
Metal induced self-assembly of designed V-shape protein into 2D wavy supramolecular nanostructure†
S. P.
Qiao
,
C.
Lang
,
R. D.
Wang
,
X. M.
Li
,
T. F.
Yan
,
T. Z.
Pan
,
L. L.
Zhao
,
X. T.
Fan
,
X.
Zhang
,
C. X.
Hou
,
Q.
Luo
,
J. Y.
Xu
and
J. Q.
Liu
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: junqiuliu@jlu.edu.cn
First published on 16th November 2015
Abstract
In order to understand and imitate the more complex bio-processes and fascinating functions in nature, protein self-assembly has been studied and has attracted more and more interest in recent years. Artificial self-assemblies of proteins have been constructed through many strategies. However, the design of complicated protein self-assemblies utilizing the special profile of building blocks remains a challenge. We herein report linear and 2D nanostructures constructed from a V shape SMAC protein and induced by metal coordination. Zigzag nanowires and wavy 2D nanostructures have been demonstrated by AFM and TEM. The zigzag nanowires can translate to a 2D nanostructure with an excess of metal ions, which reveals the step by step assembly process. Fluorescence and UV/Vis spectra have also been obtained to further study the mechanism and process of self-assembly. Upon the protein nanostructure, fluorescence resonance energy transfer (FRET) could also be detected using fluorescein modified proteins as building blocks. This article provides an approach for designing and controlling self-assembled protein nanostructures with a distinctive topological morphology.
Introduction
In nature, self-assembling proteins play key roles in biological functions. They widely control biological events from signal transduction and cell regulation to tissue formation. For example, endocytosis, the process used for packing and transporting external substances into cells, takes place with the help of a clathrin coated vesicle which is assembled by a clathrin protein.1 As the cytoskeleton and muscle composition, microfilaments and myofibrils are formed by the self-assembly of actin protein.2 Histone proteins help a large amount of DNA co-assembly to form chromosomes.3 Therefore, to understand the wisdom of nature, protein self-assembly and functionalization4 have attracted lots of interest. As a more versatile and sophisticated building block, proteins have been successfully arranged into 3D nanostructures by the mediation of protein–protein interactions,5 disulfide bridges,6 covalent bond linkers,7 electrostatic interactions8 and guest–host interactions.9 By using a computational method and designing protein–protein interfaces, David Baker et al. described the design of cage-like protein assemblies.10 Taking advantage of the guest–host interaction between heme and cytochrome b562, 1D and 2D self-assembled protein nanomaterials have been achieved by Takashi Hayashi and coworkers.11 Induced by the FGG tri-peptide tag and cucurbit[8]uril (CB[8]) guest–host interaction, our group also reported the successful construction of enzyme nanowires.12In recent years, metal-mediated interactions have received much attention for the construction of defined protein self-assemblies. Taking advantage of convenient gene mutation, metal coordinating sites could be designed on specific locations on the surface of protein building blocks, providing a strategy to control the orientation of interactions between proteins. F. Akif Tezcan and coworkers13 first reported the successful use of metal coordination interactions14 to direct protein assembly. 2D arrays of protein assembly were formed by designing the coordination motifs and interface of cytochrome b562, revealing a flexible method to control and modulate the specific and complex protein assembly.15 Up to now, metal coordination, as a stable, steerable and reversible interaction, has been proved to be a promising force to contribute towards complex nanostructures.16
Although metal-mediated interactions have been reported to construct self-assemblies, utilization of the evolved V profile of proteins in nature and stable interactions to construct more complicated 1D zigzag nanowires, and corresponding 2D wavy nanostructures which are further self-assembled from 1D zigzag nanowires, has been less well reported. We know that the profile of the building block has a great influence on the morphology of the protein assembly due to the “shape” potentially affecting the growth orientation and local topology. Therefore, more complicated structures can be constructed by simpler computer simulation by making good use of the profile. We select here the SMAC protein, which comes from the mitochondrion of human beings and controls the cell apoptosis process,17 as a building block. Complex zigzag, wavy structures can be constructed by simply arranging the V profile protein head-on and side by side, providing a way of constructing protein self-assembles with a complex morphology. In addition, compared with other fluorescence resonance energy transfer (FRET) systems constructed on protein assemblies in nature, the resulting stable nanostructures seem to be a novel artificial protein scaffold for building FRET systems.
Herein we report a supramolecular system, constructed by a V profile protein as building block and induced by metal coordination interaction. The arch of the SMAC protein is applied to construct zigzag and wavy protein assemblies, and two bis-histidine motifs are accurately designed on each SMAC monomer. Characterized by AFM, TEM and spectra measurements, the morphologies and forming process of the complex protein assemblies have been studied. Furthermore, we modified a pair of donor and acceptor chromophores onto distinguishing building blocks. Fluorescence resonance energy transfer (FRET) on the artificial protein assembly has been demonstrated successfully.
Results and discussion
Design, preparation and characterization of protein self-assembly
