes. The residue was then recrystallized from acetone to yield monomer M2 as colorless needles (12.55 g, 0.044 mol, 87%). 1H-NMR (400 MHz, CDCl3): δ/ppm 8.15 (dt, J = 7.8, 1.0 Hz, 2H), 7.46–7.42 (m, 2H), 7.37 (dt, J = 8.2, 0.9 Hz, 2H), 7.33–7.23 (m, 4H), 7.11 (d, J = 8.0 Hz, 2H), 6.66 (dd, J = 17.6, 10.9 Hz, 1H), 5.69 (dd, J = 17.6, 1.0 Hz, 1H), 5.21 (dd, J = 10.9, 0.9 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 140.76, 137.01, 136.84, 136.41, 126.75, 126.73, 125.99, 123.17, 120.53, 119.36, 114.06, 109.01, 46.49. HRMS (ESI+) m/z calcd 284.1439 [M + H] found 284.1466.
Synthesis of formyl-4,4′-bis(N-carbazolyl)-1,1′-biphenyl (formyl-CBP) 5. 4,4′-Bis(N-carbazolyl)-1,1′-biphenyl (1.25 g, 2.60 mmol) was dissolved in dry dichloromethane (100 mL) in a Schlenk flask under a nitrogen atmosphere. The solution was cooled in an ice bath and tin(IV) chloride solution (3.9 mL, 1 M in dichloromethane) was added slowly. After 25 minutes dichloromethyl methyl ether (0.35 mL, 0.45 g, 0.0039 mmol) was added dropwise. After 2 hours the ice bath was removed and the mixture was stirred at room temperature for three additional hours. Then the mixture was poured onto ice (160 g). After the ice melted the phases were separated and the organic phase was extracted once with water and three times with saturated aqueous NaHCO3 solution. NaHCO3 was added to the aqueous phase and the neutralized aqueous phase was extracted twice with dichloromethane. The combined organic phases were washed with water and brine and dried with MgSO4. The solvent was removed by rotary evaporation and the residue was purified by column chromatography (eluent hexanes/ethyl acetate) to yield formyl-CBP (693 mg, 1.35 mmol, 52%). 1H-NMR (400 MHz, CDCl3): δ/ppm 10.15 (s, 1H), 8.71 (dd, J = 1.6, 0.7 Hz, 1H), 8.25 (dt, J = 7.8, 1.1 Hz, 1H), 8.19 (dt, J = 7.7, 1.0 Hz, 2H), 8.03–7.90 (m, 5H), 7.76–7.67 (m, 4H), 7.58–7.39 (m, 8H), 7.40–7.31 (m, 2H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 191.89, 144.58, 141.94, 140.90, 140.41, 140.10, 139.10, 137.64, 136.56, 136.30, 129.76, 129.71, 128.98, 128.91, 128.72, 127.80, 127.74, 127.67, 127.24, 126.18, 124.06, 123.87, 123.66, 123.50, 121.55, 121.50, 120.90, 120.55, 120.28, 110.61, 110.33, 109.93. HRMS (ESI+) m/z calcd 513.1967 [M + H] found 513.1948.
Synthesis of acrylic acid-4,4′-bis(N-carbazolyl)-1,1′-biphenyl (acrylic acid-CBP) 6. Compound 5 (1.05 g, 2.05 mmol) and malonic acid (639 mg, 6.15 mmol) were suspended in dry pyridine (100 mL) and piperidine (cat. 20 drops) was added dropwise. The reaction mixture was stirred at 100 °C for 19 hours. Upon heating all components dissolved and a clear reaction solution was obtained. The reaction solution was allowed to cool down to room temperature and slowly added to 600 mL of 3 M HCl. The aqueous phase was repeatedly extracted with dichloromethane and the combined organic phases were washed twice with water and twice with brine solution. The solvent was removed by rotary evaporation and the residue was purified by column chromatography (eluent dichloromethane/acetone) to yield compound 6 (900 mg, 1.62 mmol, 79%). 1H-NMR (400 MHz, CDCl3): δ/ppm 8.37 (d, J = 1.6 Hz, 1H), 8.18–8.22 (m, 3H), 8.05 (d, J = 15.8 Hz, 1H), 7.97–7.90 (m, 4H), 7.77–7.66 (m, 5H), 7.58–7.30 (m, 10H), 6.55 (d, J = 15.9 Hz, 1H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 172.44, 148.17, 142.41, 141.60, 140.92, 139.99, 139.22, 137.54, 136.69, 128.81, 128.69, 127.65, 127.59, 126.89, 126.52, 126.46, 126.17, 124.11, 123.65, 123.38, 121.65, 121.04, 120.73, 120.54, 120.26, 114.52, 110.51, 110.40, 109.95. HRMS (ESI+) m/z calcd 554.1994 [M + H] found 554.2001.
