
Insights into the molecular interactions of thymoquinone with histone deacetylase: evaluation of the therapeutic intervention potential against breast cancer†
Sabnam
Parbin
,
Arunima
Shilpi
,
Swayamsiddha
Kar
,
Nibedita
Pradhan
,
Dipta
Sengupta
,
Moonmoon
Deb
,
Sandip Kumar
Rath
and
Samir Kumar
Patra
*
Epigenetics and Cancer Research Laboratory, Biochemistry and Molecular Biology Group, Department of Life Science, National Institute of Technology, Rourkela, Odisha-769008, India. E-mail: samirp@nitrkl.ac.in; skpatra_99@yahoo.com; Fax: +91-6612462681; Tel: +91-6612462683
First published on 12th October 2015
Abstract
Many HDAC inhibitors have passed through the gateway of clinical trials. However, they have limited therapeutic implications due to their pleiotropic pharmaceutical properties and off-target effects. In view of this, dietary active phytochemicals were evaluated. Based upon the chemical and structural insights of HDAC active pockets, thymoquinone (TQ) was investigated to uncover its active participation in HDAC inhibition. The synergistic analysis of docking and molecular dynamics simulation disclosed the elementary interaction and stability of TQ with human HDACs. The in silico findings were corroborated with an in vitro analysis, demonstrating the efficient role of TQ in the attenuation of global HDAC activity. Furthermore, TQ also elicited downstream effects of HDAC inhibition: reactivation of HDAC target genes (p21 and Maspin), induction of the pro-apoptotic gene Bax, down regulation of the anti-apoptotic gene Bcl-2 and arrest of the cell cycle at the G2/M phase. Finally, the result of a higher cytotoxicity of TQ towards MCF-7 breast cancer cells in comparison to normal cells indicates the potential of TQ to be an anticancer drug.
1. Introduction
The process for the acetylation and deacetylation of histones presents two sides of the same coin, regulating the activity of chromatin and a plethora of other cellular activities. Several studies have envisaged a role of HDACs in the development and progression of human cancer. These HDACs undergo an active participation in gene silencing in the context of cell cycle regulation, differentiation, apoptosis, angiogenesis, invasion, adhesion and metastasis.1,2 Thus, inhibitors against these enzymes are emerging as a promising class of anticancer agents that have been tested for the treatment of multifarious malignancies. As of now, romidepsin and vorinostat are recognized as US Food and Drug Administration approved drugs for cutaneous T-cell lymphoma, while romidepsin alone is approved for relapsed peripheral T-cell lymphoma. Indeed, most HDAC inhibitors (HDACis) are still under different phases of preclinical and clinical investigations against various relapsed and refractory lymphoid malignancies, myeloid leukemia, and solid tumors.3Unfortunately, a higher clinical dose of synthetic HDACis induces extensive DNA damage and impairs DNA break repair leading to undesired cytotoxicity.4 The most common toxicities evoked by such HDACis include fatigue, diarrhoea and nausea, while cardiac toxicity is the most prevalent among them all. Nevertheless, the usual doses of synthetic HDACis might also exert adverse effects including vomiting, anorexia, dehydration, myelosuppression, thrombocytopenia, anemia and pulmonary embolism.5 Besides, these inhibitors are expensive and have low specificity. This brings dietary chemopreventive agents into the lime light, with widespread availability, possible incorporation into diets and negligible toxic effects. Of these, some bioactive phytochemicals have the potential to alter the anomalous expression of cancer related genes by modulating epigenetic events. For example, curcumin from turmeric, epigallocatechin-3-gallate (EGCG) from green tea, procyanidine B2 from grape seeds and sulforaphane (SFN) from broccoli are effective phytoconstituents to alter aberrant DNA methylation and histone modification in different malignancies.6,7
Experimental evidence suggests that diallyl disulfide and SFN increase p21waf1/cip1 expression in human colon cancer cells by inhibiting HDACs.2,8 Likewise, polyphenols are one of the important bioactive dietary components having chemopreventive and therapeutic potential against various diseases. Among them, thymoquinone (2-isopropyl-5-methyl-1,4-benzoquinone) (TQ), the active constituent of Nigella sativa (black cumin) holds potential significance. Numerous cell line and animal experiments have demonstrated the anti-inflammatory and chemopreventive nature of TQ in different diseases. It has been reported to have antineoplastic activities in multiple forms of cancer including, breast cancer, osteosarcoma, prostate cancer, myeloblastic leukemia, squamous cell carcinoma and gastric carcinoma.9–12 Additionally, it has also been demonstrated to elicit antitumor activity in mouse tumor models xenografted with breast,13 colon14 and prostate cancer.15 In view of the above perspectives, we intended to investigate the dietary supplement TQ. Several studies have contributed to the explanation of the anticancer effects of TQ, which is attributed to the induction of PPAR-γ (peroxisome proliferator-activated receptor gamma), PTEN (phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase) expression, as well as p53-dependent and independent apoptosis.9,10,16
HDAC inhibitors have been reported to upregulate the expression of multiple genes, including the p21waf1/cip1 gene and GADD45, associated with the inhibition of proliferation, induction of differentiation and/or apoptosis of transformed cells.2,8,17 Remarkably, TQ has also been demonstrated to induce apoptosis via overexpression of the p21waf1/cip1 gene in doxorubicin resistant breast cancer cells and osteosarcoma cells.10,16 In view of this, we sought to investigate the HDAC inhibition activity of TQ. Furthermore, chemical and structural insights of the HDAC active site and the information on existing HDAC inhibitors compelled us to proclaim the inhibitory role of TQ against HDACs. Hence, in the present study, we have attempted to illuminate the elementary interactions of TQ with HDAC by an in silico approach which is complemented by an in vitro cell culture study to investigate the consequence of such interactions. We also aimed to compare the cytotoxic nature of TQ towards breast cancer cells and normal keratinocytes to establish its toxicity to cancer cell lines compared to normal cell lines.
2. Materials and methods
2.1. In silico approach to study inter-molecular interactions between thymoquinone and HDACs
The ligands included in the docking analysis were TSA, SFN and TQ. The 3D co-ordinates of these ligands were obtained from the ChEBI database (http://www.ebi.ac.uk/chebi) (Fig. 1). These ligands hold the CHEBI identification numbers 46024, 47807 and 19371, respectively.