Following the approach that the profile of protein and well-designed interactions will affect the morphology of the protein self-assembly, we selected SMAC, a V shape protein, as our building block for the construction of a wavy nanostructure, and well designed stable metal-mediated interactions were used to control the orientation and stabilize the nanostructures. The SMAC protein, consisting of three α helices in each monomer, comes from the mitochondrion of human beings and plays an important role in the apoptosis process,17 and is a C2 symmetry homodimer protein with a 115° angle. Therefore, complicated zigzag and wavy structures could been easily realized by arranging the V profile protein head-on and side by side, demonstrating that the SMAC protein is a suitable candidate for constructing wavy-like protein nanostructures. Previously, a study by Tezcan and coworkers demonstrated a principle for designing coordination components on a protein surface.18 Taking advantage of computer simulations, we designed two bis-histidine motifs: bis-histidine motif 1 (BH1) H75, H79 and bis-histidine motif 2 (BH2) H137, H141 on each monomer. The four bis-histidine motifs (in one dimer) are arranged in a quadrilateral (Fig. 1a), this assures that the protein assembly could extend in two directions to form a 2D structure when coordinating with metal ions. Based on the arch profile of the engineered SMAC protein and the stable metal-mediated interaction, the novel zigzag and 2D wavy-like nanostructure could be formed.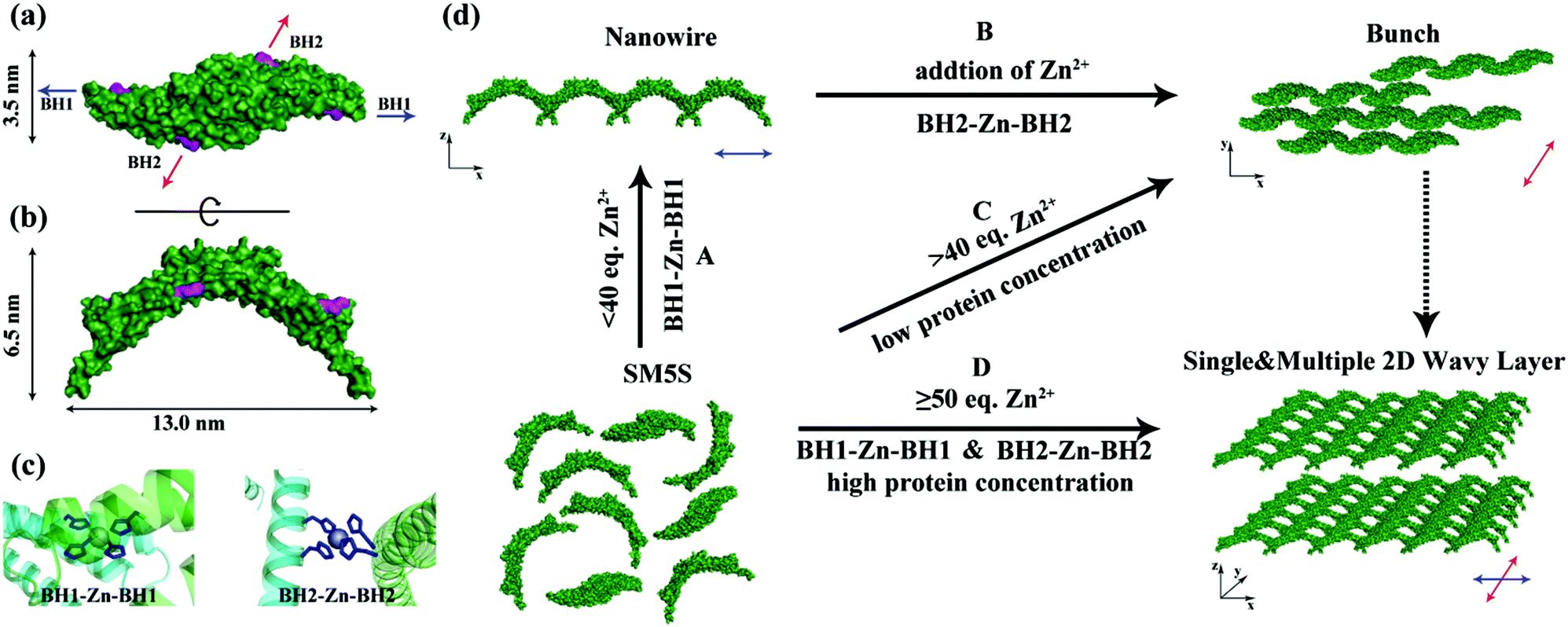 | ||
| Fig. 1 Top view (a) and side view (b) of SMAC-Mutation5Stop (SM5S) structure, four highlighted histidines on each monomer form two bis-histidine motifs: BH1 (H75, H79) and BH2 (H137, H141). The direction of the arrow shows the orientation of the bis-histidine motifs bonding with Zn2+. (c) The BH1–Zn–BH1 and BH2–Zn–BH2 centres when two SM5S are equipped with Zn2+ at the BH1 and BH2 sites. (d) Different morphologies are obtained in different conditions. 1D nanowires were found when the bis-histidine motifs were unsaturated with Zn2+, the blue arrow shows the extending direction of the nanowire benefiting from the BH1–Zn–BH1 bond (process A); the bunch assemblies germinate due to the nanowires attaching with each other through BH2–Zn–BH2 interactions, the direction of growth is shown by the red arrow (process B); when the bis-histidine motifs were saturated with Zn2+, the assemblies extended in both the directions of the blue and red arrows to form 2D nanostructures (process D). | ||
The protein with two bis-histidine motifs (SMAC-Mutation5-Stop) was engineered via site-mutation. The preparation, purification and characterization of SMAC-Mutation5-Stop (SM5S) and another protein variation, SMAC-Mutation4-Stop (SM4S) protein are reported in the SI (Fig. S1†). The final engineered protein, SM5S, was characterized by Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS, Fig. S2†): 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 689.938 Da compared with the wild type SMAC: 20429 Da, confirming the successful preparation of two variations. The circular dichroism spectrum was carried out to detect CD signals at neutral pH (Fig. S3†). The data show no difference between the three proteins, revealing that SM5S and SM4S maintain the same structure. The morphology of the SMAC, a V shape 13 nm in width, 6.5 nm in height and 3.5 nm in thickness from the crystal structure of wild type SMAC, is shown in Fig. 1a and b.17
689.938 Da compared with the wild type SMAC: 20429 Da, confirming the successful preparation of two variations. The circular dichroism spectrum was carried out to detect CD signals at neutral pH (Fig. S3†). The data show no difference between the three proteins, revealing that SM5S and SM4S maintain the same structure. The morphology of the SMAC, a V shape 13 nm in width, 6.5 nm in height and 3.5 nm in thickness from the crystal structure of wild type SMAC, is shown in Fig. 1a and b.17
To prove the formation of protein self-assembly and understand the significant role of Zn2+, we used dynamic light scattering (DLS) to analyze the size distribution of the protein assemblies. After incubation overnight at a neutral pH with different concentrations of Zn2+, the size-intensity curve of the SM5S sample without Zn2+ showed only one peak at about 10 nm, corresponding to the hydration radius of the dissociative dimer. In contrast, the distribution of the sample with 20 equivalents of Zn2+ obviously moves towards a larger size, and larger protein aggregates (above 1000 nm) are found with 50 and 100 equivalents of Zn2+ (Fig. S4b†). The DLS results demonstrate that the metal/protein ratio plays a vital role in the assembly. Another study was performed to understand the effects of reaction time on the size of assemblies. At the beginning, the protein dispersed in solution as a dissociative dimer, and after 1 h, the peak in the intensity curve moved toward 24 nm, and the size of protein aggregation was enhanced inconspicuously after longer periods of time (Fig. S4c†). Therefore, the size of protein self-assembly mainly depends on the metal/protein ratio. Interestingly, the large aggregates disappear after adding the chelator EDTA into the solution, revealing that the protein aggregates can self-assemble and disassemble reversibly through metal coordination (Fig. S4a†).
After the characterization of the size distribution by DLS measurements, the morphology of the metal induced protein assemblies were studied by atomic force microscopy (AFM). Dissociated bundled zigzag nanowires (Fig. 2), and 2D nanostructures with single, multiple layers (Fig. 3) are shown in the AFM images. Notably, the height of a typical dissociative zigzag nanowire in the AFM images is 3.1 nm, which is same as the thickness of the single protein SM5S (3.5 nm), while the height of bundled zigzag nanowires and the 2D wavy structure is about 4.3–6.6 nm, corresponding to the height of the protein (6.5 nm). We assume that the SM5S building blocks in the dissociative nanowires may lie flat on the silicon wafer (Fig. 2a), while the protein in the bundled zigzag nanowires and 2D wavy nanostructures stand on the silicon wafer (Fig. 3a). In addition, these nanowires prefer to attach with each other on identical planes rather than aggregate stochastically, indicating that there is a special interaction in the perpendicular orientation. At a high metal/protein ratio (metal:protein is 50), the protein self-assemblies are observed by a high resolution transmission electron microscope (TEM). The zoomed TEM images show the structural details and the “monomer” SM5S can also be found clearly in both 2D nanostructures. When the protein concentration is low (1 μM), the bundles of nanowires are close-packed in the same plane (Fig. 4a and b). The width of each nanowire is about 3.1 nm, which corresponds to the thickness of SM5S (3.5 nm). When the concentration of protein is 10 μM, a 2D wavy array is formed through extension in two directions of the SM5S building blocks, induced by a coordination interaction (Fig. 4c). Therefore, depending on the AFM and TEM results, we suppose that 2D nanostructures/arrays are formed by the close-packing of dissociative nanowires and protein building blocks self-assembling in two directions, respectively.
