
A high performance, visible to mid-infrared photodetector based on graphene nanoribbons passivated with HfO2†
Xuechao
Yu‡
a,
Zhaogang
Dong‡
 b,
Yanping
Liu‡
a,
Tao
Liu
a,
Jin
Tao
a,
Yongquan
Zeng
a,
Joel K. W.
Yang
cb and
Qi Jie
Wang
*ad
b,
Yanping
Liu‡
a,
Tao
Liu
a,
Jin
Tao
a,
Yongquan
Zeng
a,
Joel K. W.
Yang
cb and
Qi Jie
Wang
*ad
aCentre for OptoElectronics and Biophotonics, School of Electrical and Electronic Engineering, Nanyang Technological University, 639798, Singapore
bInstitute of Materials Research and Engineering, Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, #08-03 Innovis, 138634, Singapore
cSingapore University of Technology and Design, 8 Somapah Road, 487372, Singapore
dCentre for Disruptive Photonics Technologies, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore. E-mail: qjwang@ntu.edu.sg
First published on 13th November 2015
Abstract
Graphene has drawn tremendous attention as a promising candidate for electronic and optoelectronic applications owing to its extraordinary properties, such as broadband absorption and ultrahigh mobility. Nevertheless, the absence of a bandgap makes graphene unfavorable for digital electronic or photonic applications. Although patterning graphene into nanostructures with the quantum confinement effect is able to open a bandgap, devices based on these graphene nanostructures generally suffer from low carrier mobility and scattering losses. In this paper, we demonstrated that encapsulation of an atomic layer deposited high-quality HfO2 film will greatly enhance the carrier mobility and decrease the scattering losses of graphene nanoribbons, because this high-k dielectric layer weakens carrier coulombic interactions. In addition, a photodetector based on HfO2 layer capped graphene nanoribbons can cover broadband wavelengths from visible to mid-infrared at room temperature, exhibiting ∼10 times higher responsivity than the one without a HfO2 layer in the visible regime and ∼8 times higher responsivity in the mid-infrared regime. The method employed here could be potentially used as a general approach to improve the performance of graphene nanostructures for electronic and optoelectronic applications.
Introduction
Graphene, a two-dimensional allotrope of honeycomb carbon atoms, has been considered as an attractive building block for electronic and optoelectronic applications,1–6 such as novel photodetectors with ultrafast speed7–9 and broadband operation wavelengths ranging from visible to terahertz.10–12 Such broadband absorption emerged from the unique conic band structure and linear energy dispersion of massless Dirac fermions.13–15 Nevertheless, this zero bandgap characteristic of the monolayer graphene introduces some limitations that render its applications. First, the low absorption (∼2.3%) of monolayer graphene via interband transitions is still a critical challenge.16 Second, the zero bandgap leads to a short photogenerated carrier lifetime at the picosecond level, which causes fast electron–hole recombinations.17,18 As a result, the photoresponse of the pristine graphene photodetectors is quite weak nowadays.Several approaches have been used to enhance the photoresponse of graphene photodetectors. For example, graphene quantum dot (GQD) arrays were successfully employed to create a bandgap of around 100 meV to improve the responsivity of pure graphene photodetectors through bandgap and defect engineering.19 Although a broadband photodetector with a responsivity of up to 8.6 A W−1 was achieved in the visible range, it still remains as a challenge to obtain room temperature operation of graphene photodetectors with a relatively fast response speed in a broadband range due to the random scattering losses. Alternatively, graphene nanoribbons (GNRs)20–22 have been demonstrated to have a bandgap opening by utilizing the quantum confinement effect,23–25 which has been realized through unzipping of carbon nanotubes,26 lithographic patterning23–25 and other chemical approaches.27,28 However, disordered edges, impurities and phonon scattering of the GNR reduce the carrier mobility and increase the carrier scattering losses, which limit the device performance.29 Previously, graphene photodetectors based on ∼240 nm wide GNRs were reported with a low photoresponse in the mid-infrared regime, because graphene nanoribbons with this width cannot open a sufficient bandgap, even though the light–graphene interaction is enhanced by graphene plasmons.30 On the contrary, ultra-narrow GNRs with widths of ∼2–5 nm were used for graphene photodetectors for achieving a considerable bandgap.26 However, the performance of the device is significantly limited by the low mobility of GNR as induced by the tremendous carrier scattering losses from defects and GNR edges with such a narrow width. Therefore, it is critical to design appropriate GNR structures to obtain a suitable bandgap for broadband operation, while still maintaining its high mobility in order to achieve high photodetection performance.