Synthesis of propanoic acid-4,4′-bis(N-carbazolyl)-1,1′-biphenyl (propanoic acid-CBP) 7. Compound 6 (817 mg, 1.47 mmol) was dissolved in dry THF in a Schlenk flask and palladium on charcoal (82 mg, 10 wt% Pd) was added. The reaction mixture was purged with hydrogen and a balloon filled with hydrogen was attached to the flask via the stopcock. The reaction mixture was shaken at room temperature for 24 hours. The black residue was filtered off and the solvent was removed by rotary evaporation. Compound 7 was used in the next step without further purification. 1H-NMR (400 MHz, CDCl3): δ/ppm 8.20–8.15 (m, 3H), 8.05–8.00 (m, 1H), 7.95–7.88 (m, 4H), 7.75–7.67 (m, 4H), 7.55–7.42 (m, 7H), 7.37–7.29 (m, 4H), 3.20 (t, J = 7.8 Hz, 2H), 2.82 (t, J = 7.8 Hz, 2H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 178.02, 141.19, 140.94, 139.73, 139.41, 139.33, 137.43, 137.37, 132.56, 132.18, 130.21, 129.06, 128.64, 128.41, 127.61, 127.49, 126.65, 126.19, 126.16, 123.86, 123.63, 123.44, 120.52, 120.50, 120.21, 120.18, 119.93, 110.12, 110.04, 109.99, 109.96, 68.11, 36.50, 30.95, 25.75. HRMS (ESI+) m/z calcd 579.2049 [M + Na] found 579.2057.
Synthesis of propanol-4,4′-bis(N-carbazolyl)-1,1′-biphenyl (propanol-CBP) 8. Compound 7 (780 mg, 1.40 mmol) dissolved in THF (50 mL) was slowly added to a LiAlH4 (53.2 g, 1.4 mmol) suspension in THF (20 mL). The reaction mixture was stirred at room temperature for four hours. The reaction was quenched with ethanol. Afterward 110 mL of 10% H2SO4 solution was added and the mixture was repeatedly extracted with diethyl ether. The combined organic phases were washed with water and brine and dried over MgSO4. The solvent was removed by rotary evaporation and the residue was purified by column chromatography (eluent dichloromethane/acetone) to yield compound 8 (580 mg, 1.12 mmol, 76% (two steps)) 1H-NMR (400 MHz, CDCl3): δ/ppm 8.19 (d, J = 7.7 Hz, 2H), 8.16 (d, J = 7.7 Hz, 1H), 8.01 (dd, J = 1.7, 0.7 Hz, 1H), 7.93–7.90 (m, 4H), 7.75–7.68 (m, 4H), 7.56–7.41 (m, 7H), 7.37–7.28 (m, 4H), 3.76 (t, J = 6.4 Hz, 2H), 2.94 (t, J = 7.6 Hz, 2H), 2.10–1.96 (m, 2H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 141.15, 140.95, 139.52, 139.44, 139.26, 137.53, 137.35, 133.74, 128.63, 127.62, 127.48, 126.86, 126.16, 126.09, 123.81, 123.63, 123.50, 120.52, 120.46, 120.21, 120.10, 119.94, 109.96, 109.88, 62.53, 35.17, 32.26. HRMS (ESI+) m/z calcd 542.2358 found 542.2379.
Synthesis of monomer M3. Compound 8 (300 mg, 0.55 mmol), NaH (60% in oil, 26.5 mg, 1.11 mmol) and KI (cat.) were added to dry THF (50 mL)/DMF (5 mL). 4-Vinylbenzyl chloride (90%, 0.13 mL, 126 mg, 0.83 mmol) was added and the reaction mixture was stirred at 50 °C for 24 hours. After addition of DCM (100 mL) and water (100 mL) the organic phase was separated and washed with water and brine. The solvent was removed by rotary evaporation and the residue was purified by column chromatography (eluent hexanes/dichloromethane) to yield monomer M3 as a colorless powder (295 mg, 0.45 mmol, 83%). 1H-NMR (400 MHz, CDCl3): δ/ppm 8.19 (d, J = 7.7, 2H), 8.14 (d, J = 7.8, 1H), 7.98 (d, J = 1.6 Hz, 1H), 7.95–7.89 (m, 4H), 7.75–7.68 (m, 4H), 7.56–7.27 (m, 16H), 6.73 (dd, J = 17.6, 10.9 Hz, 1H), 5.76 (dd, J = 17.6, 1.0 Hz, 1H), 5.25 (dd, J = 10.9, 0.9 Hz, 1H), 3.56 (t, J = 6.3 Hz, 2H), 2.94 (t, J = 7.6 Hz, 2H), 2.18–1.99 (m, 2H). 13C NMR (100.6 MHz, CDCl3) δ/ppm 141.11, 140.94, 139.45, 139.21, 138.40, 137.56, 137.34, 137.05, 136.72, 133.93, 128.63, 128.61, 128.06, 127.61, 127.46, 126.96, 126.38, 126.15, 126.02, 123.74, 123.62, 123.55, 120.52, 120.45, 120.21, 120.07, 120.00, 113.87, 109.97, 109.93, 109.77, 72.84, 69.66, 32.54, 32.32. HRMS (ESI+) m/z calcd 658.2984 found 658.2972.
Polymer P1. Monomer M1 (680 mg, 1.22 mmol), CTA (10.8 mg, 0.020 mmol), AIBN (0.44 mg, 0.0029 mmol), THF (4.5 mL). Solvent for purification: methanol/diethyl ether 2/1. 380 mg polymer as a light yellow powder (51%). Mn = 10
900 g mol−1, PDI 1.1
Polymer P2. Monomer M2 (1.0 g, 3.53 mmol), CTA (30.4 mg, 0.058 mmol), AIBN (1.38 mg, 0.0084 mmol), THF (10 mL). Solvent for purification: hexanes/acetone 4/1. 480 mg of polymer as a light yellow powder (48%). Mn = 5300 g mol−1, PDI 1.2.