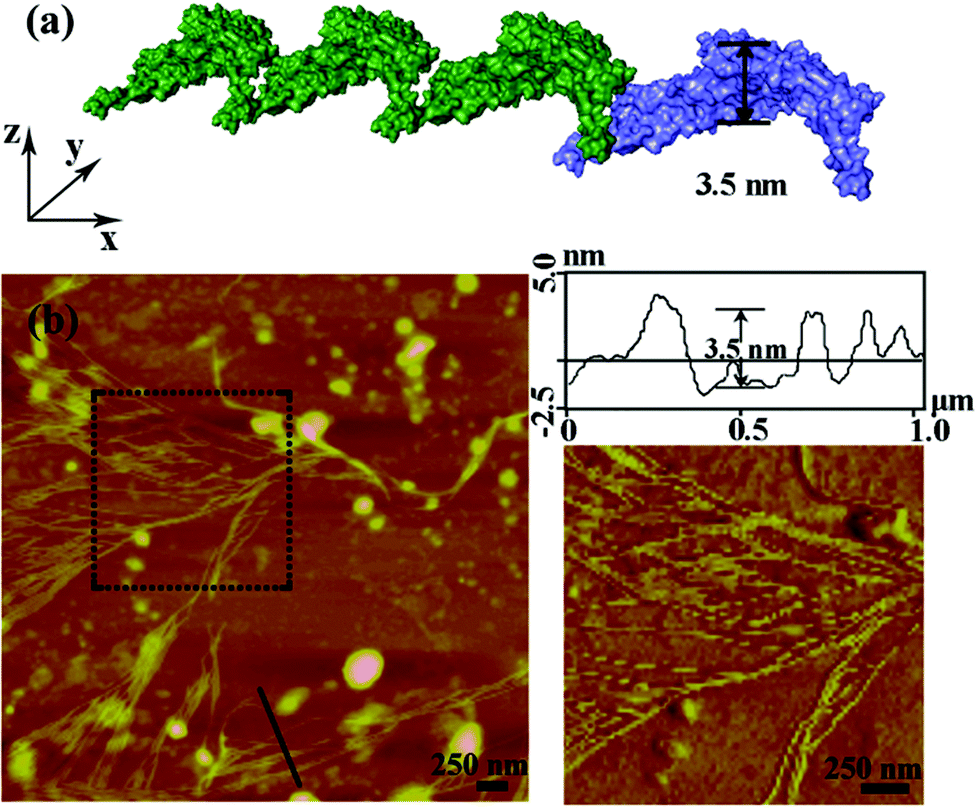 | ||
| Fig. 2 Zn2+ induced self-assembly of SM5S detected by AFM with a low metal/protein ratio. (a) The height of simulative assembly in the PyMOL software corresponds to the width of SM5S, 3.5 nm. (b) The AFM result when 20 equivalents of Zn2+ were added to SM5S, and dissociated nanowires were observed. | ||
 | ||
| Fig. 3 AFM images of Zn2+ induced 2D protein nanostructures with a high metal/protein ratio. (a) The height of simulative assembly in the PyMOL software corresponds to the height of SM5S, 6.5 nm. (b) Upon increasing the metal/protein ratio to 40:1, the linear zigzag assemblies further aggregate into 2D bundled nanowires. (c) With excess Zn2+ (50 eq.), large 2D-wavy single and multiple layers of protein assemblies were found. The concentration of SM5S in (b) is 1 μM while the concentration in (c) is 10 μM. | ||
 | ||
| Fig. 4 TEM images of protein assemblies when the Zn2+/protein ratio is 50. The structures shown in (a) and (b) consist of several strands of nanowires. The width of each strand is about 3.0 nm. (b) The monomer (SM5S) of the nanowire is observed clearly, indicating that SM5S may first form linear assemblies through the BH1–Zn–BH1 centre, and then aggregate through BH2–Zn–BH2. Figure (c) demonstrates the detail of the 2D arrays, the protein could extend in two directions and a wavy structure is clearly observed. Figure (d) shows the cartoon image illustrating the two formation modes of 2D nanostructures. All of scale bars represent 10 nm. | ||
Understanding the process of self-assembling
To understand the processes of protein assembly, we first focus on the two bis-histidine motifs of the engineered protein. With the help of two Zn2+ coordination sites, it should be possible to form protein assemblies in a proper orientation. There are mainly three modalities to form Zn2+-bonding-histidine centres between different proteins based on spatial resistance: (1) BH2 from one and BH2 from another; (2) BH1 from one protein and BH1 from another; and (3) BH1 from one and BH2 from another. Considering that the protein would form a huge helix self-assembly by the third method, CD measurement was performed (Fig. S3b†). However, the signal which could prove the existence of the helix structure was not found, revealing that it is difficult to form BH1–Zn–BH2. Thus, the protein assembly is possibly induced by BH1–Zn–BH1 and BH2–Zn–BH2 bonds.Then, the protein assembly system was analyzed by fluorescence and UV/Vis titration to further study the assembly process. We note that SM5S has intrinsic fluorescence due to the existence of Trp residues. Taking advantage of the Trp 78 residue next to BH1, fluorescence titration of SM5S can be employed to discuss the coordination and self-assembly mechanism.19 When excited at 278 nm, a fluorescence signal was recorded from 300 nm to 400 nm at a neutral pH. Fig. 5 shows that adding Zn2+ quenches the fluorescence intensity of SM5S (mutation site: D75H, Q79H, L137H, E141H), while another SMAC variant SM4S (mutation site: E76H, Q79H, L137H, E141H) quenches less (Fig. 5c and d). This indicates that the quenching of fluorescence only depends on the interaction between Zn2+ and the BH1 motif. We assume that the interaction between Zn2+ and the bis-histidine motifs may induce the combination of SM5S proteins at BH1, forming a pair of Trp 78 residues with a distance of 1.3 nm (Fig. 5a and b). The Zn2+ between the Trp pair changes the microenvironment of the Trp residue, resulting in the quenching of the fluorescence signal. In contrast, the fluorescence titration of SM4S is quenched slightly, possibly due to the longer distance between the Trp residue pair and Zn2+ or the different binding modality. Thus, all fluorescence measurements reveal that SM5S proteins prefer to connect with each other through the BH1–Zn–BH1 centre.