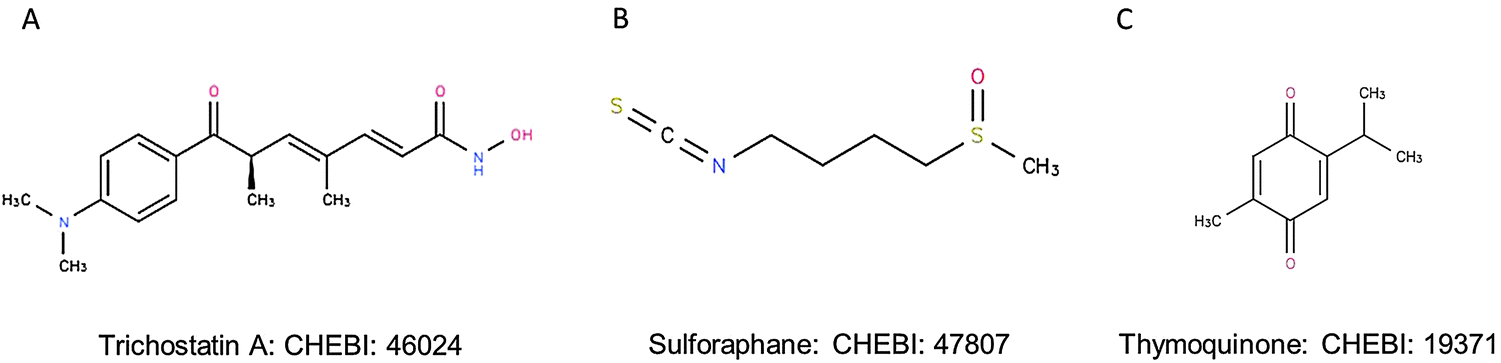 | ||
| Fig. 1 Chemical structures of (A) trichostatin A, (B) sulforaphane and (C) thymoquinone retrieved from CHEBI. | ||
2.2. In vitro cell culture analyses
| Gene | Primer sequence | Amplicon size (bp) |
|---|---|---|
| p21 | 5′-TGAGCCGCGACTGTGATG-3′ | 82 |
| 5′-GTCTCGGTGACAAAGTCGAAGTT-3′ | ||
| Maspin | 5′-GGAATGTCAGAGACCAAGGGA -3′ | 139 |
| 5′-GGTCAGCATTCAATTCATCCTT-3′ | ||
| Bax | 5′-TTCATCCAGGATCGAGCAG-3′ | 94 |
| 5′-CGCTCAGCTTCTTGGTGG-3′ | ||
| Bcl-2 | 5′-CCTGTGGATGACTGAGTACC-3′ | 128 |
| 5′-GAGACAGCCAGGAGAAATCA-3′ | ||
| GAPDH | 5′-GGAGCGAGATCCCTCCAAAAT-3′ | 197 |
| 5′-GGCTGTTGTCATACTTCTCATGG-3′ | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution in phosphate buffered saline, 1% bovine serum albumin for all antibodies). The cells were rinsed with PBS then incubated with a FITC-conjugated goat anti-rabbit secondary antibody (Santa Cruz Biotech).
:100 dilution in phosphate buffered saline, 1% bovine serum albumin for all antibodies). The cells were rinsed with PBS then incubated with a FITC-conjugated goat anti-rabbit secondary antibody (Santa Cruz Biotech).
3. Results
3.1. In silico investigations
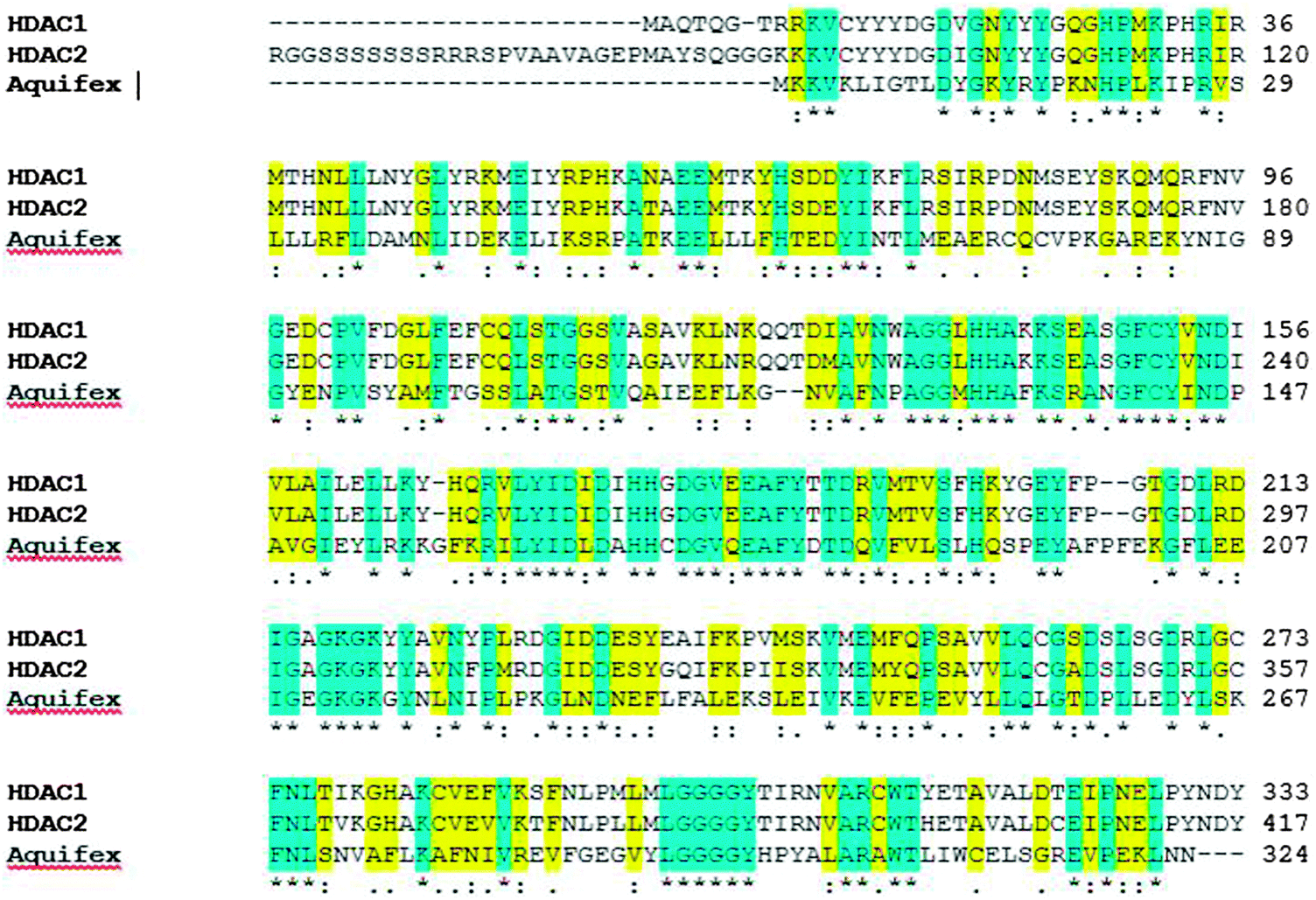 | ||
| Fig. 2 Depiction of conserved deacetylase domains among human HDAC proteins (HDAC1 and HDAC2) and with the model organism, Aquifex aeolicus. Blue highlighted regions are conserved domains within human HDAC1, HDAC2 and HDACA. The amino-acid sequence highlighted in yellow indicates the presence of similar amino acids in the domain. | ||
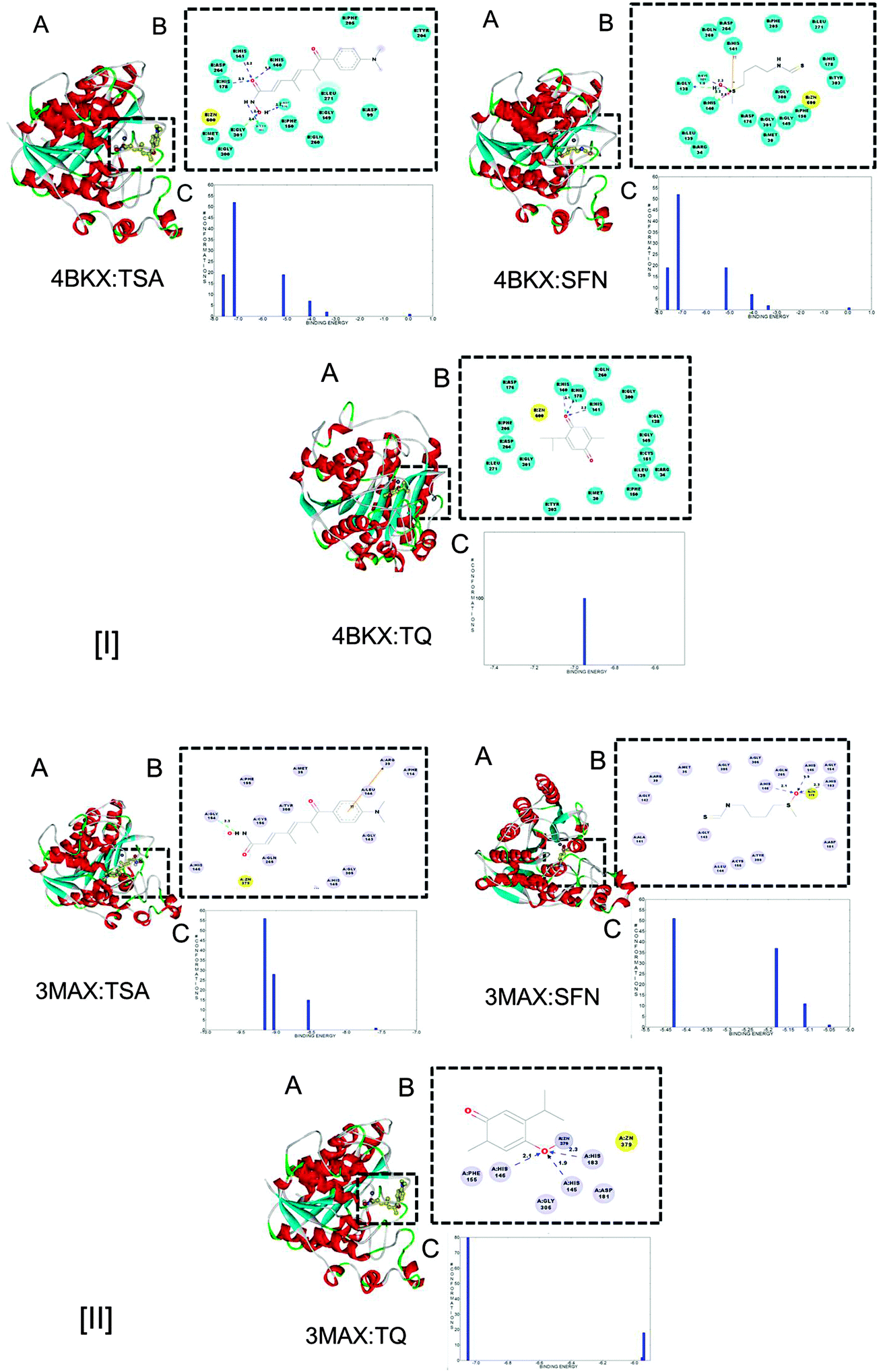 | ||
| Fig. 3 (A) Molecular interactions of HDAC1 (4BKX) and HDAC2 (3MAX) with known HDAC inhibitors (TSA, SFN) and the candidate inhibitor (TQ). (B) Detailed molecular interactions between active site amino acid residues of HDAC 1 and 2 with TSA, SFN and TQ. The hydrogen bonds are shown by dotted lines. (C) Binding energy analysis of the docked complex of HDAC1 and 2 with the respective known and candidate inhibitors. | ||
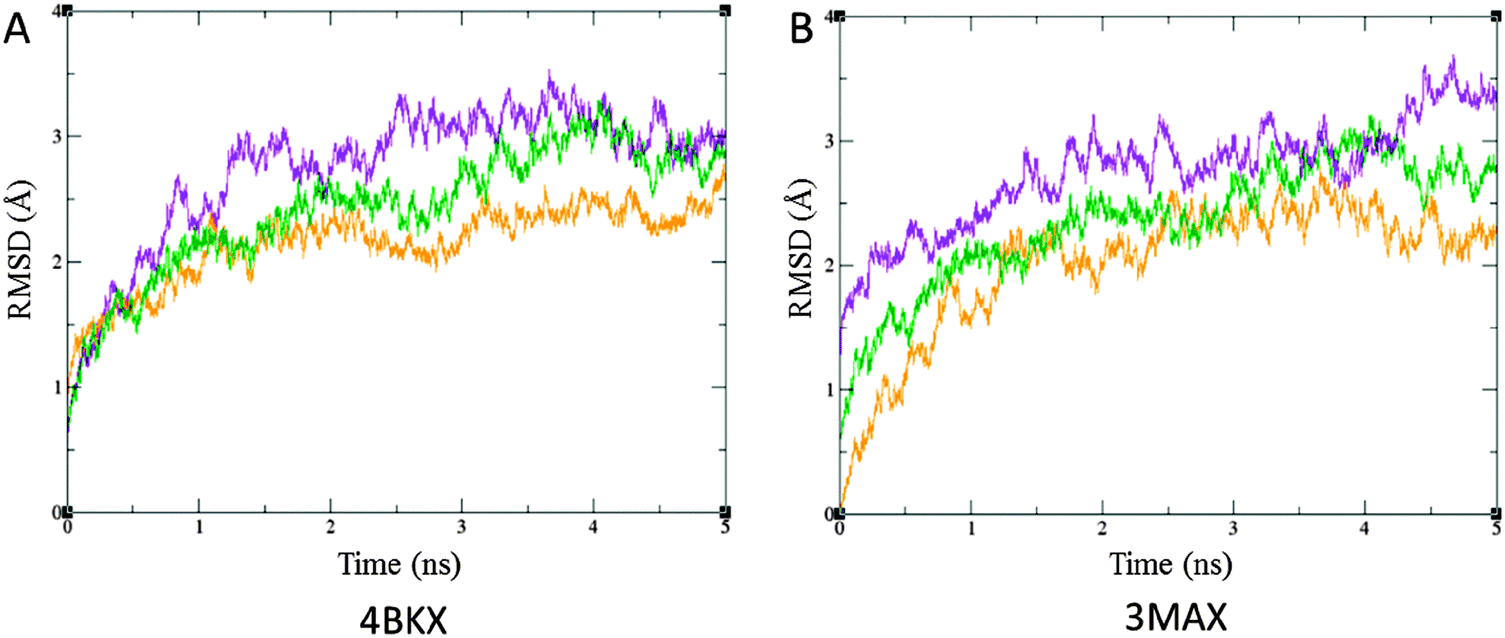 | ||
| Fig. 4 RMSD plot with respect to time in ns upon interaction of TSA (orange), SFN (violet) and TQ (green) with, (A) 4BKX and (B) 3MAX in Å. RMSD is calculated for heavy atoms with reference to their respective orientation in the crystal structures. TQ exhibits a similar deviation upon interaction with HDAC and forms a highly stable complex as that of SFN. | ||