Polymer P3. Monomer M3 (250 mg, 0.379 mmol), CTA (3.36 mg, 0.0062 mmol), AIBN (0.148 mg, 0.0009 mmol), THF (2 mL). Solvent for purification: methanol/acetone 2/1 and hexanes/acetone 3/1. 110 mg of polymer as a light yellow powder (44%). Mn = 11
600 g mol−1, PDI 1.1.
Results and discussion
The performances of hybrid semiconducting polymer/QD LED devices in previous studies have been found to be notably below the efficiencies of the contemporary OLEDs.12,28 The low performance of hybrid LEDs has been attributed to impeded hole injection from the polymer into QDs due to the large energy offset between the HOMO of the polymer and the valence band of the QDs. This makes the carbazole core a promising choice for the electroactive unit as it may provide polymers with relatively low HOMO levels and therefore a minimized hole injection barrier.Monomer synthesis
Scheme 1 illustrates the synthetic routes to carbazole-based monomers M1–M3. As mentioned above, a carbazole unit has been chosen as the core element of all three monomers due to the relatively low HOMO level as well as the widely known hole conducting ability of carbazole-based small molecules and polymers.32,33,37–39 Moreover, by the introduction of different aliphatic and/or aromatic substituents into the carbazole core the electrochemical properties of the final products can be easily tuned. A styrene group was chosen as the precursor for the chemically and electrochemically inert polystyrene backbone of the resulting polymers. The monomer M1 was synthesized in five steps starting with a copper catalyzed Ullmann coupling reaction between carbazole and 4-iodoanisole in o-dichlorobenzene which led to the formation of 9-(4-methoxyphenyl)carbazole 1. Following this the bromination of 1 with 4 eq. of N-bromosuccinimide in DMF quantitatively produced 3,6-dibromo-9-(4-methoxyphenyl)carbazole, 2, which was precipitated into water and used in the next step without any further purification. 3,6-Bis(4-methylphenyl)-9(4-methoxyphenyl)carbazole 3 was synthesized from 2 and p-tolylboronic acid using palladium-catalyzed Suzuki coupling with Pd(PPh3)4 in a toluene/2 M aqueous Na2CO3 solution/ethanol mixture. The subsequent highly efficient deprotection of the hydroxyl group with boron tribromide in dry dichloromethane was followed by a nucleophilic substitution reaction between 3,6-bis(4-methylphenyl)-9(4-hydroxyphenyl)carbazole 4 and 4-vinylbenzyl chloride in an acetonitrile/DMF mixture to yield monomer M1 as a colorless crystalline powder. The introduction of aromatic rings in the 3, 6 and 9 positions of the carbazole unit was undertaken to expand the size of the conjugated system and to increase the solubility of the monomer and thus the future polymer. Additionally, the methyl groups in the para positions of phenyl rings prevent irreversible oxidation under electrolytic conditions.39,40 Monomer M2 was synthesized in one step by a nucleophilic substitution reaction between carbazole and 4-vinylbenzyl chloride in DMF and subsequently was recrystallized from acetone to yield highly crystalline colorless needles. | ||
| Scheme 1 Synthesis of (a) M1, (b) M2 and (c) M3 monomers. | ||
The synthesis of the monomer M3 was carried out in five steps. The mono-formylation of 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) was carried out with tin(IV) chloride and dichloromethyl methyl ether in dry dichloromethane, followed by an aqueous work-up. The Knoevenagel–Doebner condensation of 5 with malonic acid in pyridine resulted in the formation of the α,β-unsaturated carboxylic acid derivative 6. In order to reconstitute the π-system of the CBP unit, the hydrogenation of the side chain double bond was performed in dry THF with palladium on charcoal producing compound 7. Subsequent reduction of the acid group with LiAlH4 led to the formation of hydroxypropyl-CBP 8. Finally, the nucleophilic substitution reaction between 8 and 4-vinylbenzyl chloride gave the monomer M3 as a colorless crystalline solid. The incorporated alkyl spacer between the styrene backbone unit and the CBP functionality facilitates the easy accessibility of vinyl double bonds during polymerization and improves monomer solubility.