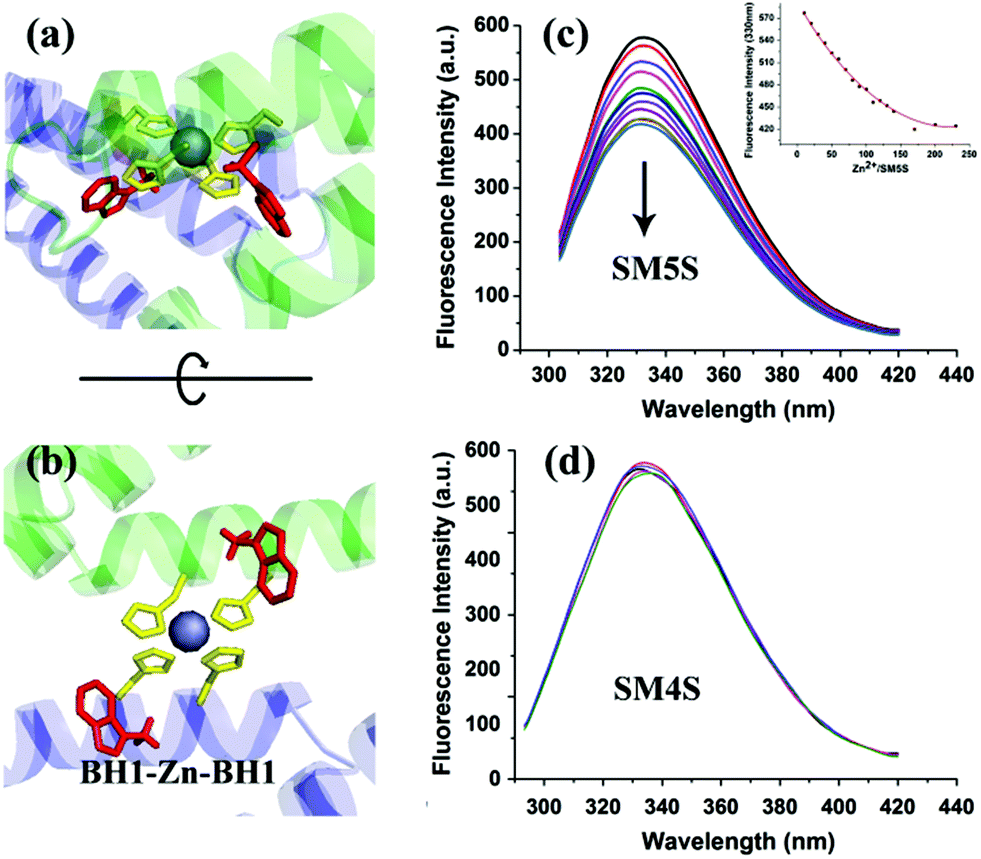 | ||
| Fig. 5 (a and b) views of BH1–Zn–BH1 centre, the highlighted residues are two BH1 sites (yellow). The distance between two Trp residues (red) is about 1.3 nm. Zn2+ in the centre, which changes the microenvironment of the Trp residues, results in the fluorescence quenching of Trp. Fluorescence titrations of SM5S (c) and SM4S (d) are performed by adding Zn2+, the fluorescence intensity of SM5S drops obviously with an increase in Zn2+, while the signal of SM4S hardly drops. | ||
The UV/Vis titration results (Fig. 6) show an increasing UV absorbance of SM5S with a higher Zn2+ concentration. We found that the increased rate of absorbance at 275 nm changes obviously when the metal/protein ratio is about 40 (Fig. 6b); this finding indicates that there are two stages of aggregation when Zn2+ induces the protein to form assemblies. Zn2+ tends to bond the BH1 motifs of two SM5S molecules first to form a BH1–Zn–BH1 centre, and then form BH2–Zn–BH2 centres.
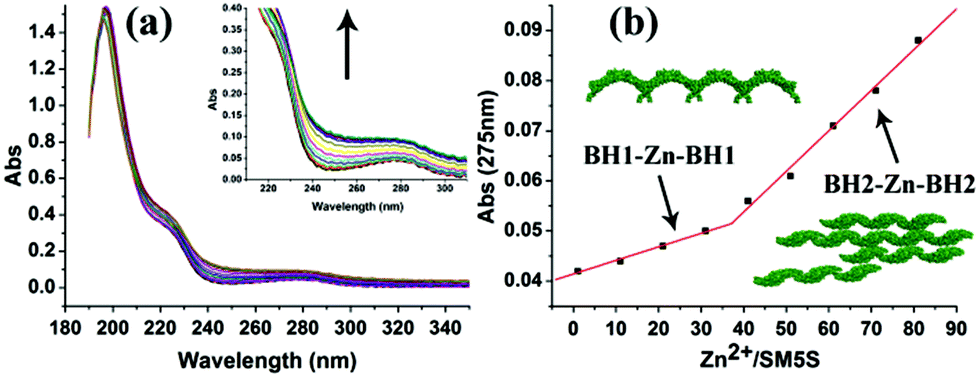 | ||
| Fig. 6 (a) UV/Vis titration spectra of SM5S employed with increasing Zn2+. The UV absorbance rises upon adding more Zn2+. (b) The curve which relates the UV absorbance intensity at 275 nm to different Zn2+/protein ratios, shows a clear turn when the Zn2+/protein ratio is above 40. The different slope reveals different bonding and aggregation modes.20 | ||
Based on all the results, we suppose that there are two assembly pathways emerging in our protein assembly system which depend on the saturation of the bis-histidine motifs; when the bis-histidine motifs are unsaturated, Zn2+ prefers to bind to the BH1 motif, and the proteins first assemble into linear zigzag nanowires through BH1–Zn–BH1 interactions. With an excess of Zn2+, the nanowires then attach to each other through BH2–Zn–BH2 linkages to form bundling nanowires. This step by step process is also proved by AFM and TEM; while the bis-histidine motifs are saturated and the protein concentration is high, the protein can extend in two directions (along the BH1 and BH2 motifs) to form 2D nanostructures, which is also observed by AFM. Due to the V profile of the building blocks, the 2D structures prefer a wavy rather than a planar morphology. The complicated protein assemblies have also been verified by TEM. (Fig. 4).