In this paper, we demonstrated a graphene field-effect transistor (FET) made of 10 nm wide GNRs encapsulated by an ultrathin high-k HfO2 layer, where the obtained GNR FET shows a semiconductor behaviour with an ON/OFF ratio of 7 ± 1. The GNR photodetector shows a high responsivity across a broadband wavelength range from visible to mid-infrared, i.e. 1.75 A W−1 in the visible, 1.5 A W−1 in the near-infrared and 0.18 A W−1 in the mid-infrared, at room temperature with a source–drain voltage as low as 10 mV. In addition, the photoresponse speed of our GNR photodetector, fabricated by electron-beam lithography (EBL) and reactive-ion etching (RIE), is relatively faster than the devices obtained by chemical methods.19,22,31 This promising performance is due to the proper design of the GNR width for broadband operation, and the increment of carrier mobility due to the reduction of carrier scattering losses in GNR via high-k HfO2 passivation.32–34 Our work reveals the importance of the dielectric environment for graphene nanostructures and it provides insights to optimize graphene-based photodetectors and transistors for optoelectronic and electronic applications.
Results and discussion
Monolayer graphene was fabricated by mechanical exfoliation of highly-ordered pyrolytic graphite (HOPG) on SiO2/Si wafer and identified by optical microscopy and Raman spectroscopy.35 We first fabricated the graphene FET devices using standard photolithography, followed by an e-beam evaporation of Ti/Au electrodes (20 nm/80 nm). The fabrication processes of GNR FET devices are shown in Fig. 1(a)–(d), where a monolayer graphene was patterned into nanoribbons by using electron beam lithography (EBL) and reactive-ion etching (RIE) oxygen plasma treatment. First, a negative e-beam resist, hydrogen silsesquioxane (HSQ), with a thickness of ∼30 nm was spin-coated onto the substrate. After the electron-beam exposure, the sample was then developed by using a NaOH/NaCl salty solution,36 and the corresponding scanning electron micrograph (SEM) of the 10 nm wide HSQ resist mask is shown in Fig. 1(e). Here, we did not obtain SEM images of GNR directly because the high energy electron beams will change the doping level of graphene so as to affect the GNR photodetector's performance. The doping effect has been reported previously.37–40 With this HSQ etching mask, patterns were transferred onto the underlying graphene monolayer via RIE oxygen plasma treatment. HSQ was then removed by immersing the sample in a hydrofluoric acid solution (HF ∼2%) for 30 seconds. Furthermore, the Raman spectrum of GNR presents a clear D-peak at ∼1350 cm−1 in Fig. 1(f), indicating that the EBL process induced defects, impurities and edge scattering in the GNR. | ||
| Fig. 1 Electron beam lithography (EBL) processes and characterization of graphene nanoribbons (GNR). (a)–(d) Schematic illustration on the process to fabricate GNR using EBL and reactive-ion etching (RIE) oxygen plasma treatment. (e) Scanning electron micrograph (SEM) of the hydrogen silsesquioxane (HSQ) resist mask prior to the RIE oxygen plasma treatment. The scale bar is 100 nm. The inset presents a close-up SEM image of the 10 nm wide HSQ resist mask. (f) Raman spectra of the exfoliated monolayer graphene and GNR. | ||
Field-effect transistor (FET) devices were fabricated for both unpatterned graphene and 10 nm wide GNRs. VDS, IDS and VG denote the source–drain bias voltage, current, and bottom gate voltage, respectively. The devices have a pair of metal pads (Ti/Au) as source/drain metal contacts, a heavily p-doped Si as the backgate, and 285 nm thick SiO2 as the gate dielectric. Moreover, Fig. S2† and Fig. 2 show the comparison of electrical characterization of the unpatterned graphene and GNR-based FETs at room temperature, where both measured IDS–VDS curves in Fig. S2(a)† and 2(a) are linear over a wide range of gate voltages. Such a linear dependence indicates that the Ti/Au electrodes have a good ohmic contact. By comparing Fig. S2(b)† and 2(b), GNR-based FET exhibits a semiconducting transport behaviour with an ON/OFF ratio of ∼7 ± 1, where the ON/OFF ratio was measured at VG − VD = −80 V, and VD denotes the neutral point.