Polymer synthesis
Reversible addition–fragmentation chain transfer (RAFT) radical polymerization was employed for the synthesis of the polymers P1–P3 (Scheme 2). S-1-Dodecyl-S′-(α,α′-dimethyl-α′′-pentafluorophenyl acetate)trithiocarbonate was chosen as the RAFT chain transfer agent because trithiocarbonates have previously been reported as efficient agents in polymerization of bulky styrene derivatives.35 Moreover, the reactive pentafluorophenyl ester group, which can be later replaced with other desired functionalities, was incorporated at the beginning of each polymer chain. To facilitate a simplified comparison of the properties of our produced QD/polymer hybrids and devices, the three polymers should have a similar number of electrochemically active groups (i.e. repeat units). Additionally, relatively short chain lengths have to be chosen to ensure the good solubility of the polymers in toluene, which is a necessary precondition during the device fabrication. Therefore, the polymerization conditions were optimized for each polymer to yield polymers with narrow molecular weight distributions and a chain length of approximately 20 repeat units (Table 1). Relevant ratios of monomer, RAFT agent, and initiator (2,2′-azobis(2-methylpropionitrile) (AIBN)) were dissolved in dry THF and degassed via three freeze–pump–thaw cycles. The polymerization was carried out under a nitrogen atmosphere at 65 °C, in a sealed Schlenk flask for 48 hours. Afterwards, the reaction solutions were cooled down to room temperature and the polymers were precipitated into hexanes. For purification, the polymers were repeatedly redissolved in THF and precipitated into suitable solvents. In the next step the reactive trithiocarbonate polymer end group was replaced with an inert 2-cyanoisopropyl functionality by reaction with an excess of AIBN in THF. Finally, the reaction of cysteamine methyl disulfide with pentafluorophenyl ester in THF at 30 °C for 24 hours led to the quantitative replacement of the pentafluorophenyl groups with cysteamine methyl disulfide and the substitution was confirmed by the disappearance of the pentafluorophenyl peaks in 19F NMR spectroscopy (Fig. S15†) and the presence of a C–N band in IR spectroscopy (Fig. S16†). The polymers were further characterized by gel permeation chromatography (GPC) and 1H NMR. Molecular weight and polydispersity values of the polymers are summarized in Table 1. The incorporated disulfide moieties enable polymers to graft onto the CdSe/CdZnS QD surface due to the preferred affinity of disulfides to the unsaturated Zn-centers.13,28,41 The polymers with disulfide functionalities can now participate in the ligand exchange procedure, replacing the weakly bound initial small molecule ligands (e.g. oleic acid) on the surface of QDs and chemically grafting on the surface via disulfide groups. The introduction of the anchor groups is a necessary step in the synthesis of the polymers which are utilized in hybrid polymer/QD systems as polymer grafting onto the QD surface is required for the uniform dispersion of QDs within polymer matrices.19,23 | ||
| Scheme 2 RAFT polymerization and post polymerization modification reactions of P1–P3. | ||
| Polymer |
M
na (D) |
PDIa | P n |
T
d5b (°C) |
T
gc (°C) |
HOMOd (eV) | LUMOe (eV) | Gapf (eV) |
|---|---|---|---|---|---|---|---|---|
| a Obtained by GPC measurements in THF with polystyrene as the standard. b Decomposition temperature at 5% weight loss. c Glass transition temperature calculated with software fit. d Values obtained from UPS measurements. e Calculated as HOMO minus optical band gap. f Optical band gap. | ||||||||
| P1 | 10900 |
1.1 | 20 | 400 | 190 | 5.52 | 2.24 | 3.28 |
| P2 | 5300 | 1.2 | 19 | 380 | 155 | 5.78 | 2.32 | 3.46 |
| P3 | 11600 |
1.1 | 18 | 390 | 160 | 5.82 | 2.45 | 3.37 |
Thermal properties
The thermal properties of the polymers prepared in the present study were characterized using thermo-gravimetric analysis (TGA) and differential scanning calorimetry (DSC). All three polymers P1, P2 and P3 possess high thermal stability with decomposition temperatures Td5 (the temperature at which 5% of weight loss occurs) above 380 °C with the highest thermal stability shown by P1 with Td5 at 400 °C (Table 1). P1 also exhibits the highest glass transition temperature at 190 °C along with 155 °C and 160 °C for P2 and P3 respectively. We note that these Tg values are higher than the glass transition temperatures of hole transport layer materials based on carbazole-based small molecules.42,43Optical properties
The normalized solution and fluorescence spectra of the polymers are shown in Fig. 1. The solution absorption spectrum of P1 in toluene exhibits a broad absorption with a maximum centered at 295 nm along with a weak shoulder with the absorption onset at 378 nm. Upon excitation of the P1 solution at 315 nm a photoluminescence (PL) spectrum with the maximum at 397 nm is observed. The absorption spectrum of P2 is consistent with previous reports of polymers containing carbazoles as side groups and exhibits the typical absorption pattern of the carbazole unit with maxima at 345, 331 and 295 nm and the absorption onset at 355 nm.34 The red-shifted absorption onset of P1 compared to P2 reflects the larger conjugated system of semiconducting side groups of the former polymer. The PL spectrum of P2 shows two maxima at 365 and 352 nm (with λex 310 nm). The absorption and PL spectra of P3 exhibit only slight deviation from the characteristics of molecular CBP with the onset of absorption at 360 nm and two absorption maxima at 325 and 295 nm and the emission maximum at 390 nm (with λex 315 nm).39 From the onsets of thin film absorption spectra, the optical band gaps of P1, P2 and P3 were estimated to be 3.28, 3.46 and 3.37 eV, respectively (Fig. S18†).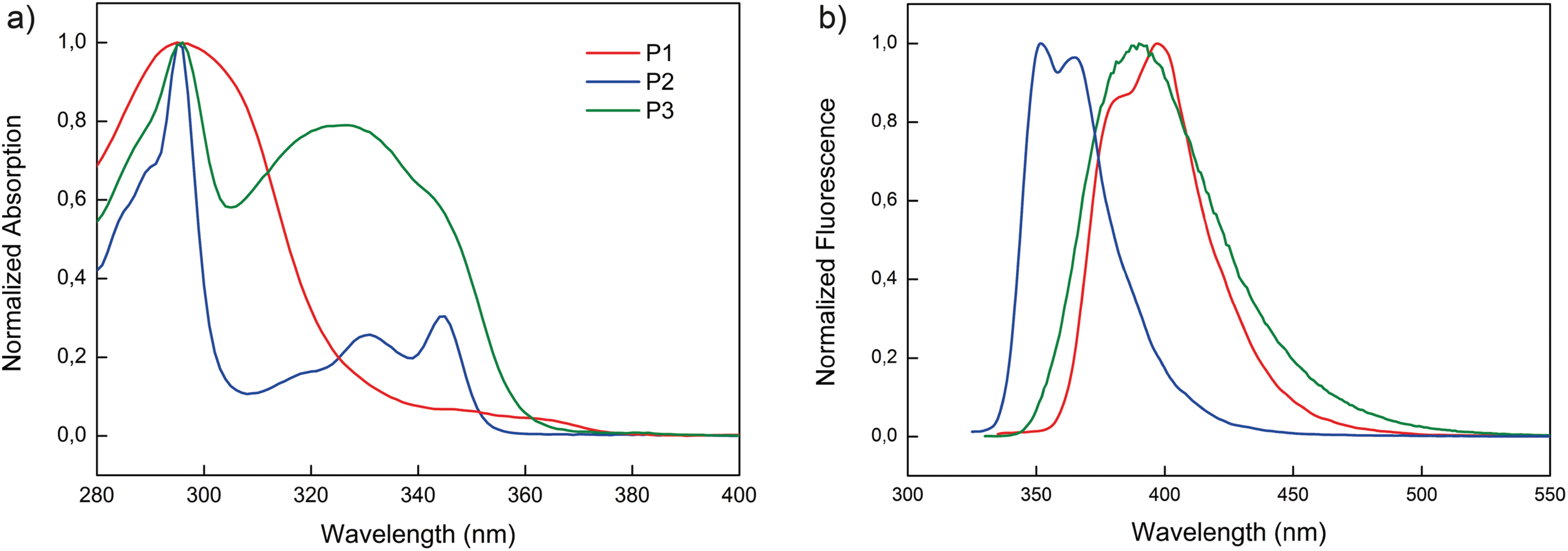 | ||