Stability of protein assembly
Because the protein assembly is induced by metal coordination interactions at multiple sites, the final compact 2D nanostructures may stabilize the protein monomers. We expected that the protein assembly would be more tolerant of a high temperature and denaturing agent. Thus, CD measurements with increasing concentrations of denaturing agent (guanidine hydrochloride) were employed with both the SM5S protein and the protein assembly induced by metal coordination to investigate the stability of the supramolecular assemblies. After incubating in guanidine hydrochloride at an objective concentration for 10 min, the CD measurement of the SM5S protein (5 μM) is recorded. Fig. S12† shows that the ratio of the CD signal at 208 nm and 222 nm begins to rise fast, which demonstrates that the α helix of the SM5S protein starts to unfold, whereas the 222 nm/208 nm ratio rises slowly when adding the denaturing agent into the metal induced protein assembly system, revealing that the protein in the self-assembling system could be harder to unfold compared with the monomer protein in the same concentration of denaturing agent. The metal induced assemblies are more stable than the protein monomer until the concentration of guanidine hydrochloride reaches 2 M. Meanwhile, the thermal stability of the protein nanostructure is also analysed by circular dichroism tests. CD spectra of SM5S and the protein assembly were recorded at intervals of 5 degrees from 30 to 95 degree. The folded-denature curves show that Tm has raised from 65 to 85 degrees, revealing an improvement in the thermal stability (Fig. S13†). These results demonstrate that assembly through metal coordination increases the stability of the system, and also makes this system a potential candidate as a biomaterial. The stability may benefit from the limitation of protein unfolding which results from the metal tidily combining between protein monomers. Notably, this phenomenon provides an approach for further application in biomaterials, without the limitation of instability.FRET of protein assembly
Light harvesting systems play a vital role in nature due to their ability to convert light to chemical substances.21 Energy transfer, which is one important process in a light harvesting system, is realized through a fluorescence resonance energy transfer (FRET) event between chromophores with a fine arrangement. To imitate the arrays of regular chromophores, Matthew B. Francis and coworkers first successfully constructed a series of outstanding light harvesting systems using a modified nature protein assembly as a scaffold.22 The highly ordered protein self-assembly could hang chromophores for optimal energy transfer and prevent contact quenching in the meantime. Therefore, a protein assembly would be an appropriate scaffold for the construction of a FRET system.The highly ordered arrangement and stability makes our artificial protein self-assembly an ideal scaffold for the construction of a light harvesting system. Thus, we constructed two kinds of protein building blocks by connecting an amino-reactive chromophore-isothiocyanate (donor and acceptor) to the SM5S protein. After being induced by metal-mediated interactions, a chromophore-modified protein self-assembly could be formed, and on account of the close space between the chromophore-modified proteins, fluorescence resonance energy transfer (FRET) could be realized. It is worth noting that we used a “modified then assembling” rather than an “assembling then modified” schema for the artificial assembly because we could accurately control the chromophore ratio for characterization of our FRET system, even though this schema would affect the assembly efficiency of the building block and the final energy transfer efficiency between the donor and acceptor.
We herein select FITC as the donor and rhodamine B as the acceptor, since the appropriate degree of overlap between the emission of FITC (maximum absorption at 497 nm, maximum emission at 510 nm) and the absorption of rhodamine B (maximum absorption at 554 nm and emission at 584 nm), allows FRET to more easily occur from the donor to the acceptor (shown in Fig. S7†). FITC and rhodamine B were attached to the proteins at pH = 9.0 for 8 h by mild reactions.23 The two modified proteins SF (SM5S-FITC) and SRB (SM5S-rhodamine B) were confirmed by Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (Fig. S8 and S9†), revealing that we obtained two kinds of chromophore-modified proteins. Mixing SF with the SRB protein at different ratios, chromophore-modified protein self-assemblies were prepared by metal-mediated interactions, which is ascertained by the excitation spectra (Fig. S10†). When characterized by TEM, the resulting protein assemblies show a similar morphology (Fig. 7e) to the 2D nanostructure obtained before (Fig. 7d).
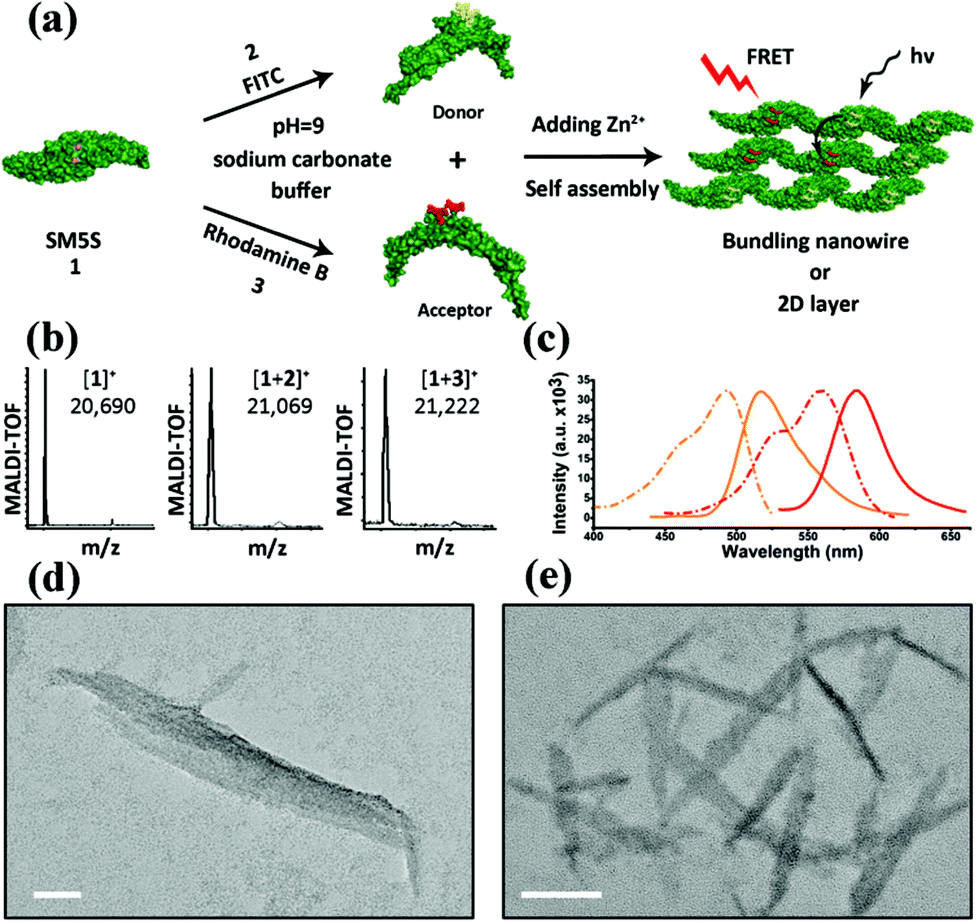 | ||
| Fig. 7 (a) Schema of constructing the FRET system: donor (FITC) and acceptor (rhodamine B) are first linked to SM5S, and protein self-assemblies are then induced by metal coordination. (b) MALDI-TOF results of SM5S, SM5S-FITC (SF) and SM5S-rhodamine B (SRB) reveal that the two chromophores have been linked onto the proteins. (c) Emission (solid line) and excitation (dash line) spectra of SF protein (orange) and SRB protein (red). An obvious overlap between the excitation of SRB and the emission of SF could be observed. (d) TEM images of the SM5S-Zn2+ protein assembly and (e) modified-SM5S-Zn2+ assembly in the FRET system. The morphologies of both assemblies are similar. The scale bars are 50 nm. | ||
To estimate the Förster radius of the two chromophores in the FRET system we constructed, the fluorescence quantum yield of the chromophore-modified proteins (SF and SRB) was determined by the method reported by Williams et al.24 (Fig. S10†). The spectral overlap integral was first calculated using the following equation:25
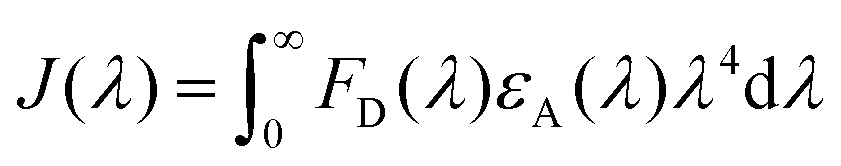
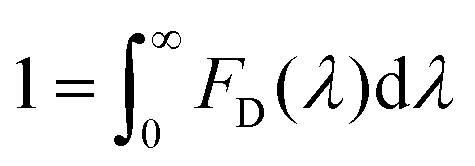
The overlap integral for transfers between FITC and rhodamine B, two FITC molecules and two rhodamine B molecules were calculated to be 4.70 × 1015, 1.92 × 1015 and 4.04 × 1015 M−1 cm−1 nm4, respectively (Table 1).