The stability of HDAC–inhibitor complexes was also characterised by the binding mode of inhibitors (TSA, SFN and TQ) inside the active site pocket. An average hydrogen bond formation was estimated between HDAC–inhibitor complexes. It is evident from our investigation that TQ forms a higher number of hydrogen bonds with the cognate donor/receptor in the proximity of the active site pocket of the HDAC enzyme. On average 3.47 and 5.2 hydrogen bonds are formed when TSA makes a complex with HDAC1 and HDAC2, respectively. Similarly, TQ interacts with the respective protein with an average of 2.35 and 2.5 hydrogen bonds. SFN also holds a similar mode of interaction as that of TQ (Fig. S1, ESI†) (Table 3).
Thus, MD simulation studies based upon RMSD and average hydrogen bonding affirm that the stability of HDAC–TQ complexes is similar to that of SFN. Later, an in vitro characterization was carried out to uncover the ability of TQ in HDAC inhibition and successfully restore the expression of tumor suppressor genes.
3.2. In vitro analyses
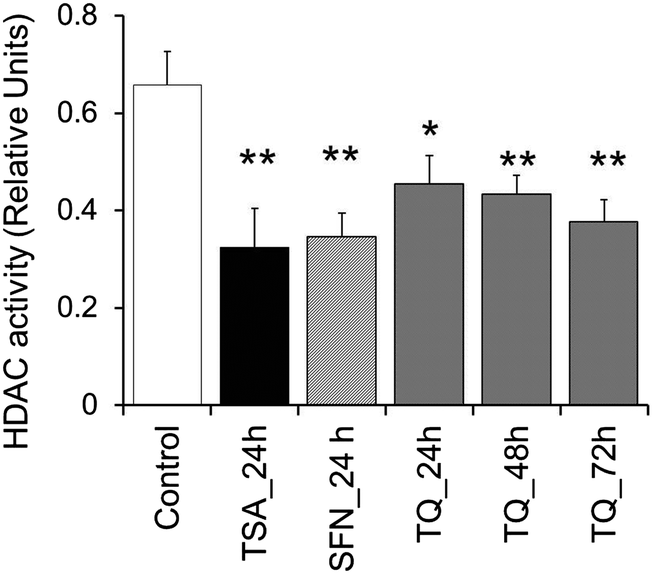 | ||
| Fig. 5 TQ exhibits HDAC inhibition activity in MCF7 cells in a time dependent manner. MCF-7 cells were treated with TQ for 24, 48 and 72 h and with TSA and SFN for 24 h. Whole cell protein lysates were prepared and incubated with the acetylated substrate peptide provided by the manufacturer. Statistical analysis of the data was performed using a one way ANOVA with a post hoc Dunnett multiple comparisons test, which compares each sample against the control. Data are expressed as the mean ± S.D., n = 3. Single asterisk (p < 0.05) and double asterisk (p < 0.01) indicate the level of significance. | ||
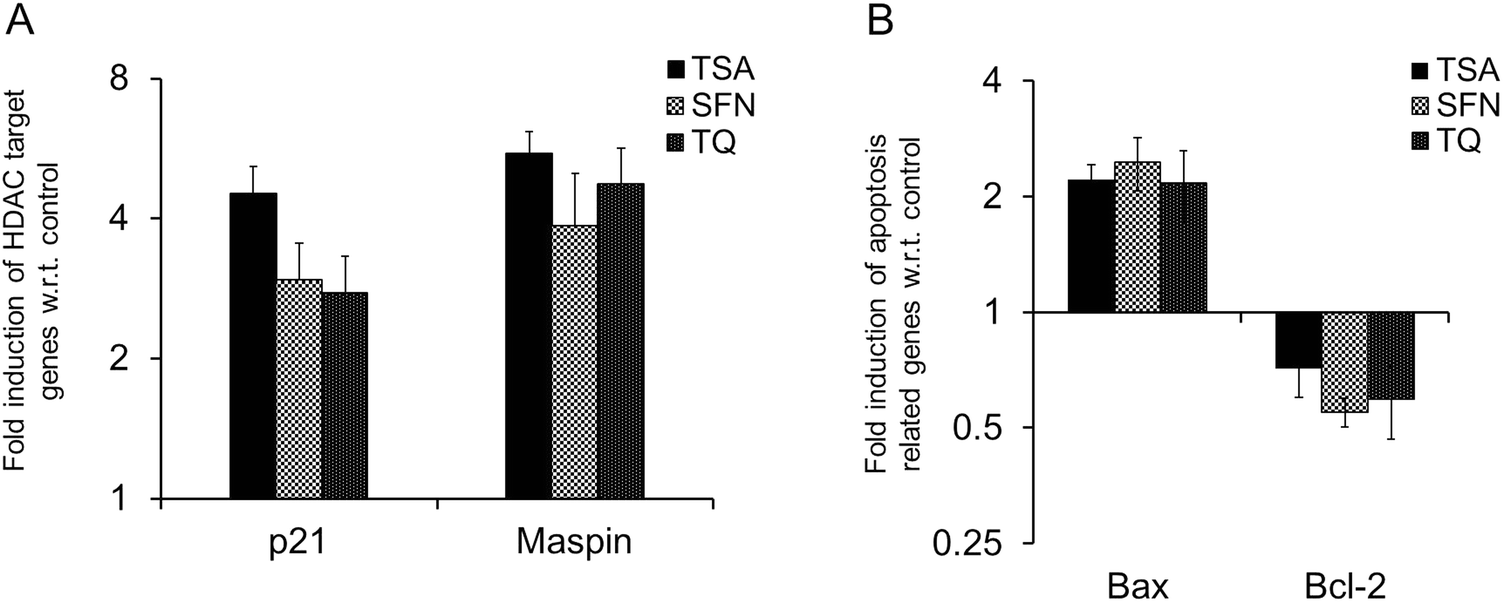 | ||
| Fig. 6 Real time RT-PCR analysis of HDAC target genes (A) p21 and Maspin, and apoptosis related genes (B) Bax and Bcl-2 after treatment with TSA, SFN and TQ in MCF-7 cells. The transcript level of p21, Maspin and Bax are upregulated after treatment with TQ as are known HDAC inhibitors (TSA and SFN), while Bcl-2 is downregulated. Data are expressed as the mean ± S.D., n = 3, and p < 0.05. | ||