 | ||
| Fig. 2 Electrical characterization of the GNR-based field-effect transistor (FET) at room temperature. (a) IDS–VDS curves recorded at different gate voltages. (b) Dependence of the resistance on the gate voltage recorded at VDS = 10 mV, indicating a semiconducting behaviour curve. | ||
The GNR FET is shown to behave like a semiconducting material with an adequate bandgap to operate in the mid-infrared regime at room temperature, and the relationship between the GNR width and its bandgap can be expressed as:24
 | (1) |
On the other hand, the mobility of the GNR device is reduced significantly compared to the unpatterned graphene devices, and the carrier mobility μ can be calculated by:
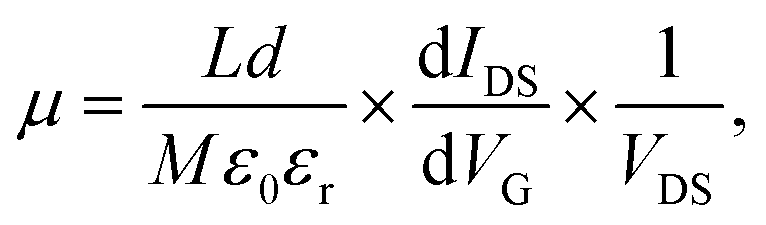 | (2) |
The carrier mobility could be enhanced by passivating a layer of high-k dielectric material, such as HfO2, on top of GNR, the detailed sample schematic structure is shown in Fig. 3(a), and this approach of using high-k dielectrics was previously employed in MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31,32 and graphene devices.33,34 The source and drain electrodes are exposed by selectively etching with a HF solution after the photolithography process. The use of this high-k dielectric material (HfO2) is that it can change the dielectric environment of GNR so as to reduce the carrier scattering from impurities, defects, and surface roughness through dielectric screening. The HfO2 layer is grown by using the atomic layer deposition (ALD) method, where an atomic force microscope (AFM) was then used to characterize the surface profile of the ALD grown HfO2 film, and the line-scan profile shows a thickness of 1.8 nm in Fig. 3(a) for 20 ALD process cycles. The thickness of the HfO2 film can be well controlled based on the number of ALD process cycles. Moreover, the high crystallization quality of the HfO2 layer was confirmed by the X-ray photoelectron spectroscopy (XPS) measurements in Fig. S1.†
31,32 and graphene devices.33,34 The source and drain electrodes are exposed by selectively etching with a HF solution after the photolithography process. The use of this high-k dielectric material (HfO2) is that it can change the dielectric environment of GNR so as to reduce the carrier scattering from impurities, defects, and surface roughness through dielectric screening. The HfO2 layer is grown by using the atomic layer deposition (ALD) method, where an atomic force microscope (AFM) was then used to characterize the surface profile of the ALD grown HfO2 film, and the line-scan profile shows a thickness of 1.8 nm in Fig. 3(a) for 20 ALD process cycles. The thickness of the HfO2 film can be well controlled based on the number of ALD process cycles. Moreover, the high crystallization quality of the HfO2 layer was confirmed by the X-ray photoelectron spectroscopy (XPS) measurements in Fig. S1.†