| Donor–donor/acceptor pair | Φ D (donor) | J (1015 M−1 cm−1 nm4) | R 0 (nm) | r (nm) | E (%) |
|---|---|---|---|---|---|
| SF-SF | 0.256 | 1.90 | 4.35 | 7.93 | 2.7 |
| SRB-SRB | 0.185 | 4.04 | 5.19 | 3.28 | 94 |
| SF-SRB | 0.256 | 4.70 | 5.32 | 4.53 | 72 |
Assuming a value of 2/3 for the orientation value κ2, an aqueous refractive index of η = 1.33, and considering the unit overlap integral J described above, the Förster radius (R0), the theoretical donor–acceptor distance at 50% energy transfer efficiency, was calculated using the following equation:26
| R0 = 0.211(κ2η−4QDJ(λ))1/6 (in Å) |
The donor–acceptor distance (r) was thus estimated to be 4.5 nm by a structural model in the software PyMOL. We finally get the efficiency of energy transfer (E) using the following equation:26
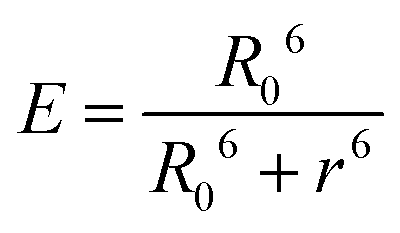
The high efficiency of energy transfer between the FITC and rhodamine B chromophores is 72%.
For detection of the FRET event from FITC to rhodamine B, the emission spectra of the SF-SRB system are detected27 before (Fig. S11†) and after self-assembly induced by metal-mediated interactions (Fig. 8). In order to affirm the quenching of FITC caused by rhodamine B, when an increasing amount of SRB protein and invariant SF protein were mixed and self-assembled through metal coordination, the emission of the donor FITC conspicuously decreased and the emission of the acceptor increased. Meanwhile, the emission of the acceptor could also be enhanced when adding the donor to the system and assembling. All the results reveal that FRET does occur in this system, with the energy transfer from donor to acceptor resulting in the quenching of donor emission and the enhancement of acceptor emission. It also demonstrates that the novel protein nanostructure we designed is a promising artificial scaffold for the construction of the FRET system.
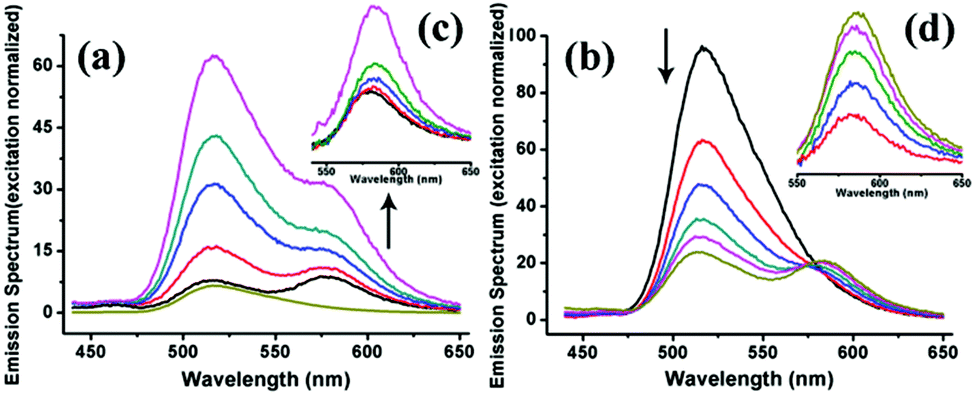 | ||
| Fig. 8 Normalized emission spectra of the FRET system constructed by using SF and SRB proteins as building blocks. (a) The emission of SRB rises with the addition of SF into the system (normalized at 570 nm of acceptor excitation), confirming that FRET events take place. (b) Increasing amounts of the acceptor results in an obvious quenching of the donor and an increase in acceptor emission due to the FRET event. (c, d) The resulting acceptor emission spectra obtained by removing the FITC emission peak from (a) and (b), showing the raised emission of the acceptor. | ||
Conclusion
We herein report the successful construction of a complicated zigzag and 2D-wavy protein self-assembling system, utilizing the V profile of a protein building block and coordination interactions. Zigzag nanowires and 2D wavy nanostructures are obtained at different metal/protein ratios. The self-assembly of proteins has been studied by DLS, AFM, TEM, fluorescent and UV/Vis spectra in order to understand the process of self-assembly and the morphology of the protein self-assemblies. The complex 2D wavy nanostructures benefit from the V profile of the building block and the designed metal coordination interactions, demonstrating an approach for the construction of a complicated protein nanostructure. Upon the greater stability of the protein assembly compared to the dissociative protein monomer, a FRET system has been successfully realized using the artificial protein self-assembly as a scaffold. This shows that the novel designed protein assembly is a promising frame for a light harvesting system, and we anticipate its further application in biomaterials.Experimental procedures
Construction and expression of SMAC variants SMAC-mutation4-stop & SMAC-mutation5-stop
Depending on the computer simulation, the bis-histidine motifs were designed on the surface of the protein, and the protein crystal structure was viewed in property distance and orientation in the software PyMOL. Two variants were obtained through four point mutations; the primers used for point mutation are shown in Table S1.† The final DNA sequence is confirmed by genetic sequencing before expression. Three kinds of protein are expressed in the Escherichia Coli strain BL21(ED3), after incubating in 5 ml LB liquid medium including Kanamycin (100 μg mL−1) overnight, cells were inoculated into 500 ml LB liquid medium containing Kanamycin (100 μg mL−1) until OD600 was about 0.1, and then incubated till OD600 was about 0.6–0.8. Then, IPTG was added to the final concentration of 1 mM and the cells were incubated for 4 hours. After being collected by centrifugation, the cells were ultrasonicated and the sediment was removed by centrifugation. The soluble fraction was purified over anion-exchange chromatography (DEAE) and further purification was performed on G75 and G25 to remove the nucleinic acid and salt.DLS measurement
DLS was performed to study the protein self-assembly process. SM5S was dissolved in a TBS buffer (10 mM Tris, pH8.5), and the final concentration was about 1 μM. Samples were made with different additions of Zn2+ concentration (0 μM, 20 μM, 40 μM, 50 μM, 100 μM) and were incubated overnight prior to the DLS measurement (Fig. S4a†). The influence of the assembly time on the size of protein self-assemblies was also studied by DLS. After being dissolved in TBS and adding 20 eq. Zn2+, the DLS signal of the sample was recorded during 4 hours.AFM imaging