| Fig. 1 Optical properties of polymers P1–P3. (a) Normalized absorption in toluene. (b) Normalized fluorescence in toluene. | ||
Electrochemical properties
The HOMO levels of the polymers P1–P3 were determined by UPS measurements and estimated to be 5.52, 5.78 and 5.82 eV for P1, P2, and P3, respectively. Additionally, the electrochemical behavior of the polymers was studied via cyclic voltammetry in dichloromethane (Fig. 2). The onset positions of the first polymer oxidation peaks are in agreement with the values obtained by UPS measurements with P1 having the lowest oxidation potential, followed by P2 and P3. In the analyzed electrochemical window P2 exhibited one broad irreversible oxidation peak which is typical for carbazole structures with unprotected 3 and 6 positions.39 A similar electrochemical behavior is observed for the polymer P3. However, in the same voltage range P1 with methyl protected 3 and 6 positions shows two distinct oxidation peaks with the first oxidation process being reversible and the second irreversible (Fig. S21†). | ||
| Fig. 2 Cyclic voltammogram of polymers P1–P3 in DCM/TBAPF6. | ||
QD/polymer hybrids as an active layer in QLEDs
The QD/polymer hybrids QH1, QH2, and QH3 were prepared using CdSe/CdxZn1−xS red QDs and polymers P1, P2 and P3 respectively. In order to prevent inorganic/organic phase separation and to obtain films with a uniform QD distribution within the polymer matrices the QDs were functionalized with polymer chains by the ligand exchange procedure.19 During ligand exchange the polymer disulfide end groups coordinate to the QD surface, substituting the oleic acid ligands initially coordinated thus leading to the formation of polymer-modified QDs.13,28,41 QDs with weakly bound oleic acid ligands are used in the ligand exchange as the initial small molecule ligands should possess weaker affinity to QD surfaces than the disulfide functionalities. The course of the ligand exchange can be monitored by the solubility change of the QDs. The solubility change of QDs is governed by the types of ligands on the surface, thus after the ligand exchange procedure QDs acquire the solubility properties of the polymers grafted on their surface (Fig. S23†). The modified QDs can now be efficiently dispersed in the polymer matrices leading to films with improved QD distribution within the polymer compared with physically blended QD/polymer systems (Fig. S25†).19,28To examine the impact of different polymers on QLED performance, devices with QH1, QH2, and QH3 films as the active layers were fabricated in the inverted structure configuration: ITO(150 nm)//ZnO nanoparticles (40 nm)//QD@polymer hybrids (30 nm)//CBP(4,4′-bis(9-carbazolyl)-1,1′-biphenyl (40 nm)//MoOx (10 nm)//Al (100 nm) (Fig. S27†). In the inverted architecture the ZnO layer provides the electron transport from the ITO cathode, while CBP and MoOx layers are responsible for the hole transport from the aluminum anode. Fig. 3 shows the characteristics (electroluminescence spectra, external quantum efficiency, current–voltage (J–V) and luminance–voltage (L–V) curves) of the devices fabricated in the present study. The results obtained show that the polymers synthesized can be successfully used in the active layer of QD/polymer hybrid LEDs leading to devices with good efficiencies while retaining the superior electroluminescence properties of QDs. As shown in Fig. 3a all the devices tested show narrow electroluminescence spectra with Gaussian fits (λmax = 630 nm, FWHM = 35 nm), indicating that light emission is solely originating from the quantum dots without parasitic emissions from the hybridized polymers or neighboring hole transport layers. A maximum external quantum efficiency (EQE) of 6.09% is achieved with the device containing the QH3 active layer. An EQE of 6.09% is, to the best of our knowledge, the highest reported EQE for LEDs with mixed QD/polymer active layers. The devices with QH2 and QH1 exhibit efficiencies of 5.05% and 2.14% respectively (Fig. 3b). Furthermore, the device with QH3 also shows better performance in terms of both driving voltage and luminance in a wide range of applied voltages followed by QH2 and QH1 (Fig. 3c and d). This tendency is in agreement with the results previously published suggesting that polymers with low lying HOMO levels provide a reduced barrier for hole injection into QDs and lead to devices with improved characteristics.28 Hybrid LED performance is typically dependent on several factors including QD/polymer ratio, active layer morphology, charge carrier mobility and charge carrier injection into QDs. The results presented here indicate that among many other factors the HOMO energy of the polymer has a significant influence on device performance. As a consequence, polymers with appropriately low HOMO levels should be used in the active layers of hybrid polymer/QD LEDs in order to fabricate efficient devices. Polymers P2 and P3 with HOMO levels of 5.78 eV and 5.82 eV, respectively, led to devices with high efficiencies which are comparable to the efficiencies of QD only devices fabricated utilizing analogous architectures.36 However, a HOMO level of 5.52 eV determined for polymer P1 is thought to be too high and led to a device with diminished performance.