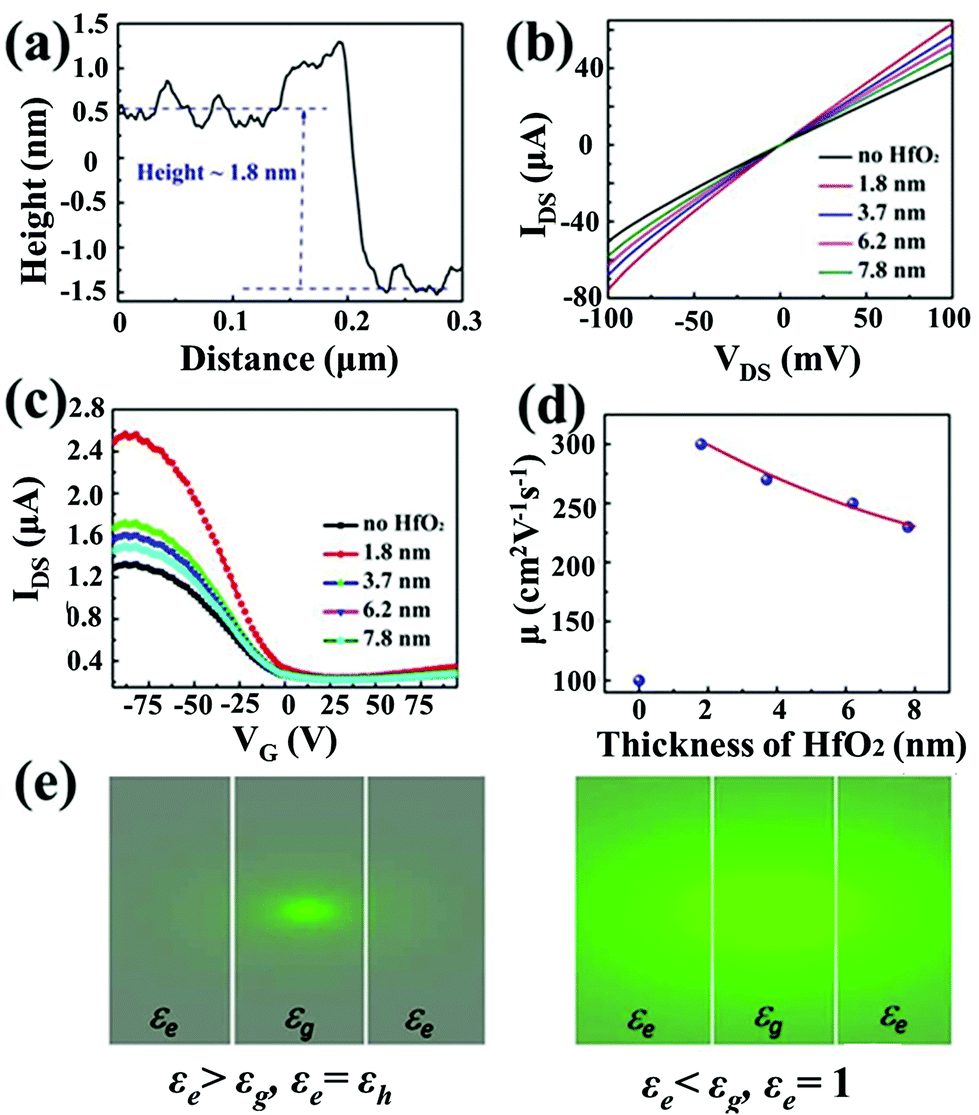 | ||
| Fig. 3 Schematic and characterization results of GNR-based FET deposited with HfO2 films. (a) Height profile of the HfO2 film as measured by using an atomic force microscope (AFM). The HfO2 film was grown using the atomic layer deposition (ALD) method with 20 cycles. (b) IDS–VDS curves of different HfO2 coated GNR-based FETs recorded at VDS = 10 mV. (c) IDS–VG curves of HfO2 coated GNR array based FET recorded at VDS = 10 mV. (d) Mobility of GNR-based FET with a different thickness of HfO2 layer. (e) Coulomb potential corresponding to two different dielectric environments, i.e. εg > εe and εg < εe. εh denotes the dielectric constant of HfO2. | ||
The electrical transport characterization was then carried out for both the GNR-based FETs with and without an HfO2 film under the same conditions. The IDS–VDS curves, as shown in Fig. 3(b), are linear and symmetric at the small bias voltages. All devices with the deposited HfO2 show a larger source–drain current than the one without HfO2, which is because of the mobility enhancement of GNR due to dielectric screening. In order to demonstrate the mechanism of the dielectric environment effect on carrier scattering as caused by the charged impurity, we compared the results on mobility by using two kinds of dielectric materials capped on GNR as shown in Fig. 3(e). For the first scenario, the capping material has a higher dielectric constant than GNR, i.e. εe > εg. εe refers to the dielectric constant of HfO2 in our case and εg refers to the dielectric constant of graphene. For the second scenario, the capping material is a low-k material with εe < εg, e.g. εe = 1. Due to the mismatch of the dielectric constant between GNR and the capping material, image charges are induced in the capping dielectric materials.32 To be more specific, the induced array of image charges are located at zn = na, where a is the thickness of GNR and n = ±1, ±2, … The n-th point charge has a magnitude of eγ|n| and γ = (εe − εg)/(εe + εg). Then, we could calculate the net electric potential seen by the electron due to these image charges as:42
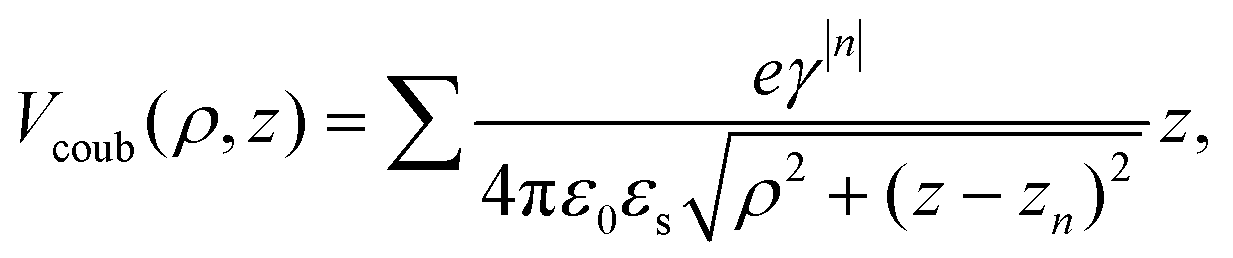 | (3) |