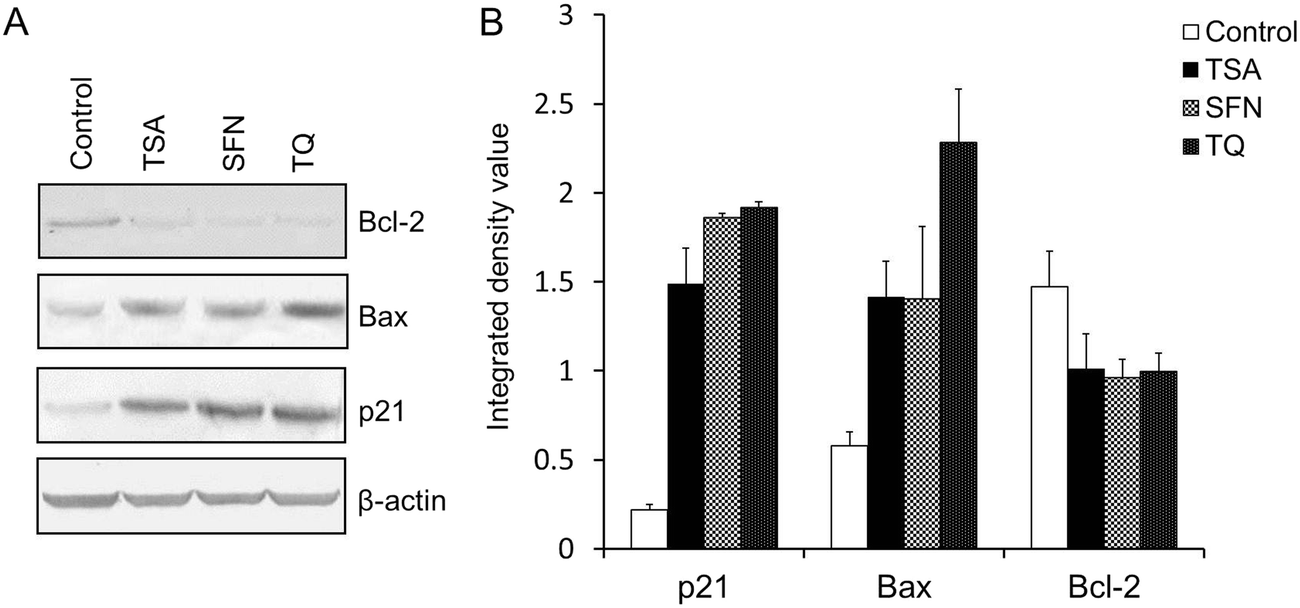 | ||
| Fig. 7 (A) Western blot analysis of p21, Bax and Bcl-2 in MCF-7 cells treated with TSA or SFN for 24 h and TQ for 48 h. The protein level of p21 and Bax is enhanced after treatment with TQ like the known HDAC inhibitors (TSA and SFN), while Bcl-2 exhibits a diminished expression. β-Actin was used as the internal control to confirm equal protein loading. (B) A histogram depicting the integrated density value for different bands obtained from the Western blot using ImageJ software. Data are expressed as the mean ± S.D., n = 3, and p < 0.05. | ||
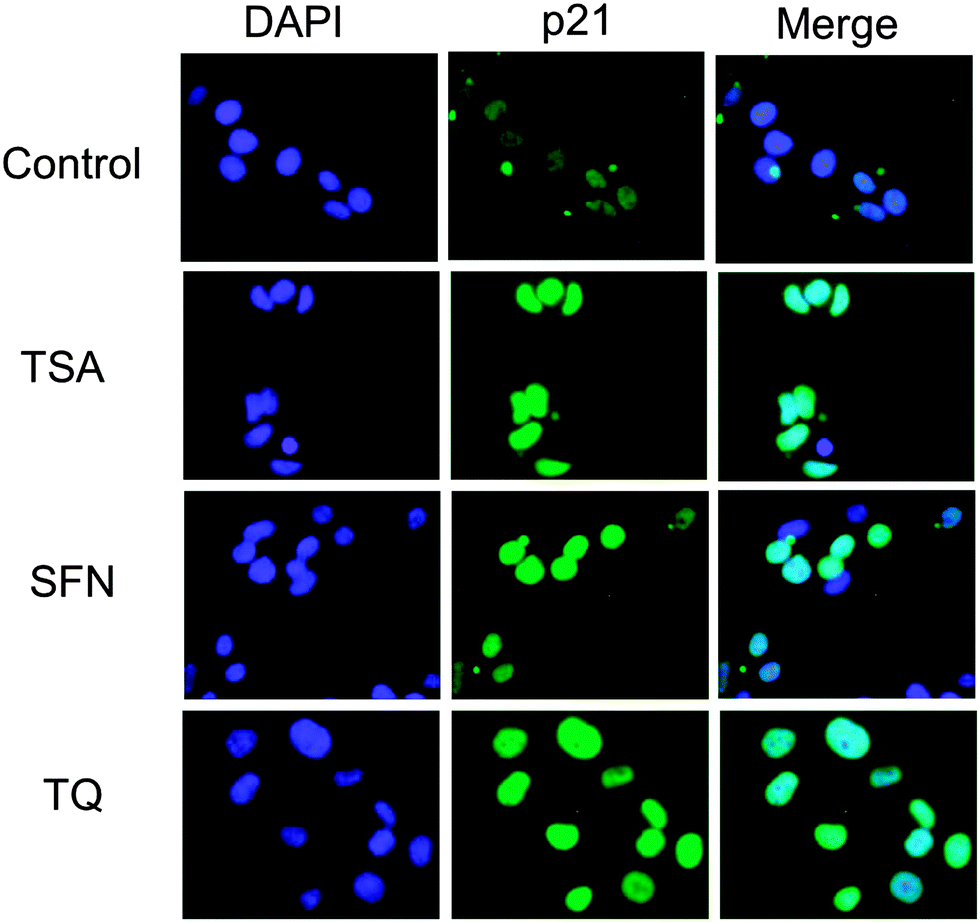 | ||
| Fig. 8 Immunofluorescence microscopy was used to detect p21 expression in untreated and TSA, SFN and TQ treated MCF-7 cells. The expression of p21 is enhanced after treatment with TQ. Each of the images is representative of 30 images obtained from three independent experiments. | ||
In addition, the HDAC inhibitor dependent increased expression of p21 can also arrest the cell cycle at the G1 and/or G2 phase.
 | ||
| Fig. 9 Cell cycle analysis of MCF-7 cells exposed to TSA and SFN for 24 h and TQ for 48 h at their respective IC50 in parallel with control (DMSO treated) cells. TQ arrests the MCF-7 cells at the G2/M interphase of cell cycle like TSA and SFN. Shown are the representative images of two independent experiments. | ||
Furthermore, in the upcoming analyses, we have demonstrated the cytotoxic effect of TQ in metastatic breast adenocarcinoma MCF-7 cells.
Furthermore, the comet assay indicates the extent of DNA damage due to apoptosis upon application of any insults to the cells. The tail length denotes the apoptosis inducing ability of drugs. The increased tail length is observed in TQ treated MCF-7 cells similar to TSA and SFN. Conversely, in normal cells TQ induces less DNA damage, as revealed by the short tail length (Fig. S5, ESI†).
Similarly, the clonogenic assay reveals the anti-proliferative effect of TQ in MCF-7 cells. It has been observed that TQ effectively inhibits the proliferation of MCF-7 cells, while it is not effective in normal cells. This is clearly revealed by the comparatively lower number of colonies formed in MCF-7 than in HaCaT cells (Fig. S7, ESI†).