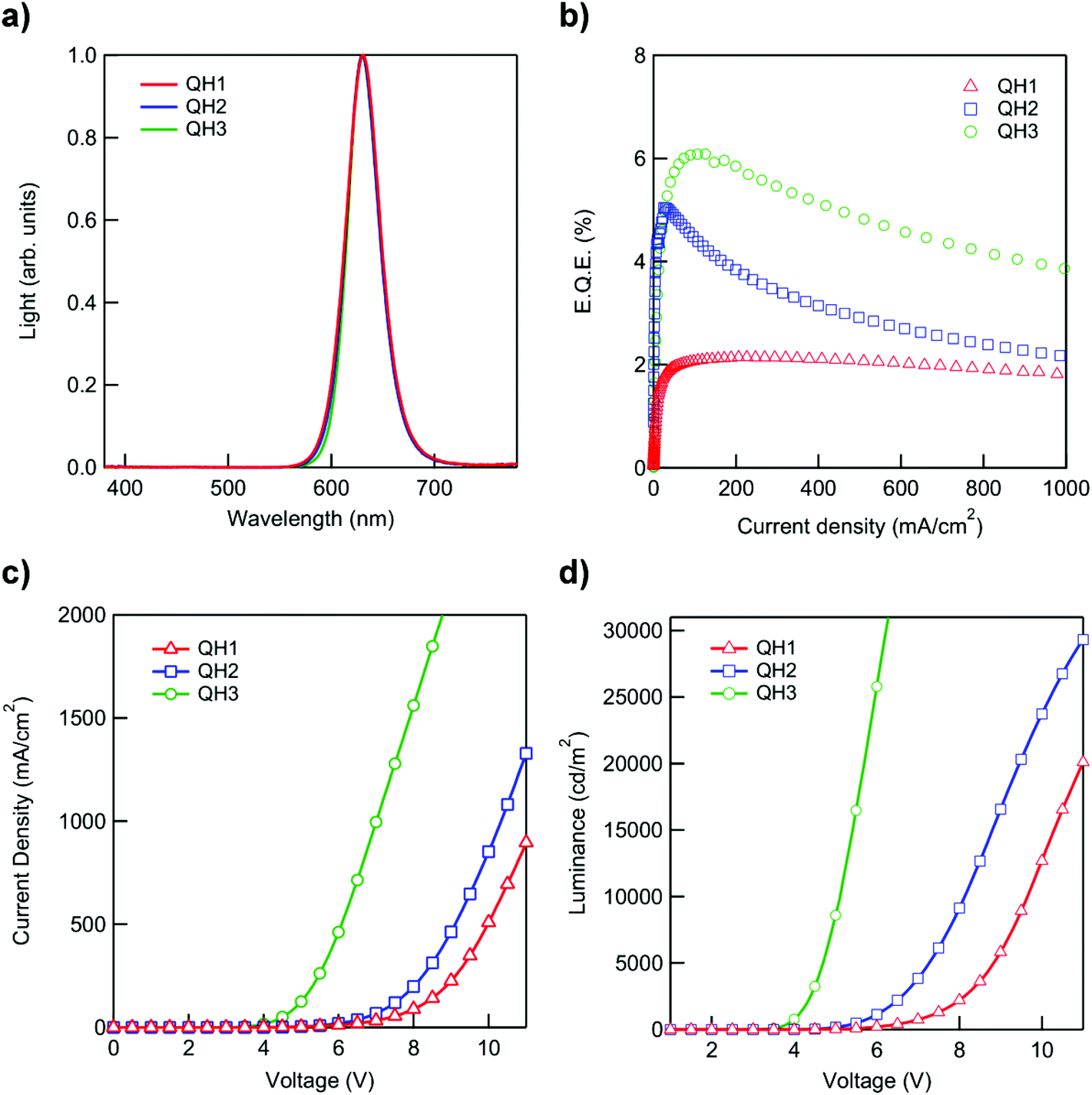 | ||
| Fig. 3 Device characteristics with QD/polymer hybrids QH1–QH3 as the active layer. (a) Normalized electroluminescence intensity versus emission wavelength, (b) external quantum efficiency versus current density, (c) J–V characteristic, and (d) luminance versus voltage characteristics. | ||
Conclusion
We have presented the synthesis of three carbazole-based monomers and their corresponding side-chain conjugated polymers for use in hybrid polymer/QD LEDs. Through RAFT polymerization, polymers with defined molecular weights, narrow polydispersities and well defined end groups were prepared. Depending on the substituents of the carbazole unit, we were able to tune the thermal, optical and, electrochemical properties of the polymers. The polymer disulfide end group enabled the chemical blending of the polymers onto a QD surface via the ligand exchange procedure leading to the formation of films with uniform QD distribution within a polymer matrix. Chemically blended QD/polymer hybrids were then tested as the active layers in LED devices and devices with high efficiencies and narrow electroluminescence profiles, solely composed of QD emission, were fabricated. Moreover, the importance of polymer HOMO level adjustment for the development of efficient hybrid LEDs has been demonstrated. We believe that the side-chain conjugated polymers presented here and the corresponding hybrids demonstrate a promising platform for hybrid LEDs. Further investigations into the role of other aspects such as active layer morphology and composition on device performance are currently underway.Acknowledgements
This work was supported by the International Research Training Group (IRTG 1404) Program (Grant No. 2014001836 funded by NRF and DFG). AF thanks Florian Menk and Younghun Sung and acknowledges the financial support from graduate school MAINZ and is a recipient of the fellowship through the Excellence Initiative (DFG/GSC 266). YL thanks Myeongjin Park and acknowledges the financial support from the Industrial Strategic Technology Development Program (10045145) funded by MOTIE, Korea and also supported by the Human Resources Development program (no. 20124010203170) of the KETEP grant funded by the Korea government Ministry of Trade, Industry, and Energy. KC acknowledges the financial support from NRF of Korean for the National Creative Research Initiative Program (Grant No. 2010-001829) and the Gutenberg Research College fellowship. WKB acknowledges KIST internal funding (2E25393).References