Fig. 3(e) shows the schematic diagram Coulomb potential contours, from which we can conclude that the Coulomb potential is enhanced in the low-k case, while it is strongly damped in the high-k case. In addition, the GNR FET with the 1.8 nm thick HfO2 layer shows a larger mobility value, where the mobility decreases with the increment of HfO2 layer thickness calculated from Fig. 3(c) as shown in Fig. 3(d). This phenomenon is consistent with eqn (3) and it can be explained by the increment of defects and surface roughness induced scattering with thicker ALD HfO2 layers.42 Here, we would like to mention that we could not carry out experiments with the thickness of the HfO2 layer being below 1.8 nm since the ALD fabricated film is not uniform with less than 20 ALD cycles. Further improvements are also possible when using other materials with a thinner layer and better uniformity, such as ALD grown Al2O343,44 or monolayer h-BN.45
We analyzed the optoelectronic properties of bare and HfO2 capped GNR photodetectors and their time-dependent photoresponse, with the excitation laser wavelengths of 632 nm, 1.47 μm and 10 μm, respectively. A parabolic mirror with a micromechanical stage was used to focus laser spots onto the sample and the estimated power intensity is ∼2.5 W cm−2. The time-dependent photocurrent responses at different wavelengths are shown in Fig. 4(a)–(c). It is shown that, in a broadband range from visible, to near-infrared, and mid-infrared, the photocurrent of HfO2 capped GNR photodetectors is about ∼8–10 times higher than the one without HfO2 capping, corresponding to a responsivity of 1.75 A W−1, 1.5 A W−1, and 0.18 A W−1, respectively, with a small source–drain bias voltage of only 10 mV. This high responsivity achieved in our devices can be attributed to the enhancement of mobility through dielectric screening. The response time is still at the second level as shown in Fig. 4(a)–(d), which is limited by the long trapping lifetime of carriers in the defect and edges states. However, the response speed demonstrated here is about one order of magnitude faster than the previous reports on a randomly distributed graphene quantum dot (GQD) photodetector19 which has complex deep surface and edge states and an electron trapping time of ∼30 seconds. The improvement was achieved by the e-beam lithography fabrication method which is used to define the edge and surface states of graphene nanoribbons with low-level chemical processes involved, thus producing graphene nanoribbons with cleaner and less surface and edge defects. The usage of high-k HfO2 as a passivated layer, apart from enhancing the carrier mobility, can also efficiently modify the edge and defect states of GNRs, leading to a faster response speed. Moreover, further improvements of the response speed are possible by using monolayer h-BN for dielectric engineering or optimizing the nanofabrication process to achieve well-defined edges and clean surfaces with less defects and residues.