Protein assemblies were made by dissolving SM5S in 15 mM Tris-HCl buffer (pH = 8.5), followed by the addition of Zn2+ and incubation overnight. All the samples were prepared by dropping 10 μl of the solution on a silicon slice which was hydroxylated and adsorbed for 10 min, then the silicon slice was washed three times using water and air dried before being employed on the AFM. During AFM imaging, the tapping mode was used with a scan speed of 0.8 Hz.TEM characterization
TEM imaging was used to study the morphology of protein self-assemblies. The sample was prepared by placing a drop of the samples on a 300-mesh, carbon-coated copper grid for 10 min, and after being negatively stained by 4% Sodium phosphotungstate for 40 s, the samples were dried in air before measurement. The observation was performed with a JEOL1011 transmission electron microscope operating with an acceleration voltage of 200 kV.Spectroscopy measurements
Fluorescence and UV/Vis titrations were performed as follows. SM5S and SM4S were dissolved in TBS with a final concentration of 1 μm, and 1 μl of 10 mM Zn2+ was added to 1 ml of the sample each time. Then, the system was incubated for 2 min before recording the fluorescence signal. The system was excited at 278 nm, both slit widths were 10 nm, and the fluorescence signal was recorded from 300 nm to 400 nm at a pH of 8.5 (10 mM Tris-HCl buffer). Similarly, the UV/Vis spectra were also recorded under the same conditions and the UV results were recorded from 190 nm to 350 nm. To detect the changes on the second structure and stability in a high temperature or organic solvent, CD measurements were employed. All the samples contained 20 mM TBS, 200 mM NaCl and protein of different concentrations. For studying the thermostability, CD signals were recorded from 190 nm to 260 nm where the information of the second structure was contained. Large range CD was also performed to detect the CD signal of protein assemblies.Acknowledgements
This work was supported by the Natural Science Foundation of China (no. 21234004, 21420102007, 21574056, 21221063), 111 project (B06009), and the Chang Jiang Scholars Program of China.Notes and references
- M. K. Higgins and H. T. McMahon, Trends Biochem. Sci., 2002, 27, 257–263 CrossRef CAS PubMed.
- G. J. Doherty and H. T. McMahon, Annu. Rev. Biophys., 2008, 37, 65–95 CrossRef CAS PubMed.
- B. Guillemette, A. R. Bataille, N. Gévry, M. Adam, M. Blanchette, F. Robert and L. Gaudreau, PLoS Biol., 2005, 3, 2100–2110 CAS.
- (a) N. J. M. Sanghamitra and T. Ueno, Chem. Commun., 2013, 49, 4114–4126 RSC; (b) B. Maity, K. Fujita and T. Ueno, Curr. Opin. Chem. Biol., 2015, 25, 88–97 CrossRef CAS PubMed; (c) T. Ueno, J. Mater. Chem., 2008, 18, 3741–3745 RSC.
- (a) J. C. Sinclair, K. M. Davies, C. Vénien-Bryan and M. E. M. Noble, Nat. Nanotechnol., 2011, 6, 558–562 CrossRef CAS PubMed; (b) J. F. Graveland-Bikker, R. I. Koning, H. K. Koerten, R. B. J. Geels, R. M. A. Heerenc and C. G. de Kruif, Soft Matter, 2009, 5, 2020–2026 RSC; (c) C. R. Bourne, S. P. Katen, M. R. Fulz, C. Packianathan and A. Zlotnick, Biochemistry, 2009, 48, 1736–1742 CrossRef CAS PubMed; (d) Y. I. Maeda and H. Matsui, Soft Matter, 2012, 8, 7533–7544 RSC; (e) M. Dalmau, S. Lim and S. Wang, Nano Lett., 2009, 9, 160–166 CrossRef CAS PubMed; (f) Z. Liu, J. Qiao, Z. W. Niu and Q. Wang, Chem. Soc. Rev., 2012, 41, 6178–6194 RSC.
- (a) K. Oohora, S. Burazerovic, A. Onoda, Y. M. Wilson, T. R. Ward and T. Hayashi, Angew. Chem., Int. Ed., 2012, 51, 3818–3821 CrossRef CAS PubMed; (b) W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS PubMed; (c) T. T. H. Pham, P. J. Skrzeszewska, M. W. T. Werten, W. H. Rombouts, M. A. C. Stuart, F. A. deWolfb and J. vander Gucht, Soft Matter, 2013, 9, 6391–6397 RSC.
- (a) M. M. Ma and D. Bong, Org. Biomol. Chem., 2011, 9, 7296–7299 RSC; (b) K. Oohora, A. Onoda and T. Hayashi, Chem. Commun., 2012, 48, 11714–11726 RSC; (c) L. Arce, M. Zosugagh, C. Arce, A. Moreno, A. Ros and M. Valcárcel, Biosens. Bioelectron., 2007, 22, 3217–3223 CrossRef CAS PubMed; (d) Z. Matharu, A. J. Bandodkar, G. Sumana, P. R. Solanki, E. M. I. MalaEkanayake, K. Kaneto, V. Gupta and B. D. Malhotra, J. Phys. Chem. B, 2009, 113, 14405–14412 CrossRef CAS PubMed; (e) A. G. Mantzila, V. Maipa and M. I. Prodromidis, Anal. Chem., 2008, 80, 1169–1175 CrossRef CAS PubMed; (f) A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- (a) L. Miao, J. S. Han, H. Zhang, L. L. Zhao, C. Y. Si, X. Y. Zhang, C. X. Hou, Q. Luo, J. Y. Xu and J. Q. Liu, ACS Nano, 2014, 4, 3743–3751 CrossRef PubMed; (b) C. Chen, K. M. Bromley, J. Moradian-Oldak and J. J. DeYoreo, J. Am. Chem. Soc., 2011, 133, 17406–17413 CrossRef CAS PubMed; (c) M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2013, 8, 52–56 CrossRef CAS PubMed.
- (a) M. Ramaekers, S. P. W. Wijnands, J. L. J. van Dongen, L. Brunsveld and P. Y. W. Dankers, Chem. Commun., 2015, 51, 3147–3150 RSC; (b) J. C. T. Carlson, S. S. Jena, M. Flenniken, T. Chou, R. A. Siegel and C. R. Wagner, J. Am. Chem. Soc., 2006, 128, 7630–7638 CrossRef CAS PubMed; (c) H. Kitagishi, K. Oohora and T. Hayashi, Biopolymers, 2009, 91, 194–200 CrossRef CAS PubMed; (d) H. Kitagishi, Y. Kakikura, H. Yamaguchi, K. Oohora, A. Harada and T. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1271–1274 CrossRef CAS PubMed; (e) K. Oohora, A. Onoda, H. Kitagishi, H. Yamaguchi, A. Haradacand and T. Hayashi, Chem. Sci., 2011, 2, 1033–1038 RSC.
- N. P. King, W. Sheffler, M. R. Sawaya, B. S. Vollmar, J. P. Sumida, I. André, T. Gonen, T. O. Yeates and D. Baker, Science, 2012, 336, 1171–1174 CrossRef CAS PubMed.
- H. Kitagishi, K. Oohora, H. Yamaguchi, H. Sato, T. Matsuo, A. Harada and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 10326–10327 CrossRef CAS PubMed.
- C. X. Hou, J. X. Li, L. L. Zhao, W. Zhang, Q. Luo, Z. Y. Dong, J. Y. Xu and J. Q. Liu, Angew. Chem., Int. Ed., 2013, 52, 5590–5593 CrossRef CAS PubMed.
- (a) R. J. Radford, M. Lawrenz, P. C. Nguyen, J. A. McCammon and F. A. Tezcan, Chem. Commun., 2011, 47, 313–315 RSC; (b) R. J. Radford and F. A. Tezcan, J. Am. Chem. Soc., 2009, 131, 9136–9137 CrossRef CAS PubMed; (c) D. N. Woolfson and Z. N. Mahmoud, Chem. Soc. Rev., 2010, 39, 3464–3479 RSC; (d) A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- (a) D. J. E. Huard, K. M. Kane and F. A. Tezcan, Nat. Chem. Biol., 2013, 9, 169–176 CrossRef CAS PubMed; (b) Y. S. Bai, Q. Luo, W. Zhang, L. Miao, J. Y. Xu, H. B. Li and J. Q. Liu, J. Am. Chem. Soc., 2013, 135, 10966–10969 CrossRef CAS PubMed.
- J. D. Brodin, J. R. Carr, P. A. Sontz and F. A. Tezcan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2897–2902 CrossRef CAS PubMed.
- J. D. Brodin, X. I. Ambroggio, C. Tang, K. N. Parent1, T. S. Baker and F. A. Tezcan, Nat. Chem., 2012, 4, 375–382 CrossRef CAS PubMed.
- (a) J. J. Chai, C. Y. Du, J. W. Wu, S. Kyin, X. D. Wang and Y. G. Shi, Nature, 2000, 406, 855–862 CrossRef CAS PubMed; (b) H. Y. Sun, Z. Nikolovska-Coleska, J. F. Lu, J. L. Meagher, C. Y. Yang, S. Qiu, Y. Tomita, Y. Ueda, S. Jiang, K. Krajewski, P. P. Roller, J. A. Stuckey and S. M. Wang, J. Am. Chem. Soc., 2007, 129, 15279–15294 CrossRef CAS PubMed; (c) D. R. Green, Cell, 2000, 102, 1–4 CrossRef CAS PubMed; (d) D. Potenza, L. Belvisi, F. Vasile, E. Moroni, F. Cossu and P. Seneci, Org. Biomol. Chem., 2012, 10, 3278–3287 RSC.
- E. N. Salgado, R. J. Radford and F. A. Tezcan, Acc. Chem. Res., 2010, 43, 661–672 CrossRef CAS PubMed.
- (a) R. Yang, L. Chen, T. Zhang, S. Yang, X. Leng and G. Zhao, Chem. Commun., 2014, 50, 481–483 RSC; (b) H. Shen, Z. Gu, K. Jian and J. Qi, J. Pharm. Biomed., 2013, 75, 86–93 CrossRef CAS PubMed; (c) F. Bou-Abdallah, G. H. Zhao, G. Biasiotto, M. Poli, P. Arosio and N. D. Chasteen, J. Am. Chem. Soc., 2008, 130, 17801–17811 CrossRef CAS PubMed; (d) R. Yang, L. Chen, S. Yang, C. Lv, X. Leng and G. Zhao, Chem. Commun., 2014, 50, 2879–2882 RSC.
- (a) C. C. Decandio, E. R. Silva, I. W. Hamley, V. Castelletto, M. S. Liberato, V. X. Oliveira Jr., C. L. P. Oliveira and W. A. Alves, Langmuir, 2015, 31, 4513–4523 CrossRef CAS PubMed; (b) C. Yuan, J. Chen, S. Yu, Y. Chang, J. Mao, Y. Xu, W. Luo, B. Zeng and L. Dai, Soft Matter, 2015, 11, 2243–2250 RSC.
- (a) X. D. Liu and Y. G. Shen, FEBS Lett., 2004, 569, 337–340 CrossRef CAS PubMed; (b) B. Andersson, D. H. Yang and H. Paulsen, FEBS Lett., 2000, 466, 385–388 CrossRef.
- (a) T. L. Schlick, Z. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS PubMed; (b) R. A. Miller, A. D. Presley and M. B. Francis, J. Am. Chem. Soc., 2007, 129, 3104–3109 CrossRef CAS PubMed; (c) R. A. Miller, N. Stephanopoulos, J. M. McFarl, A. S. Rosko, P. L. Geissler and M. B. Francis, J. Am. Chem. Soc., 2010, 132, 6068–6074 CrossRef CAS PubMed; (d) M. T. Dedeo, K. E. Duderstadt, J. M. Berger and M. B. Francis, Nano Lett., 2010, 10, 181–186 CrossRef CAS PubMed; (e) A. C. Obermeyer, S. L. Capehart, J. B. Jarman and M. B. Francis, PLoS One, 2014, 9, e100678 Search PubMed.
- V. Levi and F. L. Gonzalez Flecha, BBA, 2002, 1599, 141–148 CrossRef CAS.
- L. Mottram, S. Boonyarattanakalin, R. E. Kovel and B. R. Peterson, Org. Lett., 2006, 8, 581–584 CrossRef CAS PubMed.
- A. Montali, G. S. Harms, A. Renn, C. Weder, P. Smith and U. P. Wild, Phys. Chem. Chem. Phys., 1999, 1, 5697–5702 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 1983, pp. 446–447 Search PubMed.
- (a) N. Stephanopoulos, Z. M. Carrico and M. B. Francis, Angew. Chem., Int. Ed., 2009, 48, 9498–9502 CrossRef CAS PubMed; (b) Y. S. Nam, T. Shin, H. Park, A. P. Magyar, K. Choi, G. Fantner, K. A. Nelsonand and A. M. Belcher, J. Am. Chem. Soc., 2010, 132, 1462–1463 CrossRef CAS PubMed; (c) F. Pu, L. Wu, E. G. Ju, X. Ran, J. S. Ren and X. G. Qu, Adv. Funct. Mater., 2014, 24, 4549–4555 CrossRef CAS; (d) Y. Zhang, H. Y. Zhang, J. Hollins, M. E. Webb and D. J. Zhou, Phys. Chem. Chem. Phys., 2011, 13, 19427–19436 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, circular dichroism spectra, dynamic light scattering, additional AFM images and fluorescence spectra. See DOI: 10.1039/c5nr06378g |
| This journal is © The Royal Society of Chemistry 2016 |