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 933–933 CrossRef.
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354–357 CrossRef CAS.
- P. Anikeeva, C. Madigan, J. Halpert, M. Bawendi and V. Bulović, Phys. Rev. B: Condens. Matter, 2008, 78, 1–8 CrossRef.
- B. S. Mashford, M. Stevenson, Z. Popovic, C. Hamilton, Z. Zhou, C. Breen, J. Steckel, V. Bulovic, M. Bawendi, S. Coe-Sullivan and P. T. Kazlas, Nat. Photonics, 2013, 7, 407–412 CrossRef CAS.
- L. Qian, Y. Zheng, J. Xue and P. H. Holloway, Nat. Photonics, 2011, 5, 543–548 CrossRef CAS.
- J. Kwak, W. K. Bae, D. Lee, I. Park, J. Lim, M. Park, H. Cho, H. Woo, D. Y. Yoon, K. Char, S. Lee and C. Lee, Nano Lett., 2012, 12, 2362–2366 CrossRef CAS PubMed.
- Y. Yang, Y. Zheng, W. Cao, A. Titov, J. Hyvonen, J. R. Manders, J. Xue, P. H. Holloway and L. Qian, Nat. Photonics, 2015, 9, 259–266 CAS.
- J. Kwak, W. K. Bae, D. Lee, I. Park, J. Lim, M. Park, H. Cho, H. Woo, D. Y. Yoon, K. Char, S. Lee and C. Lee, Nano Lett., 2012, 12, 2362–2366 CrossRef CAS PubMed.
- K.-S. Cho, E. K. Lee, W.-J. Joo, E. Jang, T.-H. Kim, S. J. Lee, S.-J. Kwon, J. Y. Han, B.-K. Kim, B. L. Choi and J. M. Kim, Nat. Photonics, 2009, 3, 341–345 CrossRef CAS.
- W. K. Bae, Y.-S. Park, J. Lim, D. Lee, L. a Padilha, H. McDaniel, I. Robel, C. Lee, J. M. Pietryga and V. I. Klimov, Nat. Commun., 2013, 4, 2661 Search PubMed.
- K. S. Leck, Y. Divayana, D. Zhao, X. Yang, A. P. Abiyasa, E. Mutlugun, Y. Gao, S. Liu, S. T. Tan, X. W. Sun and H. V. Demir, ACS Appl. Mater. Interfaces, 2013, 5, 6535–6540 CAS.
- X. Dai, Z. Zhang, Y. Jin, Y. Niu, H. Cao, X. Liang, L. Chen, J. Wang and X. Peng, Nature, 2014, 515, 96–99 CrossRef CAS PubMed.
- W. K. Bae, J. Lim, M. Zorn, J. Kwak, Y.-S. Park, D. Lee, S. Lee, K. Char, R. Zentel and C. Lee, J. Mater. Chem. C, 2014, 2, 4974 RSC.
- Y. Li, A. Rizzo, M. Mazzeo, L. Carbone, L. Manna, R. Cingolani and G. Gigli, J. Appl. Phys., 2005, 97 Search PubMed.
- K. Kwak, K. Cho and S. Kim, Appl. Phys. Lett., 2014, 104, 1–5 CrossRef.