 | ||
| Fig. 4 (a) Photodetection measurement of HfO2 capped GNR-based FETs with visible, near-infrared and mid-infrared lasers. (a)–(c) Time-dependent photocurrent measurements on the GNR-based FETs under the laser illumination with the wavelength of 632 nm, 1.47 μm, and 10 μm, respectively. The power density for all the measurements was kept at 2.5 W cm−2. (d) Photocurrent of 1.8 nm thick HfO2 capped GNR FETs measured in one period of modulation with the 1.47 μm laser illumination (12 mW). (e) Power dependence of the photocurrent with 1.47 μm lasers, for every laser illumination we measured 3 different samples. (f) Photocurrent and decay time measurements of graphene with different thicknesses of HfO2 film, corresponding to thicknesses as shown in Fig. 3(e), at least 3 different samples are measured with the same thickness of the HfO2 film. | ||
Fig. 4(e) presents the detailed photoresponse as a function of the laser power at the wavelength of 1.47 μm, and it is fitted by the relationship IPC = CPγ, where C is a constant and P is the illumination power. The value of γ is calculated to be ∼1.09 and this relatively high value of γ in GNR is attributed to the 1 − d transport characteristic of GNR and the relatively high mobility of our HfO2 capped GNR devices. A further increment of the HfO2 thickness will increase the response time slightly as shown in Fig. 4(f), which is due to the increased density of defect states and trapping states with the increasing thickness of the dielectric layer.42 We note that the HfO2 capped GNR photodetector operated in the mid-infrared regime exhibits a longer photoresponse time compared to those in the visible and near-infrared regimes as shown in Fig. 4(a)–(c), the detailed mechanism for this slower decay time at the mid-infrared regime needs further investigations.
The photocurrent obtained by our HfO2 capped graphene nanoribbons is higher than that by bare graphene nanoribbons31 and chemically etched GQDs,19 because the Coulomb interactions and carrier/defect scattering are strongly decreased through the passivation of a high-k dielectric layer and e-beam lithography fabrication of the GNRs. The design of graphene nanoribbons of an appropriate width is important for optoelectronic applications. For example, we measured the photodetector with 100 nm wide GNR, in which the photoresponse is only ∼5 mA W−1 under illumination with a 10 μm laser as shown in Fig. S3.† On the other hand, the photoresponse in our work is higher than the one using ∼240 nm wide graphene nanoribbons, because the GNR width is not narrow enough to open a bandgap according to eqn (1), even though the absorption is enhanced by graphene plasmons.30 In comparison, although the ultra-narrow (<5 nm) GNR was fabricated by unzipping of carbon nanotubes (CNT) exhibiting typical semiconductor behaviour,26 the photoresponse is very weak due to the strong edge scattering losses, which increase as W−4 with W being the width of the nanoribbons.41 Furthermore, other newly proposed 2D materials with narrow bandgaps46 combined with this dielectric engineering strategy are promising in the development of novel mid-infrared photodetectors.
Conclusions
In summary, we demonstrated a novel strategy to enhance the mobility of 10 nm wide GNR FETs by capping a high-k dielectric HfO2 layer, which improves the mobility of the GNR FETs by ∼3 times, compared to the pure GNR devices. Such an improvement in mobility is attributed to the reduced carrier scattering in capped GNR. Furthermore, the optical characterization results of HfO2 capped GNR photodetectors show that the response time will be ∼5 times faster and the responsivity was enhanced by ∼10 times in the visible and near-infrared regions, and ∼8 times in the mid-infrared region. We believe that our work paves a way towards high performance and flexible graphene-based optoelectronic devices, especially with the potential combination of large-area CVD growth of graphene and the CMOS technology.47Acknowledgements
This work was supported by (MOE2011-T2-2-147 and MOE2011-T3-1-005) the Ministry of Education, Singapore. Z. D. and J. K. W. Y. would like to acknowledge the funding support from the Agency for Science, Technology and Research (A*STAR) Young Investigatorship (grant number 0926030138), SERC (grant number 092154099), and the National Research Foundation (grant number NRF-CRP 8-2011-07). X. Y., Z. D. and Y. L. contributed equally to this work.Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Falko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- F. H. L. Koppens, T. Mueller, P. Avouris, A. C. Ferrari, M. S. Vitiello and M. Polini, Nat. Nanotechnol., 2014, 9, 780–793 CrossRef CAS PubMed.