Based upon the above findings it is evident that TQ is non-toxic to normal cells and can be successfully implemented in clinical applications.
4. Discussion and conclusion
DNA methylation mediated gene silencing is invariably associated with histone deacetylase activity.29,30 The recognition that HDACs are causally implicated in cancer has exhilarated the development of small-molecule inhibitors as anticancer drugs. Despite the identification of structurally diverse classes of HDAC inhibitors, several of them hold considerable toxicity, unintended off-target effects and poor bioavailability leading to their limited therapeutic implications.31 This led us to explore the possible role of the most commonly consumed dietary phytochemical, thymoquinone (TQ), in HDAC inhibition. Previous studies have explored the anticancer activity of TQ through p53-dependent and independent apoptosis, involving the induction of PPAR-γ, PTEN, NF-κB and AKT.9,10,16,32,33 Gurung et al. explained the anticancer effect of TQ in terms of its ability to induce DNA damage and telomere attrition, by inhibiting telomerase, and cell death.34 However, the molecular mechanism of TQ induced growth inhibition and its pro-apoptotic effects in cancer are still in its infancy.14,34 The present investigation, for the first time, illustrates a plausible insight into this question by demonstrating that TQ inhibits HDAC activity in human breast adenocarcinoma MCF-7 cells.We began our investigation with an in silico approach using docking analyses followed by a molecular dynamics (MD) simulation study. Known inhibitors of HDACs, TSA and SFN were considered as reference molecules in our analyses. The higher binding affinity of TQ with HDAC1 and HDAC2 compared to SFN and its apparent similarity to that of TSA reveal its potential to inhibit HDACs (Fig. 3, panels [I] and [II] C). Furthermore, the stability of TQ inside the binding pocket of HDACs has been validated by RMSD and average hydrogen bonding (Fig. 4 and Fig. S1, ESI†). The formation of such a stable complex is also underpinned by the fact that TQ undergoes covalent bond formations with the nucleophilic amino acid side chains of Cys and His residues. It occurs through a highly reactive α,β-unsaturated carbonyl group (a Michael acceptor) of TQ.35,36 This clearly