- J. Zhou, W. Q. Tang, C. F. Wang, L. Chen, Q. Chen and S. Chen, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3736–3742 CrossRef CAS.
- H. Jung, W. Chung, C. H. Lee and S. H. Kim, J. Nanosci. Nanotechnol., 2012, 12, 5407–5411 CrossRef CAS PubMed.
- P. T. K. Chin, R. a. M. Hikmet and R. a. J. Janssen, J. Appl. Phys., 2008, 104, 013108 CrossRef.
- J. Kwak, W. K. Bae, M. Zorn, H. Woo, H. Yoon, J. Lim, S. W. Kang, S. Weber, H.-J. Butt, R. Zentel, S. Lee, K. Char and C. Lee, Adv. Mater., 2009, 21, 5022–5026 CrossRef CAS PubMed.
- L. Zhao, Z.-L. Zhou, Z. Guo, G. Gibson, J. A. Brug, S. Lam, J. Pei and S. S. Mao, J. Mater. Res., 2012, 27, 639–652 CrossRef.
- J. Y. Park and R. C. Advincula, Phys. Chem. Chem. Phys., 2014, 16, 8589–8593 RSC.
- M. Zorn, W. K. Bae, J. Kwak, H. Lee, C. Lee, R. Zentel and K. Char, ACS Nano, 2009, 3, 1063–1068 CrossRef CAS PubMed.
- Z.-S. Guo, L. Zhao, J. Pei, Z.-L. Zhou, G. Gibson, J. Brug, S. Lam and S. S. Mao, Macromolecules, 2010, 43, 1860–1866 CrossRef CAS.
- F. Mathias, A. Fokina, K. Landfester, W. Tremel, F. Schmid, K. Char and R. Zentel, Macromol. Rapid Commun., 2015, 36, 959–983 CrossRef CAS PubMed.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4218–4233 CrossRef CAS PubMed.
- S. Gong, C. Yang and J. Qin, Chem. Soc. Rev., 2012, 41, 4797–4807 RSC.
- L. zur Borg, D. Lee, J. Lim, W. K. Bae, M. Park, S. Lee, C. Lee, K. Char and R. Zentel, J. Mater. Chem. C, 2013, 1, 1722 RSC.
- A. K. Bansal, M. T. Sajjad, F. Antolini, L. Stroea, P. Gečys, G. Raciukaitis, P. André, A. Hirzer, V. Schmidt, L. Ortolani, S. Toffanin, S. Allard, U. Scherf and I. D. W. Samuel, Nanoscale, 2015, 7, 11163–11172 RSC.
- J. Liu, L. Li and Q. Pei, Macromolecules, 2011, 44, 2451–2456 CrossRef CAS.
- J. H. Park, T. W. Koh, J. Chung, S. H. Park, M. Eo, Y. Do, S. Yoo and M. H. Lee, Macromolecules, 2013, 46, 674–682 CrossRef CAS.
- J. Ha Park, T.-W. Koh, Y. Do, M. Hyung Lee and S. Yoo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2356–2365 CrossRef CAS.
- J. H. Park, C. Yun, T.-W. Koh, Y. Do, S. Yoo and M. H. Lee, J. Mater. Chem., 2011, 21, 5422 RSC.
- D. D. Evanoff, J. B. Carroll, R. D. Roeder, Z. J. Hunt, J. R. Lawrence and S. H. Foulger, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7882–7897 CrossRef CAS.
- F. Mathias, M. N. Tahir, W. Tremel and R. Zentel, Macromol. Chem. Phys., 2014, 215, 604–613 CrossRef CAS.
- J. Lim, B. G. Jeong, M. Park, J. K. Kim, J. M. Pietryga, Y.-S. Park, V. I. Klimov, C. Lee, D. C. Lee and W. K. Bae, Adv. Mater., 2014, 26, 8034–8040 CrossRef CAS PubMed.
- Q. Zhang, J. Chen, Y. Cheng, L. Wang, D. Ma, X. Jing and F. Wang, J. Mater. Chem., 2004, 14, 895 RSC.
- J. Kwak, Y.-Y. Lyu, H. Lee, B. Choi, K. Char and C. Lee, J. Mater. Chem., 2012, 22, 6351 RSC.
- P. Schrögel, A. Tomkevičienė, P. Strohriegl, S. T. Hoffmann, A. Köhler and C. Lennartz, J. Mater. Chem., 2011, 21, 2266–2273 RSC.
- A. Iraqi, D. F. Pickup and H. Yi, Chem. Mater., 2006, 18, 1007–1015 CrossRef CAS.
- P. J. Roth, K.-S. Kim, S. H. Bae, B.-H. Sohn, P. Theato and R. Zentel, Macromol. Rapid Commun., 2009, 30, 1274–1278 CrossRef CAS PubMed.
- M.-H. Tsai, Y.-H. Hong, C.-H. Chang, H.-C. Su, C.-C. Wu, A. Matoliukstyte, J. Simokaitiene, S. Grigalevicius, J. V. Grazulevicius and C.-P. Hsu, Adv. Mater., 2007, 19, 862–866 CrossRef CAS.
- S.-J. Yeh, M.-F. Wu, C.-T. Chen, Y.-H. Song, Y. Chi, M.-H. Ho, S.-F. Hsu and C. H. Chen, Adv. Mater., 2005, 17, 285–289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01492a |
| This journal is © The Royal Society of Chemistry 2016 |