- L. Shao, X. Wang, H. Xu, J. Wang, J.-B. Xu, L.-M. Peng and H.-Q. Lin, Adv. Opt. Mater., 2014, 2, 162–170 CrossRef.
- B. J. Schultz, R. V. Dennis, V. Lee and S. Banerjee, Nanoscale, 2014, 6, 3444–3466 RSC.
- W. Jie and J. Hao, Nanoscale, 2014, 6, 6346–6362 RSC.
- K. J. Tielrooij, L. Piatkowski, M. Massicotte, A. Woessner, Q. Ma, Y. Lee, K. S. Myhro, C. N. Lau, P. Jarillo Herrero, N. F. van Hulst and F. H. L. Koppens, Nat. Nanotechnol., 2015, 10, 437–443 CrossRef CAS PubMed.
- T. Mueller, F. Xia and P. Avouris, Nat. Photonics, 2010, 4, 297–301 CrossRef CAS.
- T. J. Echtermeyer, P. S. Nene, M. Trushin, R. V. Gorbachev, A. L. Eiden, S. Milana, Z. Sun, J. Schliemann, E. Lidorikis, K. S. Novoselov and A. C. Ferrari, Nano Lett., 2014, 14, 3733–3742 CrossRef CAS PubMed.
- Q. Bao and K. P. Loh, ACS Nano, 2012, 6, 3677–3694 CrossRef CAS PubMed.
- Z. Sun and H. Chang, ACS Nano, 2014, 8, 4133–4256 CrossRef CAS PubMed.
- X. Cai, A. B. Sushkov, R. J. Suess, M. M. Jadidi, G. S. Jenkins, L. O. Nyakiti, R. L. Myers-Ward, S. Li, J. Yan, D. K. Gaskill, T. E. Murphy, H. D. Drew and M. S. Fuhrer, Nat. Nanotechnol., 2014, 9, 814–819 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- J. Li, L. Niu, Z. Zheng and F. Yan, Adv. Mater., 2014, 26, 5239–5273 CrossRef CAS PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308–1308 CrossRef CAS PubMed.
- P. A. George, J. Strait, J. Dawlaty, S. Shivaraman, M. Chandrashekhar, F. Rana and M. G. Spencer, Nano Lett., 2008, 8, 4248–4251 CrossRef CAS PubMed.
- K. J. Tielrooij, J. C. W. Song, S. A. Jensen, A. Centeno, A. Pesquera, A. Zurutuza Elorza, M. Bonn, L. S. Levitov and F. H. L. Koppens, Nat. Phys., 2013, 9, 248–252 CrossRef CAS.
- Y. Zhang, T. Liu, B. Meng, X. Li, G. Liang, X. Hu and Q. J. Wang, Nat. Commun., 2013, 4, 1811 CrossRef PubMed.
- Y. F. Zhu, Q. Q. Dai, M. Zhao and Q. Jiang, Sci. Rep., 2013, 3, 1524 CAS.
- V. Barone, O. Hod and G. E. Scuseria, Nano Lett., 2006, 6, 2748–2754 CrossRef CAS PubMed.
- B. Chitara, L. S. Panchakarla, S. B. Krupanidhi and C. N. R. Rao, Adv. Mater., 2011, 23, 5419–5424 CrossRef CAS PubMed.
- M. Y. Han, B. Özyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef PubMed.
- F. Sols, F. Guinea and A. H. C. Neto, Phys. Rev. Lett., 2007, 99, 166803 CrossRef CAS PubMed.
- X. Liang, Y.-S. Jung, S. Wu, A. Ismach, D. L. Olynick, S. Cabrini and J. Bokor, Nano Lett., 2010, 10, 2454–2460 CrossRef CAS PubMed.
- D. Wei, L. Xie, K. K. Lee, Z. Hu, S. Tan, W. Chen, C. H. Sow, K. Chen, Y. Liu and A. T. S. Wee, Nat. Commun., 2013, 4, 1374 CrossRef PubMed.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229–1232 CrossRef CAS PubMed.