shows that TQ forms a stable and energetically favored complex with HDACs. Such property of TQ can be successfully employed in the development of irreversible drugs for the treatment of cancer.37 Corroborating our in silico findings, the in vitro examination explains the potential role of TQ in HDAC inhibition. Generally, enhanced expression of HDACs has been reported in various cancers38 and inhibitors have been identified to successfully impede their activity.1,23 Previously, the phytochemicals, such as butyrate and SFN are known to inhibit enzymatic activity of HDAC.2 Intriguingly, our study has revealed TQ as a potential candidate that ameliorated the total cellular HDAC activity in MCF-7 cells in a time dependent manner. Our observation that TQ is capable of decreasing HDAC activity in treated cells is consistent with the existing HDAC inhibitors (Fig. 5). This finding is further substantiated by the enhanced expression of HDAC target genes upon application of TQ. Previous reports have shown that the p21 promoter is epigenetically repressed by HDAC activity via their Sp1 sites and HDAC inhibitors, such as TSA, apicidin, phenylbutyrate, SAHA and Helminthosporium carbonum toxin, could induce p21 expression in MCF-7 cells.1,26,39 Similarly, the histone hypoacetylation mediated epigenetic silencing of Maspin can be successfully reversed by TSA in MCF-7 cells.25 In agreement with this, we observed that TQ markedly enhanced the expression of p21 and Maspin both at the transcript and protein level. Moreover, we demonstrated that the ability of TQ to restore the expression of these HDAC target genes is almost similar to TSA and SFN treatment. As previously seen by other groups, it was found that upon treatment with known HDAC inhibitors (TSA and SFN), the expression of pro-apoptotic Bax is upregulated and that of anti-apoptotic Bcl-2 is down-regulated.40,41 A consistent expression profile of Bax and Bcl-2 was observed in the case of TQ treated MCF-7 cells (Fig. 6–8). The differential expression profile of the HDAC target and apoptosis related genes are the universal consequences of the downstream effects of the HDAC inhibitor. In our investigation, TQ elicits a similar expression profile; this apparently reflects the HDAC inhibition potential of TQ.
To further support our findings, we analyzed the cell cycle progression of MCF-7 cells treated with TSA, SFN and TQ. In line with previous reports of HDAC inhibitor induced G2/M phase arrest in cancer cells,42,43 we found an increased percentage of cells in the G2/M interface following TSA, SFN and TQ treatment, compared to the untreated control (Fig. 9). It is well known that p21 is implicated in the induction of G1/S phase arrest through its interactions with cyclin E/cdk2, however, in p53 wild type cells, such as LnCaP prostate cancer cells, p21 induction by an HDAC inhibitor has also been demonstrated to induce a G2/M arrest.43 Collectively, TQ mediated HDAC activity attenuation leads to p21 induction, which in turn arrests MCF-7 cells at the G2/M interface ultimately resulting in reduced cell proliferation.
Identification of HDAC inhibitors with a high therapeutic efficacy and minimal cytotoxicity continues to be an unmet challenge. In a quest to address this, after unraveling the role of TQ in HDAC inhibition, our next attempt was to inspect its cytotoxic nature towards normal cells. TQ, at its sub-lethal concentration (IC50 34 μM) induces the formation of cytoplasmic vacuoles in MCF-7 breast cancer cells, while HaCat cells exhibit no obvious abnormalities (Fig. S2, ESI†). This may stem from the cytotoxic property of TQ, as shown previously by Racoma et al. in glioblastoma cells.44 It is also apparent from the cytotoxic MTT assay that TQ, unlike TSA, possesses no cytotoxic effect in normal HaCat cells while it exerts an extensive toxic effect in MCF-7 cells (Fig. S3, ESI†). Supporting these findings, in normal cells, TQ was found to induce less chromatin condensation and DNA damage (Fig. S4 and S5, ESI†), which are hallmarks of apoptosis. In agreement with this finding, TQ was also demonstrated to exert a decreased anti-proliferative and anti-migratory effect in HaCat cells compared to MCF-7 cells (Fig. S6 and S7, ESI†). Thus, our findings illustrate well that TQ exhibits specific cytotoxicity in MCF-7 cells, while eliciting no such effect in normal cells.
In conclusion, this investigation reveals a new role for TQ in inducing an anticancer threat on metastatic breast adenocarcinoma cells via the attenuation of global HDAC activity and the ensuing prevalent epigenetic changes. Indeed, such changes are implicated in the regulation of genes, including p21, Maspin, Bax and Bcl-2, which eventually elicits a strong anticancer effect. Moreover, TQ is a natural dietary component with no obvious cytotoxicity to normal cells. In line with this, further investigations may be executed in experimental models and clinical settings to enlist TQ as a chemo-preventive drug, which may lead to a lasting cure for breast cancer.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
S. Parbin is thankful to DST, Govt. of India for INSPIRE fellowship awarded to her. A. Shilpi, S. Kar, D. Sengupta, M. Deb and S. K. Rath are thankful to NIT-Rourkela for granting them fellowships under the Institute Research Scheme. We apologize to those, whose works and related publications we have not been able to discuss and cite due to space limitations.References
- M. A. Glozak and E. Seto, Oncogene, 2007, 26, 5420–5432 CrossRef CAS PubMed.
- M. C. Myzak and R. H. Dashwood, Curr. Drug Targets, 2006, 7, 443–452 CrossRef CAS PubMed.
- M. Dokmanovic, C. Clarke and P. A. Marks, Mol. Cancer Res., 2007, 5, 981–989 CrossRef CAS PubMed.
- N. Azad, C. A. Zahnow, C. M. Rudin and S. B. Baylin, Nat. Rev. Clin. Oncol., 2013, 10, 256–266 CrossRef CAS PubMed.
- A. A. Lane and B. A. Chabner, J. Clin. Oncol., 2009, 27, 5459–5468 CrossRef CAS PubMed.
- S. Shukla, S. M. Meeran and S. K. Katiyar, Cancer Lett., 2014, 355, 9–17 CrossRef CAS PubMed.
- A. Shilpi, S. Parbin, D. Sengupta, S. Kar, M. Deb, S. K. Rath, N. Pradhan, M. Rakshit and S. K. Patra, Chem.-Biol. Interact., 2015, 233, 122–138 CrossRef CAS PubMed.
- N. Druesne, A. Pagniez, C. Mayeur, M. Thomas, C. Cherbuy, P. H. Duee, P. Martel and C. Chaumontet, Carcinogenesis, 2004, 25, 1227–1236 CrossRef CAS PubMed.
- C. C. Woo, S. Y. Loo, V. Gee, C. W. Yap, G. Sethi, A. P. Kumar and K. H. Tan, Biochem. Pharmacol., 2011, 82, 464–475 CrossRef CAS PubMed.
- M. Roepke, A. Diestel, K. Bajbouj, D. Walluscheck, P. Schonfeld, A. Roessner, R. Schneider-Stock and H. Gali-Muhtasib, Cancer Biol. Ther., 2007, 6, 160–169 CrossRef CAS PubMed.
- X. Lei, X. Lv, M. Liu, Z. Yang, M. Ji, X. Guo and W. Dong, Biochem. Biophys. Res. Commun., 2012, 417, 864–868 CrossRef CAS PubMed.
- S. Das, K. K. Dey, G. Dey, I. Pal, A. Majumder, S. MaitiChoudhury, S. C. kundu and M. Mandal, PLoS One, 2012, 7, e46641 CAS.
- C. C. Woo, A. Hsu, A. P. Kumar, G. Sethi and K. H. Tan, PLoS One, 2013, 8, e75356 CAS.
- H. Gali-Muhtasib, M. Ocker, D. Kuester, S. Krueger, Z. El-Hajj, A. Diestel, M. Evert, N. El-Najjar, B. Peters, A. Jurjus, A. Roessner and R. Schneider-Stock, J. Cell. Mol. Med., 2008, 12, 330–342 CrossRef PubMed.
- A. O. Kaseb, K. Chinnakannu, D. Chen, A. Sivanandam, S. Tejwani, M. Menon, Q. P. Dou and G. P. Reddy, Cancer Res., 2007, 67, 7782–7788 CrossRef CAS PubMed.
- S. A. Arafa el, Q. Zhu, Z. I. Shah, G. Wani, B. M. Barakat, I. Racoma, M. A. El-Mahdy and A. A. Wani, Mutat. Res., 2011, 706, 28–35 CrossRef PubMed.
- Z. Chen, S. Clark, M. Birkeland, C. M. Sung, A. Lago, R. Liu, R. Kirkpatrick, K. Johanson, J. D. Winkler and E. Hu, Cancer Lett., 2002, 188, 127–140 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- G. M. Morris, R. Huey and A. J. Olson, in Current protocols in bioinformatics, ed. A. D. Baxevanis, et al., 2008, ch. 8, unit 8.14 Search PubMed.
- A. W. Schuttelkopf and D. M. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- M. Deb, D. Sengupta, S. K. Rath, S. Kar, S. Parbin, A. Shilpi, N. Pradhan, S. K. Bhutia, S. Roy and S. K. Patra, Biochim. Biophys. Acta, 2015, 1852, 1630–1645 CrossRef CAS PubMed.
- S. Parbin, S. Kar, A. Shilpi, D. Sengupta, M. Deb, S. K. Rath and S. K. Patra, J. Histochem. Cytochem., 2014, 62, 11–33 CrossRef PubMed.
- A. L. Gartel and A. L. Tyner, Exp. Cell Res., 1999, 246, 280–289 CrossRef CAS PubMed.
- X. H. Liao, Y. Q. Li, N. Wang, L. Zheng, W. J. Xing, D. W. Zhao, T. B. Yan, Y. Wang, L. Y. Liu, X. G. Sun, P. Hu, H. Zhou and T. C. Zhang, Cell. Signalling, 2014, 26, 1335–1346 CrossRef CAS PubMed.
- R. Varshochi, F. Halim, A. Sunters, J. P. Alao, P. A. Madureira, S. M. Hart, S. Ali, D. M. Vigushin, R. C. Coombes and E. W. Lam, J. Biol. Chem., 2005, 280, 3185–3196 CrossRef CAS PubMed.
- L. J. Juan, W. J. Shia, M. H. Chen, W. M. Yang, E. Seto, Y. S. Lin and C. W. Wu, J. Biol. Chem., 2000, 275, 20436–20443 CrossRef CAS PubMed.
- S. V. Singh, A. Herman-Antosiewicz, A. V. Singh, K. L. Lew, S. K. Srivastava, R. Kamath, K. D. Brown, L. Zhang and R. Baskaran, J. Biol. Chem., 2004, 279, 25813–25822 CrossRef CAS PubMed.
- S. K. Patra and M. Szyf, FEBS J., 2008, 275, 5217–5235 CrossRef CAS PubMed.
- S. Kar, S. Parbin, M. Deb, A. Shilpi, D. Sengupta, S. K. Rath, M. Rakshit, A. Patra and S. K. Patra, Cell. Mol. Life Sci., 2014, 71, 1017–1032 CrossRef CAS PubMed.
- O. H. Kramer, M. Gottlicher and T. Heinzel, Trends Endocrinol. Metab., 2001, 12, 294–300 CrossRef CAS PubMed.
- G. Sethi, K. S. Ahn and B. B. Aggarwal, Mol. Cancer Res., 2008, 6, 1059–1070 CrossRef CAS PubMed.
- T. Yi, S. G. Cho, Z. Yi, X. Pang, M. Rodriguez, Y. Wang, G. Sethi, B. B. Aggarwal and M. Liu, Mol. Cancer Ther., 2008, 7, 1789–1796 CrossRef CAS PubMed.
- R. L. Gurung, S. N. Lim, A. K. Khaw, J. F. Soon, K. Shenoy, S. Mohamed Ali, M. Jayapal, S. Sethu, R. Baskar and M. P. Hande, PLoS One, 2010, 5, e12124 Search PubMed.
- J. M. Chalker, G. J. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- C. Liao, J. E. Park, J. K. Bang, M. C. Nicklaus and K. S. Lee, ACS Med. Chem. Lett., 2010, 1, 110–114 CrossRef CAS PubMed.
- A. Wissner and T. S. Mansour, Arch. Pharm., 2008, 341, 465–477 CrossRef CAS PubMed.
- S. K. Patra, A. Patra and R. Dahiya, Biochem. Biophys. Res. Commun., 2001, 287, 705–713 CrossRef CAS PubMed.
- V. M. Richon, T. W. Sandhoff, R. A. Rifkind and P. A. Marks, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10014–10019 CrossRef CAS PubMed.
- L. Wang, D. Liu, T. Ahmed, F. L. Chung, C. Conaway and J. W. Chiao, Int. J. Oncol., 2004, 24, 187–192 Search PubMed.
- J. Ma, X. Guo, S. Zhang, H. Liu, J. Lu, Z. Dong, K. Liu and L. Ming, Mol. Med. Rep., 2015, 11, 4525–4531 CAS.
- S. J. Jackson and K. W. Singletary, J. Nutr., 2004, 134, 2229–2236 CAS.
- S. Roy, K. Packman, R. Jeffrey and M. Tenniswood, Cell Death Differ., 2005, 12, 482–491 CrossRef CAS PubMed.
- I. O. Racoma, W. H. Meisen, Q. E. Wang, B. Kaur and A. A. Wani, PLoS One, 2013, 8, e72882 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5mb00412h |
| This journal is © The Royal Society of Chemistry 2016 |