- A. N. Abbas, G. Liu, A. Narita, M. Orosco, X. Feng, K. Müllen and C. Zhou, J. Am. Chem. Soc., 2014, 136, 7555–7558 CrossRef CAS PubMed.
- E. Ulrich Stützel, T. Dufaux, A. Sagar, S. Rauschenbach, K. Balasubramanian, M. Burghard and K. Kern, Appl. Phys. Lett., 2013, 102, 043106 CrossRef.
- M. Freitag, T. Low, W. Zhu, H. Yan, F. Xia and P. Avouris, Nat. Commun., 2013, 4, 1951 Search PubMed.
- J. G. Son, M. Son, K.-J. Moon, B. H. Lee, J.-M. Myoung, M. S. Strano, M.-H. Ham and C. A. Ross, Adv. Mater., 2013, 25, 4723–4728 CrossRef CAS PubMed.
- Z.-Y. Ong and M. V. Fischetti, Phys. Rev. B: Condens. Matter, 2013, 88, 165316 CrossRef.
- L. Liao, J. Bai, R. Cheng, Y.-C. Lin, S. Jiang, Y. Huang and X. Duan, Nano Lett., 2010, 10, 1917–1921 CrossRef CAS PubMed.
- F. Chen, J. Xia, D. K. Ferry and N. Tao, Nano Lett., 2009, 9, 2571–2574 CrossRef CAS PubMed.
- X. Yu, J. Tao, Y. Shen, G. Liang, T. Liu, Y. Zhang and Q. J. Wang, Nanoscale, 2014, 6, 9925–9929 RSC.
- Z. Dong, M. Asbahi, J. Lin, D. Zhu, Y. M. Wang, K. Hippalgaonkar, H.-S. Chu, W. P. Goh, F. Wang, Z. Huang and J. K. W. Yang, Nano Lett., 2015, 15, 5976–5981 CrossRef CAS PubMed.
- I. Childres, L. A. Jauregui, M. Foxe, J. Tian, R. Jalilian, I. Jovanovic and Y. P. Chen, Appl. Phys. Lett., 2010, 97, 173109 CrossRef.
- D. Teweldebrhan and A. A. Balandin, Appl. Phys. Lett., 2009, 94, 013101 CrossRef.
- M. M. Ugeda, I. Brihuega, F. Hiebel, P. Mallet, J.-Y. Veuillen, J. M. Gómez-Rodríguez and F. Ynduráin, Phys. Rev. B: Condens. Matter, 2012, 85, 121402 CrossRef.
- L. Tao, C. Qiu, F. Yu, H. Yang, M. Chen, G. Wang and L. Sun, J. Phys. Chem. C, 2013, 117, 10079–10085 CAS.
- T. Fang, A. Konar, H. Xing and D. Jena, Phys. Rev. B: Condens. Matter, 2008, 78, 205403 CrossRef.
- D. Jena and A. Konar, Phys. Rev. Lett., 2007, 98, 136805 CrossRef PubMed.
- C. Goo Kang, S. Kyung Lee, T. Jin Yoo, W. Park, U. Jung, J. Ahn and B. Hun Lee, Appl. Phys. Lett., 2014, 104, 161902 CrossRef.
- C. G. Kang, S. K. Lee, S. Choe, Y. G. Lee, C.-L. Lee and B. H. Lee, Opt. Express, 2013, 21, 23391–23400 CrossRef CAS PubMed.
- H. Zeng, C. Zhi, Z. Zhang, X. Wei, X. Wang, W. Guo, Y. Bando and D. Golberg, Nano Lett., 2010, 10, 5049–5055 CrossRef CAS PubMed.
- S. Zhang, Z. Yan, Y. Li, Z. Chen and H. Zeng, Angew. Chem., Int. Ed., 2015, 54, 3112–3115 CrossRef CAS PubMed.
- X. Zhang, J. Ning, X. Li, B. Wang, L. Hao, M. Liang, M. Jin and L. Zhi, Nanoscale, 2013, 5, 8363–8366 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Method, characterization results, calculation of photoresponsivity, and calculation of photoconductive gain. See DOI: 10.1039/c5nr06869j |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |